

Abstract
Identifying Cu-exchanged zeolites able to activate C–H bonds and selectively convert methane to methanol is a challenge in the field of biomimetic heterogeneous catalysis. Recent experiments point to the importance of trinuclear [Cu3(μ-O)3]2+ complexes inside the micropores of mordenite (MOR) zeolite for selective oxo-functionalization of methane. The electronic structures of these species, namely, the oxidation state of Cu ions and the reactive character of the oxygen centers, are not yet fully understood. In this study, we performed a detailed analysis of the electronic structure of the [Cu3(μ-O)3]2+ site using multiconfigurational wave-function-based methods and density functional theory. The calculations reveal that all Cu sites in the cluster are predominantly present in the Cu(II) formal oxidation state with a minor contribution from Cu(III), whereas two out of three oxygen anions possess a radical character. These electronic properties, along with the high accessibility of the out-of-plane oxygen center, make this oxygen the preferred site for the homolytic C–H activation of methane by [Cu3(μ-O)3]2+. These new insights aid in the construction of a theoretical framework for the design of novel catalysts for oxyfunctionalization of natural gas and suggest further spectroscopic examination.
Introduction
Natural gas is considered the main source of conventional energy for the future due to recent technological advances in the excavation techniques and the lower environmental impact of its utilization compared to other fossil fuels.1 The primary component of natural gas is methane. Its low price and large availability have motivated scientists and engineers to explore new routes for the efficient conversion of methane to liquid fuels and chemicals.2 The selective catalytic oxyfunctionalization of methane to methanol is considered one of the most viable conversion routes.1,3,4 Methanol is a reactive and easily condensable chemical that can serve both as the energy carrier and as a versatile platform chemical, and it thus plays a central role in future chemical and energy technologies.5 An important advantage of methanol over other chemicals is the ease of integration into the existing chemistry infrastructure and the possibility to derive it from a wide range of potential feedstocks. The selective activation of methane and its direct conversion to methanol represent an important challenge for catalysis and chemical engineering. The established technology to obtain methanol is only profitable at a very large scale. It involves first the steam reforming of methane over a nickel catalyst to produce syngas, which is then converted to methanol over a Cu/ZnO/Al2O3 catalyst in a separate process. The overall conversion scheme is optimized for the large-scale operation under high temperature and pressure conditions.6 The availability of a cheaper and more flexible process at smaller scale not only would allow developing relatively small remote natural gas fields such as shale gas and clathrate reserves but would also constitute the game changer in the chemical industry as it would enable the production of a wide range of chemicals and fuels.
Methane monoxygenases (MMOs) are a family of enzymes that nature employs for selective oxidation of the inert C–H bond of methane.7 The MMOs are sorted in two different categories based on their position in the cell. The cytoplasmic, soluble MMOs (sMMOs) contain a bis(μ-oxo)diiron core,8 while the membrane-bound MMOs (particulate MMOs or pMMOs) oxidize the C–H bond over a Cu-based active site. Different handling conditions in the purification of the pMMOs resulted in different Cu concentrations giving rise to a substantial uncertainty about the nature of the Cu-containing active site. Whereas dimeric Cu complexes have been postulated as the active centers by Rosenzweig et al.,9−11 an alternative hypothesis of the trinuclear speciation of the reactive Cu sites is advocated by Chan et al.12
Biomimetic catalysis is a promising approach to the design of functional materials, but it also provides new insights into how enzymes work. Nanoporous inorganic matrices such as zeolites and metal–organic frameworks (MOFs) provide well-defined confinement space and coordination environments suitable for the stabilization of highly reactive intermediates often regarded as enzymatic analogues. Cu-containing reactive species have been the synthetic target for homogeneous13,14 and heterogeneous15−25 catalysis. Cationic Cu complexes stabilized in zeolite micropores have been shown to be efficient catalysts for the selective oxyfunctionalization of methane.1 However, the exact structure of the catalytic sites in such materials is still not fully understood.15,26−35 Originally, a binuclear bis-oxo [Cu(μ-O)2Cu]2+ ion was proposed as the active site in Cu-ZSM-5 zeolite.16 In later studies, Woertink et al. employed Raman spectroscopy to characterize the working Cu-ZSM-5 catalyst and suggested the mono-(μ-O) dicopper site [Cu(μ-O)Cu]2+ to be the active complex, which does not have analogues in biological or organometallic systems.35 Similar dicopper sites were proposed for Cu-SSZ-13 and Cu-SSZ-39 zeolites based on synchrotron X-ray diffraction, in situ UV–vis, Raman spectroscopy, and theory.36 Recent theoretical studies addressed the stability and catalytic performance of mono-CuOH,37 dicopper,38 and polynuclear CuxOy (x = 1–5) sites39 as possible catalytic sites responsible for methane oxidation. An alternative structural proposal suggesting the trinuclear active site formulation [Cu3(μ-O)3]2+ (Figure 1) has been put forward by Grudner et al. on the basis of a combined experimental and computational study on well-defined Cu-exchanged mordenite catalysts.21,40,41 The active site is the ring-type [Cu3(μ-O)3]2+ extraframework cluster, in which the positive charge is balanced by two Al sites of the zeolite framework. A similar active site has been proposed for pMMO,13 and it has also been isolated in a molecular complex.42,43 The importance of the trinuclear structure for the activity of Cu-ZSM-5 catalysts has also been recently discussed.44
Figure 1.
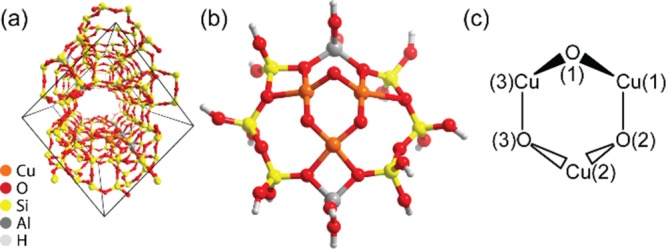
(a) Optimized periodic structure of Cu/MOR containing a [Cu3(μ-O)3]2+ cationic cluster in the S = 1/2 spin state and (b) the respective cluster with (c) the schematic representation and atom numbering within the trinuclear copper-oxo core.
Understanding the relationships between the electronic structure, geometry, and reactivity of such multinuclear clusters is essential not only to explain the mechanism of selective oxidation of methane by zeolites but also to design new and improved catalyst systems. The key challenge here is that the electronic configuration of the [Cu3(μ-O)3]2+ cluster as well as other potential multinuclear Cu-oxo complexes cannot be readily rationalized based on two-electron Lewis models. The presence of at least three paramagnetic centers and of covalent bonds may give rise to a multiconfigurational system, whose description needs more than one (“multiple”) electronic configuration. Most computational studies on Cu–zeolite systems reported up to date have been performed using single-determinant density functional theory (DFT) that has intrinsic limitations for multiconfigurational systems.45 The multiconfigurational character of the Cu-oxo clusters that may be responsible for metal-to-ligand charge transfer was claimed to be crucial for their ability to selectively activate methane.21 The evidence of such effects further comes from X-ray absorption spectroscopy, indicating the predominance of Cu(II) in the active materials, whereas the formal oxidation state of Cu in the trimer is Cu(III).
