

Abstract
Background:
Obesity/hyperlipidemia is closely related to both atrial and ventricular arrhythmias. CaMKII, a multifunctional serine/threonine kinase, has been involved in cardiac arrhythmias of different etiologies. However, its role in obesity/hyperlipidemia-related cardiac arrhythmia is unexplored. The aim of this was to determine the involvement of CaMKII in the process.
Methods:
Adult male APOE−/− mice were fed a high-fat diet (HFD), administrated with KN93 (10 mg·kg−1·2d−1), a specific inhibitor of CaMKII. Serum lipid and glucose profile, cardiac function, and surface electrocardiogram were determined. Electrophysiological study and epicardial activation mapping were performed in Langendorff-perfused heart. Expression of cardiac ion channels, gap junction proteins, Ca2+ handling proteins, and CaMKII were evaluated, coupled with histological analysis.
Results:
A hyperlipidemia condition was induced by HFD in the APOE−/− mice, which was associated with increased expression and activity of CaMKII in the hearts. In Langendorff-perfused hearts, HFD-induced heart showed increased arrhythmia inducibility, prolonged action potential duration, and decreased action potential duration alternans thresholds, coupled with slow ventricular conduction, connexin-43 upregulation, and interstitial fibrosis. Downregulation of ion channels including Cav1.2 and Kv4.2/Kv4.3 and disturbed Ca2+ handling proteins were also observed in HFD-induced heart. Interestingly, all these alterations were significantly inhibited by KN93 treatment.
Conclusion:
Our results demonstrated an adverse effect of metabolic components on cardiac electrophysiology and implicated an important role of CaMKII underlying this process.
Key Words: CaMKII, high-fat diet, hyperlipidemia, arrhythmias, Ca2+ handling
INTRODUCTION
Obesity has been demonstrated to be an independent risk factor for cardiac arrhythmia, which has been established by both clinical investigation and experimental animal studies using mice fed a high-fat diet (HFD).1–3 Increased atrial and ventricular arrhythmia (VA) susceptibility, sympathetic hyperinnervation, aberrant pattern of gap junctional protein expression, prolonged action potential duration (APD), longer QTc intervals, increased repolarization dispersion, and increased Ica+ have been observed in the heart of mice with a HFD.4,5 Furthermore, in vitro experimental study also showed that isolated adipocytes and free fatty acids can directly modulate the electrophysiological properties and ion currents causing highly arrhythmogenesis in left atrial myocytes and ventricular cardiomyocytes.6,7 Taken together, these data from both clinical and experimental studies provide strong evidence that obesity or high lipid component has the capability to promote electrical remodeling and the pathogenesis of cardiac arrhythmia.8 Despite evidence for the arrhythmogenic action of obesity or lipid, the underlying molecular mechanism of obesity/hyperlipid-related arrhythmias are still poorly understood.
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a multifunctional serine/threonine kinase that is abundant expressed in various tissues including the heart. Recently, increasing studies have implicated an important role of elevated CaMKII signaling pathways in the detrimental remodeling of ion channels and in the genesis of cardiac arrhythmias under stressed conditions, such as myocardial hypertrophy, ischemia, heart failure, diabetes mellitus.9 And CaMKII has been implicated in atrial fibrillation, sinus node dysfunction, ventricular tachyarrhythmias in structural heart diseases and inherited tachyarrhythmias,10 suggesting a common CaMKII signal in cardiac arrhythmogenesis. CaMKII can target a large number of substrates in the cell, including ion channels, pumps, Ca2+ cycling proteins, and transcription factors (reviewed in Ref. 11–14), thus regulating cardiac electrophysiology and structure rebuilding. Thus, CaMKII has emerged as a highly validated molecular mechanism with the potential to connect “upstream” pro-arrhythmic factors with “downstream” responses such as ion channel hyperactivity, defective intracellular Ca2+ homeostasis, and scar formation that promote arrhythmias. However, the involvement of CaMKII under obesity/hyperlipidemia-related arrhythmias was still unknown.