In this study, we employed a high-level ab initio methodology based on multiconfigurational wave function theory to obtain detailed information about the ground spin state of the [Cu3(μ-O)3]2+ cluster, the partial oxidation state of each Cu center, and the oxo/oxyl character of the three (μ-O) atoms. High-level quantum chemical calculations have previously been employed to investigate Cu-exchanged zeolites46 and Fe-oxo sites in zeolitic materials4,47 and elucidate their complex electronic structure. The results of the multiconfigurational calculations are compared with those obtained with a range of popular density functionals providing thus a basis for calibrating the choice of suitable methods for applied computational catalysis studies on related systems. An important finding is that the substantial anisotropy activity of the (μ-O) centers correlates with both their accessibility and their radical character.
Computational Details
Periodic DFT Calculations
All spin-polarized periodic DFT (spin-polarized pDFT) calculations were performed using the VASP software with a generalized gradient-approximated PBE exchange-correlation functional.48,49 The projected-augmented wave method and a plane-wave basis set with a cutoff of 400 eV were employed.50 Brillouin zone sampling was restricted to the Γ point. A supercell of all-silica MOR, constructed by doubling of the monoclinic primitive cell along the c axis with lattice parameters of a = b = 13.648, c = 15.015 Å, and γ = 97.2° as optimized by DFT, was used as the initial model (Figure 1(a)).51,52 To compensate for the positive charge of the extra-framework cationic Cu complexes, two framework Si4+ ions in the MOR supercell were substituted by two Al3+ at the side-pocket positions AlISP. The other two [AlO2]− units at the remote side-pocket positions (AlIISP) were charge compensated by two Brønsted acid sites.53 The resulting MOR model had a Si/Al ratio of 11. The climbing image nudged elastic band method54 was used to determine the minimum energy path and locate the transition state for the homolytic C–H activation step resulting in a hydroxylated Cu site and a CH3 radical species.40,44 The maximum energy geometry along the reaction path obtained by the climbing image nudged elastic band method was further optimized using a quasi-Newton algorithm, as implemented in VASP. In this step, only the extra-framework atoms were relaxed. Spin-polarized calculations were performed throughout this study. Vibrational frequencies for optimized structures of reaction intermediates and transition states were calculated using the finite-difference method, as implemented in VASP. Small displacements (0.02 Å) were used to estimate the numerical Hessian matrix. The transition states were confirmed by the presence of a single imaginary frequency corresponding to the reaction path.
Multiconfigurational Calculations
For all multiconfigurational calculations a cluster model of the [Cu3(μ-O)3]2+ core stabilized inside the mordenite zeolite micropores was constructed from the periodic DFT optimized structure with spin S = 1/2. The cluster was saturated with hydrogen atoms (Figure 1b). Cu(1), O(2), Cu(3), and O(3) (Figure 1c) are located on the same plane; Cu(2) is out-of-plane and closer to the framework of the MOR zeolite; and O(1) is located also out-of-plane and points to the main channel of MOR. All calculations were performed without any symmetry restrictions (point group C1). The structure reported in Figure 1b was used in the multiconfigurational calculations to elucidate the electronic effects of the [Cu3(μ-O)3]2+ core on the methane functionalization, while it was not employed to model the oxidation reaction.
The complete active space self-consistent field (CASSCF) and restricted active space self-consistent field (RASSCF) methods55,56 and their extension through second-order perturbation theory (CASPT257 and RASPT2,58 respectively) were employed for the cluster model calculations. In CASSCF, a multiconfigurational wave function is considered by performing a full configuration interaction (full CI) expansion within a limited orbital space. The notation CAS(n,m) is followed for the description of the active spaces, where n is the number of electrons and m the number of orbitals considered for the CI expansion. In the RASSCF method, three individual orbital subspaces are considered, RAS1, RAS2, and RAS3, and a CI expansion is formed by applying restrictions between the three RAS spaces. A full CI is performed in RAS2, similarly to CAS. RAS1 includes doubly occupied orbitals, and a maximum number of holes is allowed; RAS3 includes unoccupied orbitals, and a maximum number of particles (electrons) is allowed. Since only two holes and two particles are considered in this work for RAS1 and RAS3, respectively, a simple RAS(n,m) notation is used, similar to the CAS notation.
Apart from state-specific (SS) CAS/RAS calculations, multiple states of the same spin multiplicity were examined by the state-averaged (SA) CASSCF/RASSCF methods. For the SA variants, the multistate (MS)-CASPT2/RASPT2 extensions were applied to include dynamical electron correlation. The nomenclature followed is SA(n)-CASSCF/RASSCF and MS(n)-CASPT2/RASPT2, where n is the number of the optimized states.
For the choice of a proper active space, a formal charge of 2+ for each Cu was initially assumed, corresponding to a formal 3d9 electronic configuration per Cu. Based on this assumption, and in order to have a total charge of 2+ for the [Cu3(μ-O)3]2+ core, one O2– and two O•– atoms are expected. The energetically most stable states were first explored using a minimal active space of size (5,5), namely, the bonding (σ)/antibonding (σ*) combinations of the 3dx2-y2 orbitals of two Cu atoms with the 2p of two neighboring O atoms, respectively, and a singly occupied 3dx2-y2 atomic orbital of the third Cu atom.
After a systematic examination of active spaces of various sizes (see Supporting Information), we concluded that a CAS(11,11) and a RAS(19,21) are adequate to describe the low-lying S = 1/2, 3/2, and 5/2 states of the [Cu3(μ-O)3]2+ model system. CAS(11,11) includes the five key orbitals of CAS(5,5), the three 2p orbitals of O(3), and the three 3p orbitals of O(3). RAS(19,21) includes all 2p/3p orbitals of the three O atoms and the 3dx2-y2 orbitals of the three Cu atoms. The correlated orbital space of RASSCF(19,21) was organized as such: RAS2 includes the (5,5) minimal space described above. RAS1 includes the remaining seven 2p orbitals of the three O atoms, which do not participate in the bonding/antibonding combinations with the 3dx2-y2 orbitals of Cu atoms. RAS3 includes the nine unoccupied 3p orbitals of the three O atoms. All configurations that involve up to two electron excitations from RAS1 to RAS2 and RAS3, and from RAS1 and RAS2 to RAS3 are considered in the CI expansion. It was found that the doubly occupied 3d atomic orbitals of the three Cu atoms do not participate in the bonding between the Cu and the (μ-O) atoms. Therefore, they were not further considered in this study.
The triple-ζ quality atomic natural orbital basis set59,60 (ANO-RCC-VTZP) was used for the three Cu and three O atoms of the [Cu3(μ-O)3]2+ core, the double-ζ basis set analogue (ANO-RCC-VDZP) for the six O atoms of the zeolite framework coordinated to the three Cu atoms, and a minimal basis (ANO-RCC-MB) for all remaining atoms (Si, Al, O, and H). In all calculations, scalar relativistic effects were included using the all-electron relativistic basis sets of ANO-RCC type and a second-order Douglas–Kroll–Hess Hamiltonian.61,62 A shifted zeroth-order Hamiltonian63 (IPEA shift) with the default value of 0.25 au and an imaginary shift64 of 0.2 au were applied to all CASPT2/RASPT2 calculations. The two-electron integral evaluation was simplified by using the Cholesky decomposition technique.65 All CAS/RAS calculations were performed using the MOLCAS 8.0 program package.66
Results and Discussion
Electronic Structure of [Cu3(μ-O)3]2+
Table 1 reports the energy differences among the three lowest states, namely, the doublet (S = 1/2), quartet (S = 3/2), and sextet (S = 5/2) states, calculated at the CASSCF/CASPT2(11,11) and RASSCF/RASPT2(19,21) levels of theory. The multiconfigurational SCF methods (CASSCF and RASSCF) are used to generate a multideterminant zeroth-order wave function and provide only qualitative energy differences between different spin states since they lack dynamical correlation. This type of correlation energy is recovered by second-order perturbation theory (PT2). Therefore, in this section we analyze the nature of the CASSCF/RASSCF wave function, but we discuss only the energy differences obtained at the CASPT2/RASPT2 levels.
Table 1. Relative Energy Differences (in kJ/mol) from the CASSCF/CASPT2(11,11) and RASSCF/RASPT2(19,21) Calculations.
| CASSCF(11,11)a | RASSCF(19,21)a | CASPT2(11,11) | RASPT2(19,21) | |
|---|---|---|---|---|
| sextet | 22.6 | 57.1 | 78.1 | 108.6 |
| quartet | 6.2 | 13.1 | 20.7 | 22.1 |
| doublet | 0 | 0 | 0 | 0 |
The weights of the dominant configurations demonstrate the multiconfigurational character of the doublet state, and they are 46%, 25%, 15%, and 8% (CASSCF(11,11)) and 53%, 18%, 14%, and 5% (RASSCF(19,21)).
The doublet state is the lowest in energy, followed by the quartet, 20.7 kJ/mol higher, and by the sextet, 78.1 kJ/mol higher, at the CASPT2(11,11) level of theory. These energy differences are in good agreement with the values obtained from PBE (21.1 and 67.8 kJ/mol, respectively) and BLYP67,68 functionals (22.7 and 71.2 kJ/mol, respectively). The results obtained with different popular density functionals are summarized in the Supporting Information. The RASPT2(19,21) doublet/quartet energy difference (22.1 kJ/mol) is in excellent agreement with the CASPT2(11,11) result (20.7 kJ/mol), while the doublet/sextet difference is predicted to be slightly higher by RASPT2(19,21), 108.6 kJ/mol, than by CASPT2(11,11), 78.1 kJ/mol.
The five molecular orbitals (MOs) of the [Cu3(μ-O)3]2+ cluster with occupation numbers between 0.04 and 1.96 are shown in Figure 2, as they were obtained from the RASSCF(19,21) calculation. While they are singly occupied in the sextet state, they are responsible for the multiconfigurational character of the doublet and quartet states. RASSCF(19,21) shows that Cu(3) has a pure d9 electronic configuration, while Cu(1) and Cu(2) form bonding (σ)/antibonding (σ*) combinations with the 2p orbitals of O(1) and O(3), respectively. The O(1) and O(3) atoms have a partially occupied 2p atomic orbital and some O•– (oxyl) character. On the other hand, O(2) has the three 2p orbitals doubly occupied (O2– character). Similar orbitals were obtained in the CASSCF(11,11) calculation (see Supporting Information).
Figure 2.
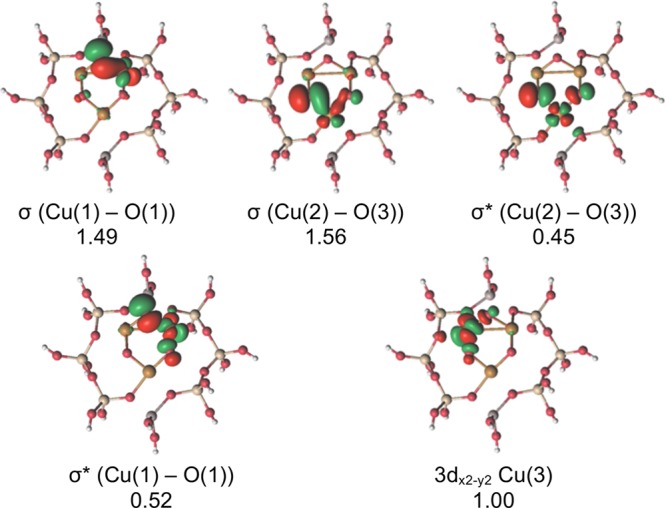
Five most relevant MOs of the doublet state of the [Cu3(μ-O)3]2+ cluster, as calculated from the RASSCF(19,21) level of theory. A description and their corresponding occupation numbers are given below each MO. The atom labels are as on Figure 1c.
The first five low-lying doublet states were examined using SA(5)-RASSCF(19,21)/MS(5)-RASPT2(19,21) level of theory (for the choice of these five states, see Supporting Information). For the first three states, one MO is characterized by a single 3d orbital of only one Cu center: Cu(3) for state 1, Cu(1) for state 2, and Cu(2) for state 3, respectively. The other four MOs are bonding (σ)/antibonding (σ*) combinations between the remaining two Cu centers and the O(1) and O(3) atoms (Cu(1)–O(1)/Cu(2)–O(3) for state 1, Cu(3)–O(1)/Cu(2)–O(3) for state 2, and Cu(1)–O(1)/Cu(3)–O(3) for state 3). The corresponding five MOs of states 4 and 5 are combinations of one 3d orbital of the three Cu centers and a 2p atomic orbital of O(1) and O(3). The occupation numbers of these MOs are close to one.