In this study, we aimed to determine the involvement of CaMKII in cardiac arrhythmogenesis under obesity/hyperlipidemia conditions. Using APOE−/− mouse model of mice fed a HFD, administrated with KN93, a specific CaMKII inhibitor, we found that HFD-induced heart exhibited increased cardiac susceptibility to arrhythmia induction and electrical remodeling evidenced by prolonged APD90, coupled with downregulation of ions channels, slowed conduction velocity (CV), and cardiac fibrosis. In addition, increased CaMKII activation and disturbed Ca2+ handling proteins were observed in the HFD-induced heart. Interestingly, all these abnormalities were significantly attenuated by KN93 treatment. In conclusion, our results suggested an important role of CaMKII in mediating obesity/hyperlipidemia-related cardiac arrhythmias.
MATERIALS AND METHODS
Animal Studies
Male APOE−/− mice on a C57BL/6 background weighting 18–22 g were used in this study. The animals were purchased from HFK Bio-Technology Company (Bejing, China). All the animal experiment protocols were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University and were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. A total of 36 mice were housed in an environmentally controlled room at 22°C ± 2.0°C and 50% ± 5% humidity with a 12-h:12-h light/dark cycle and fed food and water ad libitum. Twelve mice were fed standard animal chow and served as control group (Ctrl). The remaining 24 mice were fed with a HFD (45% fat; Beijing HFK Bio-Technology, Bejing, China) for 8 weeks. After 8 weeks of feeding, HFD-fed mice were further divided into 2 groups: HFD group (n = 12) and HFD plus KN93-treated group (n = 12). KN93 was given daily by peritoneal injection at the dose of 10 mg·kg−1·2d−1 for 8 weeks. Mice in the Ctrl and HFD groups were gavaged with vehicle only. At the end of the experiment, blood samples were collected in anaesthetized mice and centrifuged at 4°C for 10 minutes for collecting serum after an over-night fast.
Echocardiography and Histological Analysis
Echocardiography was performed to evaluate cardiac function at the end of animal experiment. In brief, echocardiography was performed under continuous anesthesia with 1.5%–2% isoflurane using a Mylab30CV (ESAOTE) ultrasound system with a 15-Mz probe. M-mode tracings derived from the short axis of the LV at the level of the papillary muscles were recorded. Fractional shortening (%) and left ventricular ejection fraction were determined. Heart was fixed in 4% paraformaldehyde solution, embedded in paraffin, and sectioned at 5 μm. After dehydration, sections were stained with Masson trichrome staining and sirius red, respectively. The stained sections were then viewed under a light microscope (×200 amplification; Nikon, Japan).
Surface Electrocardiograms
Mice were anaesthetized using isoflurane inhalation (0.8%–1.0% volume in oxygen) and efficacy of the anesthesia was monitored by watching breathing speed and tail suspension. Three lead surface electrocardiograms (ECGs) were recorded from subcutaneous 23-gauge needle electrodes attached to each limb using the Powerlab acquisition system (AD Instruments). Lead II was analyzed for heart rate (RR interval) and PR, QRS, and QT duration using Chart 5 Pro analysis software (AD Instruments). QT intervals in mice were corrected for heart rate using the following formula: QTc = QT/(RR/100)1/2 (RR in millisecond).