In the three most stable doublet states, all three Cu centers are equivalent and have an overall 3d9 atomic configuration characteristic for the +2 (Cu(II)) oxidation state. The O(2) has a closed-shell (2p6) electronic configuration. In other words, O(2) has an O2– character, whereas the O(1) and O(3) centers “share” an electron and a neighboring Cu resulting in σ/σ* MO combinations. We can assign to these O atoms a 2p5 open-shell electronic configuration corresponding to an oxyl (O•–) radical. A similar interpretation can be given by considering the lowest two states (states 1 and 2) as near-degenerate states, since they differ by less than 18 kJ/mol (0.2 eV, Table S2). The configuration of the two states differs by one σ/σ* pair (Cu(1)–O(1) for state 1, Cu(3)–O(1) for state 2). A transition between these two states would involve the spontaneous formation of O2– for O(1) and the oxidation of either Cu(1) or Cu(3) to Cu3+, as shown in Figure 3. On the other hand, Cu(2) is accessible in this “resonance” mechanism only through O(3) since O(2) was found to have constantly an oxo character (O2–). Therefore, state 3 is higher in energy (35.1 kJ/mol or 0.4 eV at the MS(5)-RASPT2 level). The nature of the oxygen centers is crucial for understanding the fundamental factors that control the reactivity of the intrazeolite Cu sites. Given the importance of the electrophilicity of the reactive oxygen centers for C–H activation,69−71 one expects that these two O atoms would provide low-energy paths for C–H activation. However, if the reactivity is dominated by the basicity of the proton-accepting site, the C–H cleavage should be more facile over the electron-rich closed-shell O(2) center.
Figure 3.
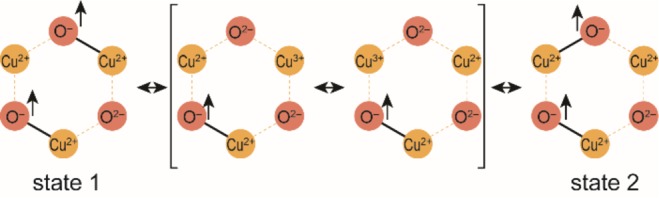
Proposed resonance mechanism between states 1 and 2. The arrows represent the radical character of the O•– atoms.
The above analysis is based only on one conformer of the [Cu3(μ-O)3]2+ cluster with an asymmetric geometry, in which only Cu(3) is closer to the walls of the MOR zeolite: Cu(3)–O(MOR-1) and Cu(3)–O(MOR-2) bond distances are 2.105 and 2.044 Å, respectively (Table 2). (The labels MOR-1 and MOR-2 correspond to the two different oxygen atoms of the zeolite coordinated with the Cu centers.) Similarly, the out-of-plane Cu(2) points to the zeolitic framework and is located 1.955 and 2.016 Å from the two oxygen atoms of the framework. Cu(1) lies significantly further from the zeolite since the bond distance between Cu(1) and one of the oxygen atoms of the framework is 2.628 Å, about 0.5 Å longer than the equivalent distance for Cu(3). On the other hand, we expect an equal probability of the cluster conformers that are located closer to MOR, from either the Cu(1) or the Cu(3) site due to the symmetric nature of the MOR pore and the [Cu3(μ-O)3]2+ ring cluster. In other words, there should be an isostructural and isoenergetic conformer of the [Cu3(μ-O)3]2+ site where Cu(1) is closer to the walls of the zeolite, while Cu(3) is further from the oxygen atoms of the zeolite (about 2.6 Å). For this conformer the electronic configurations of O(2) and O(3) are interchanged, with O(2) having a radical, oxyl-character, and O(3) a closed-shell O2– character. The two in-plane oxygens (O(2) and O(3)) are thus equivalent, and they resonate between O•– and O2–. Therefore, their reactivity toward homolytic C–H cleavage should be at most comparable but most likely less significant than O(1), which provides the optimal accessibility for the reacting alkane molecules.
Table 2. Bond Distances (in Å) between the Three Cu Centers and the Oxygen Atoms of the Mordenite Zeolite from the Structure Optimized by Periodic DFT (Doublet Spin State) and Used for the Multiconfigurational Calculations.
| Cu(1) | Cu(2) | Cu(3) | |
|---|---|---|---|
| O(MOR-1) | 1.987 | 1.955 | 2.105 |
| O(MOR-2) | 2.628 | 2.016 | 2.044 |
Active Site Anisotropy for Homolytic C–H Cleavage
The above considerations suggest that the oxidation of methane is more facile over the more electronegative O(1) center which forms a stronger O–H bond. In order to verify this hypothesis, we calculated the C–H activation barrier (electronic energies) over three different oxygen sites of the Cu trimer stabilized in the MOR model by means of periodic DFT calculations (Figure 4) to capture all effects from the full pore of the zeolite. An activation barrier of 37 kJ/mol was obtained for the hydrogen transfer from methane to O(1), resulting in the formation of a hydroxo species and a methyl radical confined in the main channel of mordenite. The same process is energetically less favorable for O(2) and O(3); the corresponding barriers from periodic DFT are 78 and 74 kJ/mol, respectively. These results indicate that O(1) is catalytically more active for methane C–H bond activation than the other two O sites, which is consistent with our conclusions drawn by multiconfigurational analysis. Notice that the anisotropy will be substantially altered after the activation of the first methane molecule and consumption of the O(1) site as this would result in the formation of a partially reduced [Cu3O2]2+ with electronic structure substantially different from the original active site.
Figure 4.

(a) Relative electronic energies of the C–H activation barriers (in kJ/mol, S = 1/2) from the three oxygen atoms of the [Cu3(μ-O)3]2+ cluster confined in MOR zeolite. (b) Optimized local geometry of the transition state of the C–H activation for O(1).
We propose that this reactivity difference can only manifest itself within a continuous catalytic process. The catalyst will be exposed to methane feed at elevated temperature commonly employed within the sequential process schemes21,72 with separate methane activation/zeolite regeneration. This exposure will level the intrinsic reactivity differences of the various O sites within the trinuclear clusters and other potentially reactive intrazeolite Cu complexes.
Our calculations provide a convincing evidence of the importance of the radical anion character of the reactive O sites in the zeolite-stabilized extraframework Cu-oxo clusters for the activation of the C–H bonds in methane. Although we have explored only one type of active sites,73 which can be selectively generated in MOR zeolite,21 the theoretical insights emerging from this study allow rationalizing reactivity trends predicted for other types of Cu-oxo complexes. Moreover, they provide guidelines for active site design to enable a low-temperature catalytic process for methane oxofunctionalization. Previous DFT studies37,39 showed that the activity of Cu complexes in C–H bond activation of methane follows the sequence CuOH+ < [CuO2Cu]2+ < [CuOCu]2+ < [Cu3O3]2+ ≈ [Cu4Ox]2+ ≈ [Cu5Ox]2+. This trend confirms our finding that the initial reactivity increase with larger nuclearity is associated with the number of spin domains in the cluster. This is directly connected to the radical character of the ligand oxygens. The effect of spin delocalization and, accordingly, reactivity enhancement level off for clusters containing more than three copper sites. We expect that further increase of the cluster size would result in a complete localization of the spin density on the Cu centers, as observed in bulk CuO.