Preparation of Langendorff-perfused Hearts, Monophasic Action Potential Recordings, and Ventricular and Atrial Arrhythmia Inducibility
Hearts were quickly isolated, excised, and transferred to ice-cold (4°C) HEPES-buffered Tyrode's solution (mM: NaCl 130; KCl 5.4; CaCl2 1.8; MgCl2 1; Na2HPO4 0.3; HEPES 10; glucose 10; pH adjusted to 7.4 with NaOH), bubbled with 95% O2–5% CO2. Hearts were then rapidly cannulated with tailor-made 21-gauge cannula in aorta and perfused with 37°C HEPES-buffered Tyrode's solution at 2–2.5 mL/min using a Langendorff-perfusion system (AD Instruments, Australia). Each heart was perfused for 20 minutes before electrophysiology tests, and a pseudoelectrogram was monitored. Hearts that did not recover to the regular spontaneous rhythm or had irreversible myocardial ischemia were discarded.15 A pair of platinum-stimulating electrodes were positioned on the basal surface of the right ventricle to deliver regular pacing and a custom-made Ag–AgCl electrode consisting of two 0.25-mm, Teflon-coated, silver wires was located at LV to record the monophasic action potential (MAP). The pacing cycle length (PCL) ranged from 150 to 30 ms, with successive 10 ms decreases. The pacing at each PCL lasts for 30 seconds, followed by a 30-second resting period to avoid pacing memory. The 90% action potential duration (APD90) was defined as the average 90% repolarization time for at least 6 successive MAPs. The APD alternans (ALT) was determined by 2 consecutive beats whose APD90 differed by at least 5% over 10 beats. The APD ALT threshold was defined as the maximal PCL (PCLmax) that induced an APD ALT. Burst pacing (2 ms pulses at 50 Hz, 2 seconds burst duration), which was repeated for 20 times separated by 2-second intervals, was used to induce VA. VA was defined as continuous rapid ventricular contractions of 2 seconds or more.16 Inducibility of atrial arrhythmias (AAs) was tested using the burst-pacing protocol described by Verheule et al.17 Burst pacing was applied in the right atrial locations, and MAP was recorded from left atrial epicardium. AAs were defined as a period of rapid atrial rhythm lasting for at least 2 seconds. The episodes of AA or VA were analyzed and quantified. All the electrophysiological studies were performed and analyzed with randomization and blinding.
Multiple Electrode Array Recording
To monitor excitation spread, multiple electrode arrays (MEAs; Multi Channel Systems, Reutlingen, Germany) were used for field potential recordings, MEA consisted of 60 electrodes arranged in 8 × 8 matrix each with a diameter of 30 μm and an interelectrode distance of 200 μm. MEA was placed against the epicardium of left ventricular apex. Experiments were conducted at 37°C and data acquisition and analysis was performed using Cardio 2D and Cardio 2D+ software (Multi Channel Systems, Reutlingen, Germany), respectively. The raw data were recorded in 16-bit resolution at a sampling rate of 10 kHz. The activation mapping and CV of the excitation transmission within the MEA registration area were recorded.
Western Blot Analysis
The common procedure for western blot analysis was described in a previous publication.18 The density cof the immunoreactive bands was analyzed using ImageJ software (NIH, Bethesda, MD). In the western analysis, β-actin was used as a loading control. Primary antibodies used in this study are as follows: CaMKII (santa Cruze Biotech, SC-9035), p-CaMKII (T287, Thermo Fisher Scientific, PA5-37833), RyR2 (Millipore, AB9080), p-RyR2 (S2808, Abcam, ab59225), p-RyR2 (S2814, badrilla, A010-31), PLB (Cell signaling Technology, 8495), p-PLB (Thr17, Badrilla, A010-13), SERCA2 (Abcam, ab150435), Kv4.2 (Sigma, SAB5200070), Kv4.3 (Sigma, SAB5200076), Cav1.2 (Thermo Fisher Scientific, PA5-23015), CX43 (Abcam, ab11370), and β-actin (Boster, BM0627).
Statistical Analysis
Data were presented as mean ± SEM. The statistical significance of differences between groups was obtained by analysis of variance multiple comparisons in GraphPad Pro5.0 (GraphPad, San Diego, CA). Differences were considered significant at P < 0.05.
RESULTS
Characterization of the HFD-induced APOE−/− Mice
We used the HFD-fed APOE−/− mouse model to investigate whether pharmacological inhibition of CaMKII with KN93 exhibits benefits in the cardiac electrophysiology under obesity/hyperlipidemia condition. As many studies use KN93 at 5 mg·kg−1·d−1 by intraperitoneal injection,19–21 and some reports use an even higher dose at 12.5 mg/kg,22 to reduce stress induced by repetitive intraperitoneal injection in mice, we used KN93 at 10 mg·kg−1·2d−1 and our preliminary experiments shows that this dosage did not elicit apparent effects on the body weight decrease (data not shown). At the end of the experiments, mice were processed by echocardiography analysis, and serum lipid indices and glucose levels were measured. As shown in Table 1, the serum free fatty acids, triglyceride, total cholesterol, and glucose levels were all significantly increased in HFD-fed mice. Interestingly, administration with KN93 improved all the metabolic indicators in HFD-fed mice. In addition, echocardiography analysis showed no apparent difference in cardiac ejection fraction and fractional shortening between control, HFD, and HFD + KN93 groups, suggesting that HFD for 16 weeks had no apparent effects on the heart function. We also performed surface ECG recording in anaesthetized mice and found no statistically significant difference in heart rate (RR interval), QRS duration, PR interval, QT, and QTc interval between Ctrl, HFD, and HFD + KN93 groups (Fig. 1).
TABLE 1.
The Summary Data for Serum Lipid and Glucose Profiles, and Echocardiography Analysis at the End of the Experiments in APOE−/− Mice Fed a HFD
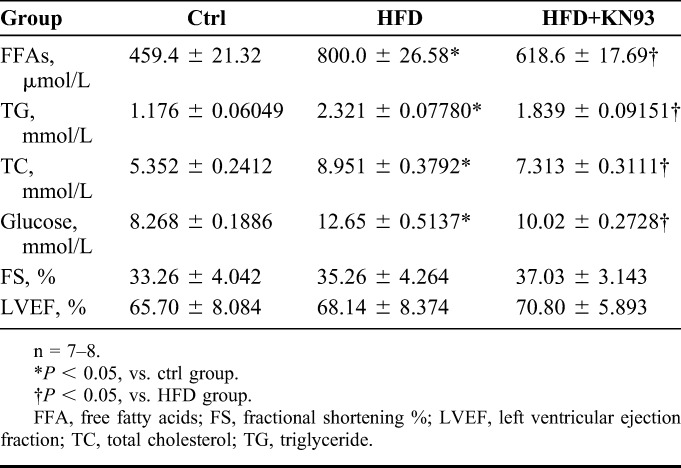
FIGURE 1.

The characteristics of surface ECG recording in the HFD-fed mice model. Representative ECG recordings (A) and the comparison of ECG parameters (B) under anesthesia at 16th weeks. n = 7–8.
KN93 Administration Inhibited Prolongation of APD90, Increased the Threshold of APD ALT and Decreased the Susceptibility to Arrhythmia Induction in HFD-induced Heart
Then Langendorff-perfused hearts were used to characterize the changes in the electrophysiological parameters (APD90, threshold of APD ALT, incidence of VA and AA). APD90 was measured under the conditions in which Langendorff-perfused hearts were administrated a programmed electrical stimulation with a different PCL. In HFD-induced heart, APD90 was significantly prolonged, but it became much shorter in the HFD + KN93 group (Figs. 2A, B). APD ALT started at a significantly slower pacing rate in hearts with HFD than in the control heart, whereas this was markedly inhibited in HFD + KN93 group (Figs. 2C, D). We then tested the cardiac susceptibility to arrhythmia inducibility. Burst stimulation was applied to both ventricle and atria, MAP were recorded at the corresponding sites. Signals from ventricular activity and atrial activity were identified by the deflection present on the pseudo-ECG monitored simultaneously, as shown in the upside images of Figures 2E, F. The summary data for arrhythmia episodes in ventricle and atria in each mouse induced by a series of bust stimulation are shown below. Our results showed that both VA episodes and AA episodes were all significantly increased in HFD-induced heart, whereas KN93 treatment decreased these changes. These results suggested an increased arrhythmic susceptibility in HFD-induced heart, which can be inhibited by CaMKII inhibition.