Conclusions and Outlook
The electronic structure of the [Cu3(μ-O)3]2+ complex deposited inside the pores of the MOR zeolite was studied by means of multiconfigurational wave function methods. The [Cu3(μ-O)3]2+ site can activate strong C–H bonds and oxidize methane to methanol, a catalytic process with great industrial interest due to the large deposits of methane. Understanding the underlying details that drive this reaction will allow us to optimize catalytic systems with enhanced performance for the functionalization of C–H bonds.
The analysis of the low-lying energy states confirms the presence of mixed valence Cu(II)/Cu(III) oxidation states of the three Cu cations. For the geometrical conformer that was studied here, Cu(2) has a more pure Cu(II) character, while Cu(1) and Cu(3) have a mixed Cu(II/III) composition. The two oxygen atoms with an oxyl character, O(1) and O(3), are expected to activate the C–H bond of methane. Despite the fact that two oxygen atoms have both radical character, O(1) is expected to be catalytically more active than O(3) because of the lower symmetry of the structural conformer used in our high-level calculations. Cu(2) and Cu(3) are closer to the zeolitic walls than Cu(1), an effect that perturbs the electronic structure of the cluster. Due to the higher symmetry of the [Cu3(μ-O)3]2+ site, the in-plane O(2) and O(3) are expected to be equivalent, and only O(1) is expected to be more catalytically active. These conclusions were verified by periodic DFT. The optimized transition state geometries adopt the proper symmetry and equalize the two in-plane O atoms. This suggestion was reflected on the calculated C–H activation barriers: the out-of-plane O(1) was found about 30 kJ/mol lower than the respective barriers from the in-plane oxygen atoms. These conclusions can be further examined by EPR spectroscopy on the activated catalyst. This technique has been already successfully applied in mono-Cu sites deposited on zeolites46,74,75 and can provide further insight on the electronic structure of the cupper cluster. Future experimental examination of the reaction with operando spectroscopy can provide further insight into the evolution of the catalyst and help us to accurately model the next reaction steps and better understand the activation of the second methane molecule.
Acknowledgments
K.D.V. would like to acknowledge the University of Tennessee for financial support of this work (start-up grant). G.L. acknowledges financial support from NWO for her personal VENI grant (no. 016.Veni.172.034) and NWO-SurfSARA for providing access to supercomputer resources. E.J.M.H. and E.A.P. acknowledge MCEC, a Gravitation programme of The Netherlands Organization for Scientific Research (NWO) funded by the Ministry of Education, Culture and Science of the government of The Netherlands. E.A.P thanks the Ministry of Education and Science of Russian Federation (GosZadanie no. 11.1706.2017PP). K.D.V. and L.G. thank the Inorganometallic Catalyst Design Center, an EFRC funded by the DOE, Office of Basic Energy Sciences (DE-SC0012702). The authors acknowledge the Minnesota Supercomputing Institute (MSI) at the University of Minnesota and the Newton High-Performance Computing program at the University of Tennessee for providing resources that contributed to the research results reported within this paper.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcc.7b08714.
Evaluation of the computational details, comparison of different density functionals, Cartesian coordinates, and cif files from periodic DFT (PDF)
Author Present Address
∇ (E.A.P.) Inorganic Systems Engineering group, Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
The authors declare no competing financial interest.
Supplementary Material
References
- Saha D.; Grappe H. A.; Chakraborty A.; Orkoulas G. Postextraction Separation, On-Board Storage, and Catalytic Conversion of Methane in Natural Gas: A Review. Chem. Rev. 2016, 116, 11436–11499. 10.1021/acs.chemrev.5b00745. [DOI] [PubMed] [Google Scholar]
- Lin R.; Amrute A. P.; Pérez-Ramírez J. Halogen-Mediated Conversion of Hydrocarbons to Commodities. Chem. Rev. 2017, 117, 4182–4247. 10.1021/acs.chemrev.6b00551. [DOI] [PubMed] [Google Scholar]
- Olivos-Suarez A. I.; Szécsényi À.; Hensen E. J. M.; Ruiz-Martinez J.; Pidko E. A.; Gascon J. Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities. ACS Catal. 2016, 6, 2965–2981. 10.1021/acscatal.6b00428. [DOI] [Google Scholar]
- Snyder B. E. R.; Vanelderen P.; Bols M. L.; Hallaert S. D.; Böttger L. H.; Ungur L.; Pierloot K.; Schoonheydt R. A.; Sels B. F.; Solomon E. I. The Active Site of Low-Temperature Methane Hydroxylation in Iron-Containing Zeolites. Nature 2016, 536, 317–321. 10.1038/nature19059. [DOI] [PubMed] [Google Scholar]
- Olah G. A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem., Int. Ed. 2005, 44, 2636–2639. 10.1002/anie.200462121. [DOI] [PubMed] [Google Scholar]
- Guo Z.; Liu B.; Zhang Q.; Deng W.; Wang Y.; Yang Y. Recent Advances in Heterogeneous Selective Oxidation Catalysis for Sustainable Chemistry. Chem. Soc. Rev. 2014, 43, 3480–3524. 10.1039/c3cs60282f. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Heppner D. E.; Johnston E. M.; Ginsbach J. W.; Cirera J.; Qayyum M.; Kieber-Emmons M. T.; Kjaergaard C. H.; Hadt R. G.; Tian L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig A. C.; Frederick C. A.; Lippard S. J.; Nordlund P. Crystal Structure of a Bacterial Non-Haem Iron Hydroxylase that Catalyses the Biological Oxidation of Methane. Nature 1993, 366, 537–543. 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- Lieberman R. L.; Rosenzweig A. C. Crystal Structure of a Membrane-Bound Metalloenzyme that Catalyses the Biological Oxidation of Methane. Nature 2005, 434, 177–182. 10.1038/nature03311. [DOI] [PubMed] [Google Scholar]