FIGURE 2.

KN93 administration improved electrophysiological properties of the heart in HFD-fed mice. A–B, Representative action potential figures and statistical analysis of 90% APDs at different a PCL in Langendorff-perfused isolated heart (n = 7). C–D, Representative electric ALT figures and statistical analysis of the ALT thresholds (n = 6). E–F, Representative images of MAP recording and arrhythmia induction in ventricle and atria by burst-pacing stimulation. The summary data for VA episodes and AA episodes were showed below (n = 8). *P < 0.05.
KN93 Administration Inhibited the Decline in Ventricular CV, CX43 Upregulation, and Cardiac Fibrosis in HFD-induced Heart
We performed ex vivo epicardial activation mapping of the left ventricle in Langendorff-perfused heart by MEA recording to analyze local conduction velocities. As shown in Figure 3A, the excitation spread quickly in the MEA registration area in the heart of ctrl group with a CV at about 80 cm/s. Compared with ctrl mice, significant slower CV in left ventricle could be registered in HFD-induced heart, which was remarkably corrected by KN93 treatment (Fig. 3A). We further analyzed ventricular CX43 expression, which plays an important role in electrical conduction. As shown from Figure 3B, compared with ctrl group, HFD-induced heart presented significantly upregulated level of CX43 protein level. However, the increased expression of Cx43 was apparently inhibited by treatment with KN93 (Fig. 3B). We also evaluated cardiac fibrosis, which can also impact on heart conduction.23 Masson trichrome and sirius red staining showed increased interstitial fibrosis in both ventricle and atria from HFD-fed mice and treatment with KN93 significantly attenuated this phonotype (Figs. 3C, D). In addition, we also observed apparent lipid accumulation in the atria of HFD group as evidenced by the existence of lipid droplets in various sizes, which were rarely seen in the atrial of HFD + KN93 group (Fig. 3D).
FIGURE 3.

KN93 administration improved the ventricular CV, CX43 upregulation, and cardiac fibrosis in HFD-induced hearts. A, Left ventricle epicardium activation mapping and ventricular CV analyzed by MEA in Langendorff-perfused isolated hearts. Activation starts with red and propagates towards blue. The summary data for CV is shown. B, Western blot analysis of Cx43 protein level in the heart tissue. C–D, Representative images of masson trichrome staining and sirius red staining of ventricle and atria, the summary data for interstitial fibrosis area were shown. n = 5. *P < 0.05.
KN93 Administration-corrected Downregulation of Cav1.2 and Kv4.2/Kv4.3 in HFD-induced Heart
To clarify whether there is a combination of altered channel expression changes underling the prolongation of APD, we assessed the protein expression levels of ion channels. Western blot showed that the protein levels of Cav1.2, Kv4.2, and Kv4.3 were markedly reduced in HFD-induced heart, compared with control group, and the reduced channels expression were significantly attenuated in HFD + KN93 group (Fig. 4).
FIGURE 4.
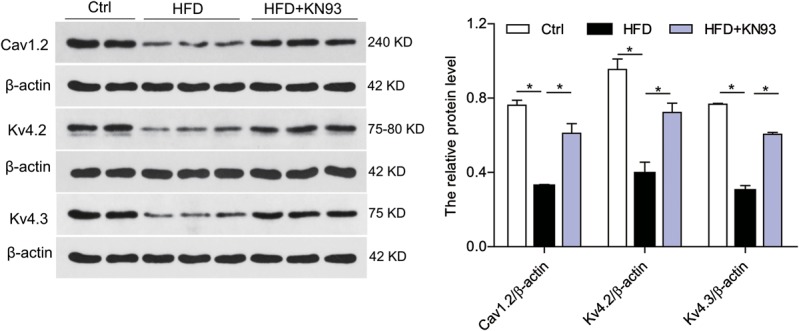
KN93 administration attenuated the downregulation of Cav1.2 and Kv4.2/Kv4.3 in HFD-induced heart. Western blot analysis of protein level of Cav1.2, Kv4.2 and Kv4.3 in the heart tissues. n = 5. *P < 0.05.