- Balasubramanian R.; Smith S. M.; Rawat S.; Yatsunyk L. A.; Stemmler T. L.; Rosenzweig A. C. Oxidation of Methane by a Biological Dicopper Centre. Nature 2010, 465, 115–119. 10.1038/nature08992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himes R. A.; Barnese K.; Karlin K. D. One is Lonely and Three is a Crowd: Two Coppers Are for Methane Oxidation. Angew. Chem., Int. Ed. 2010, 49, 6714–6716. 10.1002/anie.201003403. [DOI] [PubMed] [Google Scholar]
- Chan S. I.; Chen K. H.-C.; Yu S. S.-F.; Chen C.-L.; Kuo S. S.-J. Toward Delineating the Structure and Function of the Particulate Methane Monooxygenase from Methanotrophic Bacteria. Biochemistry 2004, 43, 4421–4430. 10.1021/bi0497603. [DOI] [PubMed] [Google Scholar]
- Chan S. I.; Wang V. C.-C.; Lai J. C.-H.; Yu S. S.-F.; Chen P. P.-Y.; Chen K. H.-C.; Chen C.-L.; Chan M. K. Redox Potentiometry Studies of Particulate Methane Monooxygenase: Support for a Trinuclear Copper Cluster Active Site. Angew. Chem., Int. Ed. 2007, 46, 1992–1994. 10.1002/anie.200604647. [DOI] [PubMed] [Google Scholar]
- Nagababu P.; Maji S.; Kumar M. P.; Chen P. P.-Y.; Yu S. S.-F.; Chan S. I. Efficient Room-Temperature Oxidation of Hydrocarbons Mediated by Tricopper Cluster Complexes with Different Ligands. Adv. Synth. Catal. 2012, 354, 3275–3282. 10.1002/adsc.201200474. [DOI] [Google Scholar]
- Smeets P. J.; Groothaert M. H.; Schoonheydt R. A. Cu Based Zeolites: A UV–vis Study of the Active Site in the Selective Methane Oxidation at Low Temperatures. Catal. Today 2005, 110, 303–309. 10.1016/j.cattod.2005.09.028. [DOI] [Google Scholar]
- Groothaert M. H.; Smeets P. J.; Sels B. F.; Jacobs P. A.; Schoonheydt R. A. Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. 10.1021/ja047158u. [DOI] [PubMed] [Google Scholar]
- Vanelderen P.; Hadt R. G.; Smeets P. J.; Solomon E. I.; Schoonheydt R. A.; Sels B. F. Cu-ZSM-5: A Biomimetic Inorganic Model for Methane Oxidation. J. Catal. 2011, 284, 157–164. 10.1016/j.jcat.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alayon E. M.; Nachtegaal M.; Ranocchiari M.; van Bokhoven J. A. Catalytic Conversion of Methane to Methanol over Cu–Mordenite. Chem. Commun. 2012, 48, 404–406. 10.1039/C1CC15840F. [DOI] [PubMed] [Google Scholar]
- Alayon E. M. C.; Nachtegaal M.; Kleymenov E.; van Bokhoven J. A. Determination of the Electronic and Geometric Structure of Cu Sites During Methane Conversion over Cu-MOR with X-ray Absorption Spectroscopy. Microporous Mesoporous Mater. 2013, 166, 131–136. 10.1016/j.micromeso.2012.04.054. [DOI] [Google Scholar]
- Vanelderen P.; Vancauwenbergh J.; Tsai M.-L.; Hadt R. G.; Solomon E. I.; Schoonheydt R. A.; Sels B. F. Spectroscopy and Redox Chemistry of Copper in Mordenite. ChemPhysChem 2014, 15, 91–99. 10.1002/cphc.201300730. [DOI] [PubMed] [Google Scholar]
- Grundner S.; Markovits M. A. C.; Li G.; Tromp M.; Pidko E. A.; Hensen E. J. M.; Jentys A.; Sanchez-Sanchez M.; Lercher J. A. Single-Site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol. Nat. Commun. 2015, 6, 7546. 10.1038/ncomms8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narsimhan K.; Iyoki K.; Dinh K.; Román-Leshkov Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016, 2, 424–429. 10.1021/acscentsci.6b00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le H. V.; Parishan S.; Sagaltchik A.; Göbel C.; Schlesiger C.; Malzer W.; Trunschke A.; Schomäcker R.; Thomas A. Solid-State Ion-Exchanged Cu/Mordenite Catalysts for the Direct Conversion of Methane to Methanol. ACS Catal. 2017, 7, 1403–1412. 10.1021/acscatal.6b02372. [DOI] [Google Scholar]
- Tomkins P.; Ranocchiari M.; van Bokhoven J. A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond. Acc. Chem. Res. 2017, 50, 418–425. 10.1021/acs.accounts.6b00534. [DOI] [PubMed] [Google Scholar]
- Latimer A. A.; Kulkarni A. R.; Aljama H.; Montoya J. H.; Yoo J. S.; Tsai C.; Abild-Pedersen F.; Studt F.; Nørskov J. K. Understanding Trends in C–H Bond Activation in Heterogeneous Catalysis. Nat. Mater. 2017, 16, 225–229. 10.1038/nmat4760. [DOI] [PubMed] [Google Scholar]
- Groothaert M. H.; van Bokhoven J. A.; Battiston A. A.; Weckhuysen B. M.; Schoonheydt R. A. Bis(μ-oxo)dicopper in Cu-ZSM-5 and Its Role in the Decomposition of NO: A Combined in Situ XAFS, UV–Vis–Near-IR, and Kinetic Study. J. Am. Chem. Soc. 2003, 125, 7629–7640. 10.1021/ja029684w. [DOI] [PubMed] [Google Scholar]
- Groothaert M. H.; Pierloot K.; Delabie A.; Schoonheydt R. A. Identification of Cu(II) Coordination Structures in Cu-ZSM-5, Based on a DFT/ab initio Assignment of the EPR Spectra. Phys. Chem. Chem. Phys. 2003, 5, 2135–2144. 10.1039/b301120h. [DOI] [Google Scholar]
- Beznis N. V.; Weckhuysen B. M.; Bitter J. H. Cu-ZSM-5 Zeolites for the Formation of Methanol from Methane and Oxygen: Probing the Active Sites and Spectator Species. Catal. Lett. 2010, 128, 14–22. 10.1007/s10562-010-0380-6. [DOI] [Google Scholar]
- Vanelderen P.; Vancauwenbergh J.; Sels B. F.; Schoonheydt R. A. Coordination Chemistry and Reactivity of Copper in Zeolites. Coord. Chem. Rev. 2013, 257, 483–494. 10.1016/j.ccr.2012.07.008. [DOI] [Google Scholar]
- Vanelderen P.; Snyder B. E. R.; Tsai M.-L.; Hadt R. G.; Vancauwenbergh J.; Coussens O.; Schoonheydt R. A.; Sels B. F.; Solomon E. I. Spectroscopic Definition of the Copper Active Sites in Mordenite: Selective Methane Oxidation. J. Am. Chem. Soc. 2015, 137, 6383–6392. 10.1021/jacs.5b02817. [DOI] [PubMed] [Google Scholar]
- Itadani A.; Sogawa Y.; Oda A.; Ohkubo T.; Yumura T.; Kobayashi H.; Sato M.; Kuroda Y. Possibility of Copper-Ion-Exchanged MFI-Type Zeolite as C–H Bond Activation Material for Propane and the Driving Force for Activation. J. Phys. Chem. C 2015, 119, 21483–21496. 10.1021/acs.jpcc.5b05577. [DOI] [Google Scholar]
- Itadani A.; Yumura T.; Ohkubo T.; Kobayashi H.; Kuroda Y. Existence of Dual Species Composed of Cu+ in CuMFI Being Bridged by C2H2. Phys. Chem. Chem. Phys. 2010, 12, 6455–6465. 10.1039/c000967a. [DOI] [PubMed] [Google Scholar]
- Alayon E. M. C.; Nachtegaal M.; Bodi A.; van Bokhoven J. A. Reaction Conditions of Methane-to-Methanol Conversion Affect the Structure of Active Copper Sites. ACS Catal. 2014, 4, 16–22. 10.1021/cs400713c. [DOI] [Google Scholar]
- Hammond C.; Forde M. M.; Rahim M. H. A.; Thetford A.; He Q.; Jenkins R. L.; Dimitratos N.; Lopez-Sanchez J. A.; Dummer N. F.; Murphy D. M.; et al. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by using Copper-Promoted Fe-ZSM-5. Angew. Chem., Int. Ed. 2012, 51, 5129–5133. 10.1002/anie.201108706. [DOI] [PubMed] [Google Scholar]
- Woertink J. S.; Smeets P. J.; Groothaert M. H.; Vance M. A.; Sels B. F.; Schoonheydt R. A.; Solomon E. I. A [Cu2O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18908–18913. 10.1073/pnas.0910461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipek B.; Wulfers M. J.; Kim H.; Göltl F.; Hermans I.; Smith J. P.; Booksh K. S.; Brown C. M.; Lobo R. F. Formation of [Cu2O2]2+ and [Cu2O]2+ toward C–H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39. ACS Catal. 2017, 7, 4291–4303. 10.1021/acscatal.6b03005. [DOI] [Google Scholar]
- Kulkarni A. R.; Zhao Z.-J.; Siahrostami S.; Nørskov J. K.; Studt F. Monocopper Active Site for Partial Methane Oxidation in Cu-Exchanged 8MR Zeolites. ACS Catal. 2016, 6, 6531–6536. 10.1021/acscatal.6b01895. [DOI] [Google Scholar]
- Mahyuddin M. H.; Staykov A.; Shiota Y.; Miyanishi M.; Yoshizawa K. Roles of Zeolite Confinement and Cu–O–Cu Angle on the Direct Conversion of Methane to Methanol by [Cu2(μ-O)]2+-Exchanged AEI, CHA, AFX, and MFI Zeolites. ACS Catal. 2017, 7, 3741–3751. 10.1021/acscatal.7b00588. [DOI] [Google Scholar]
- Palagin D.; Knorpp A. J.; Pinar A. B.; Ranocchiari M.; van Bokhoven J. A. Assessing the Relative Stability of Copper Oxide Clusters as Active Sites of a CuMOR Zeolite for Methane to Methanol Conversion: Size Matters?. Nanoscale 2017, 9, 1144–1153. 10.1039/C6NR07723D. [DOI] [PubMed] [Google Scholar]
- Grundner S.; Luo W.; Sanchez-Sanchez M.; Lercher J. A. Synthesis of Single-Site Copper Catalysts for Methane Partial Oxidation. Chem. Commun. 2016, 52, 2553–2556. 10.1039/C5CC08371K. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Kim T. Y.; Lee H.; Yi J. Distinct Activation of Cu-MOR for Direct Oxidation of Methane to Methanol. Chem. Commun. 2017, 53, 4116–4119. 10.1039/C7CC00467B. [DOI] [PubMed] [Google Scholar]
- Chen P. P.-Y.; Yang R. B.-G.; Lee J. C.-M.; Chan S. I. Facile O-atom Insertion into C-C and C-H Bonds by a Trinuclear Copper Complex Designed to Harness a Singlet Oxene. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14570–14575. 10.1073/pnas.0707119104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P. P.-Y.; Nagababu P.; Yu S. S.-F.; Chan S. I. Development of the Tricopper Cluster as a Catalyst for the Efficient Conversion of Methane into MeOH. ChemCatChem 2014, 6, 429–437. 10.1002/cctc.201300473. [DOI] [Google Scholar]
- Li G.; Vassilev P.; Sanchez-Sanchez M.; Lercher J. A.; Hensen E. J. M.; Pidko E. A. Stability and Reactivity of Copper Oxo-Clusters in ZSM-5 Zeolite for Selective Methane Oxidation to Methanol. J. Catal. 2016, 388, 305–312. 10.1016/j.jcat.2016.03.014. [DOI] [Google Scholar]
- Ghosh A.; Taylor P. R. High-Level ab initio Calculations on the Energetics of Low-Lying Spin States of Biologically Relevant Transition Metal Complexes: A First Progress Report. Curr. Opin. Chem. Biol. 2003, 7, 113–124. 10.1016/S1367-5931(02)00023-6. [DOI] [PubMed] [Google Scholar]
- Pierloot K.; Delabie A.; Groothaert M. H.; Schoonheydt R. A. A Reinterpretation of the EPR Spectra of Cu(II) in Zeolites A, Y and ZK4, Based on ab initio Cluster Model Calculations. Phys. Chem. Chem. Phys. 2001, 3, 2174–2183. 10.1039/b100531f. [DOI] [Google Scholar]
- Göltl F.; Michel C.; Andrikopoulos P. C.; Love A. M.; Hafner J.; Hermans I.; Sautet P. Computationally Exploring Confinement Effects in the Methane-to- Methanol Conversion Over Iron-Oxo Centers in Zeolites. ACS Catal. 2016, 6, 8404–8409. 10.1021/acscatal.6b02640. [DOI] [Google Scholar]
- Kresse G.; Hafner J. Ab initio Molecular Dynamics for Open-Shell Transition Metals. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 48, 13115–13118. 10.1103/PhysRevB.48.13115. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Blöchl P. E. Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50, 17953–17978. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- van Koningsveld H.; Jansen J. C.; van Bekkum H. The Monoclinic Framework Structure of Zeolite H-ZSM-5. Comparison with the Orthorhombic Framework of as-Synthesized ZSM-5. Zeolites 1990, 10, 235–242. 10.1016/0144-2449(94)90134-1. [DOI] [Google Scholar]
- Pidko E. A.; van Santen R. A.; Hensen E. J. M. Multinuclear Gallium-Oxide Cations in High-Silica Zeolites. Phys. Chem. Chem. Phys. 2009, 11, 2893–2902. 10.1039/b815943b. [DOI] [PubMed] [Google Scholar]
- Pidko E. A.; van Santen R. A. Structure–Reactivity Relationship for Catalytic Activity of Gallium Oxide and Sulfide Clusters in Zeolite. J. Phys. Chem. C 2009, 113, 4246–4249. 10.1021/jp8113607. [DOI] [Google Scholar]
- Henkelman G.; Uberuaga B. P.; Jónsson H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. 10.1063/1.1329672. [DOI] [Google Scholar]
- Olsen J.; Roos B. O.; Jørgensen P.; Jensen H. J. A. Determinant Based Configuration Interaction Algorithms for Complete and Restricted Configuration Interaction Spaces. J. Chem. Phys. 1988, 89, 2185. 10.1063/1.455063. [DOI] [Google Scholar]
- Malmqvist P. Å.; Rendell A.; Roos B. O. The Restricted Active Space Self-Consistent-Field Method, Implemented with a Split Graph Unitary Group Approach. J. Phys. Chem. 1990, 94, 5477–5482. 10.1021/j100377a011. [DOI] [Google Scholar]
- Andersson K.; Malmqvist P.-Å.; Roos B. O. Second-Order Perturbation Theory with a Complete Active Space Self-Consistent Field Reference Function. J. Chem. Phys. 1992, 96, 1218. 10.1063/1.462209. [DOI] [Google Scholar]
- Malmqvist P. Å.; Pierloot K.; Shahi A. R. M.; Cramer C. J.; Gagliardi L. The Restricted Active Space Followed by Second-Order Perturbation Theory Method: Theory and Application to the Study of CuO2 and Cu2O2 systems. J. Chem. Phys. 2008, 128, 204109. 10.1063/1.2920188. [DOI] [PubMed] [Google Scholar]
- Roos B. O.; Lindh R.; Malmqvist P.-Å.; Veryazov V.; Widmark P.-O. Main Group Atoms and Dimers Studied with a New Relativistic ANO Basis Set. J. Phys. Chem. A 2004, 108, 2851–2858. 10.1021/jp031064+. [DOI] [Google Scholar]
- Roos B. O.; Lindh R.; Malmqvist P.-Å.; Veryazov V.; Widmark P.-O. New Relativistic ANO Basis Sets for Transition Metal Atoms. J. Phys. Chem. A 2005, 109, 6575–6579. 10.1021/jp0581126. [DOI] [PubMed] [Google Scholar]
- Douglas M.; Kroll N. M. Quantum Electrodynamical Corrections to the Fine Structure of Helium. Ann. Phys. 1974, 82, 89–155. 10.1016/0003-4916(74)90333-9. [DOI] [Google Scholar]
- Hess B. A. Relativistic Electronic-Structure Calculations Employing a Two-Component No-Pair Formalism with External-Field Projection Operators. Phys. Rev. A: At., Mol., Opt. Phys. 1986, 33, 3742. 10.1103/PhysRevA.33.3742. [DOI] [PubMed] [Google Scholar]
- Ghigo G.; Roos B. O.; Malmqvist P.-Å. A Modified Definition of the Zeroth-Order Hamiltonian in Multiconfigurational Perturbation Theory (CASPT2). Chem. Phys. Lett. 2004, 396, 142–149. 10.1016/j.cplett.2004.08.032. [DOI] [Google Scholar]
- Forsberg N.; Malmqvist P. Å. Multiconfiguration Perturbation Theory with Imaginary Level Shift. Chem. Phys. Lett. 1997, 274, 196–204. 10.1016/S0009-2614(97)00669-6. [DOI] [Google Scholar]
- Aquilante F.; Lindh R.; Pedersen T. B. Unbiased Auxiliary Basis Sets for Accurate Two-Electron Integral Approximations. J. Chem. Phys. 2007, 127, 114107. 10.1063/1.2777146. [DOI] [PubMed] [Google Scholar]
- Aquilante F.; Autschbach J.; Carlson R. K.; Chibotaru L. F.; Delcey M. G.; Vico L. D.; Galván I. F.; Ferré N.; Frutos L. M.; Gagliardi L.; et al. Molcas 8: New Capabilities for Multiconfigurational Quantum Chemical Calculations Across the Periodic Table. J. Comput. Chem. 2016, 37, 506–541. 10.1002/jcc.24221. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Rosa A.; Ricciardi G.; Baerends E. J. Is [FeO]2+ the Active Center Also in Iron Containing Zeolites? A Density Functional Theory Study of Methane Hydroxylation Catalysis by Fe-ZSM-5 Zeolite. Inorg. Chem. 2010, 49, 3866–3880. 10.1021/ic1000073. [DOI] [PubMed] [Google Scholar]
- Kazaryan A.; Baerends E. J. Ligand Field Effects and the High Spin–High Reactivity Correlation in the H Abstraction by Non-Heme Iron(IV)–Oxo Complexes: A DFT Frontier Orbital Perspective. ACS Catal. 2015, 5, 1475–1488. 10.1021/cs501721y. [DOI] [Google Scholar]
- Verma P.; Vogiatzis K. D.; Planas N.; Borycz J.; Xiao D. J.; Long J. R.; Gagliardi L.; Truhlar D. G. Mechanism of Oxidation of Ethane to Ethanol at Iron(IV)–Oxo Sites in Magnesium-Diluted Fe2(dobdc). J. Am. Chem. Soc. 2015, 137, 5770–5781. 10.1021/jacs.5b00382. [DOI] [PubMed] [Google Scholar]
- Sushkevich V. L.; Palagin D.; Ranocchiari M.; Bokhoven J. A. v. Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol. Science 2017, 356, 523–527. 10.1126/science.aam9035. [DOI] [PubMed] [Google Scholar]
- Pidko E. A. Toward the Balance between the Reductionist and Systems Approaches in Computational Catalysis: Model versus Method Accuracy for the Description of Catalytic Systems. ACS Catal. 2017, 7, 4230–4234. 10.1021/acscatal.7b00290. [DOI] [Google Scholar]
- Baute D.; Arieli D.; Neese F.; Zimmermann H.; Weckhuysen B. M.; Goldfarb D. Carboxylate Binding in Copper Histidine Complexes in Solution and in Zeolite Y: X- and W-band Pulsed EPR/ENDOR Combined with DFT Calculations. J. Am. Chem. Soc. 2004, 126, 11733–11745. 10.1021/ja047761c. [DOI] [PubMed] [Google Scholar]
- Moreno-González M.; Blasco T.; Góra-Marek K.; Palomares A. E.; Corma A. Study of Propane Oxidation on Cu-Zeolite Catalysts by in-situ EPR and IR Spectroscopies. Catal. Today 2014, 227, 123–129. 10.1016/j.cattod.2013.10.055. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.