KN93 Administration Inhibited CaMKII Activation, Improved the Disturbance of Ca2+ Handling Proteins in HFD-induced Heart
To determine whether CaMKII was activated in the heart tissues of HFD-fed mice, we analyzed the protein level of phosphorylated-CaMKII (p-CaMKII, at site T287) and total CaMKII in the heart. As shown in Figure 5A, both p-CaMKII and total CaMKII were significantly increased in HFD-induced heart but normalized in HFD + KN93 group, suggesting that CaMKII was activated in the heart under hyperlipidemia condition. Alteration of CaMKII expression and activation state could mediate CaMKII-dependent phosphorylation in ryanodine receptor 2 gene (RyR2), which encodes a cardiac sarcoplasmic reticulum Ca2+ release channel, and phosphorylated RyR2 will increase its opening probability. We then test the activation state of RyR2. RyR2 contains multiple phosphorylation sites including RyR2-Ser2814 (phosphorylated by CaMKII) and RyR2-Ser2808 (which may be phosphorylated by both CaMKII and PKA). We used 2 phospho-specific antibodies (for RyR2-S2814 and RyR2-S2808) in the western blots. In the HFD-induced heart versus control heart homogenates, RyR2 phosphorylation was increased at both Ser2814 and Ser2808, suggesting an increased Ca2+ release property of RyR2 (Fig. 5B). However, all these alterations were significantly attenuated by KN93 treatment (Fig. 5B). In addition, we also examined the total protein level of RyR2 in these groups and no difference was observed. We then evaluated the other 2 CaMKII targets, SERCA2 (sarcoplasmic reticulum Ca2+-ATPase 2), which transports calcium into the sarcoplasmic reticulum and PLB (phospholamban), an inhibitor of SERCA2, which reversibly inhibits cardiac SERCA2 activity through intramembrane interactions. Phosphorylation of PLN by CaMKII at Thr17 site will relieve its inhibition on SERCA2 and consequently increase SERCA2 calcium uptake activity. As shown in Figure 5C, HFD-induced heart showed decreased protein level of phosphorylated PLB (p-PLB, at Thr17 site) and total SERCA2 without any changes in total PLB expression, suggesting a decreased SR Ca2+-ATPase function in ventricular myocytes in HFD model. However, all these dysregulated Ca2+ handling proteins were markedly attenuated by KN93 treatment (Fig. 5C).
FIGURE 5.

KN93 administration reversed CaMKII activation and the disturbed Ca2+ handling proteins in HFD-induced heart. A, Western blot analysis of phosphorylated CaMKII (p-CaMKII) at T287 site and the total protein CaMKII level in the heart tissue. B, Western blot analysis of phosphorylated RyR2 (p-RyR2) at S2807 and S2814 site, and the total protein level of RyR2 in the heart tissue. C, Western blot analysis of phosphorylated phospholamban (p-PLB) at T17 site and the total protein level of SERCA2 and PLB in the heart tissues. n = 5. *P < 0.05.
DISCUSSION
Obesity is associated with metabolic syndromes and is linked to an increased likelihood of atrial and VAs and sudden cardiac death.6,24,25 Despite the increasing prevalence of obesity, we have a limited understanding of the contributions of metabolic abnormalities to the pathogenesis of cardiac arrhythmias and SCD. Recently, increasing experimental studies using HFD-induced obese or hyperlipidemia animal model have indicated several potential mechanisms contributing to cardiac electrical abnormalities, including dysregulated ionic channels expression, prolonged APD, sympathetic hyperinnervation, and aberrant pattern of gap junctional expression and regulation, and decreased CV in the heart.3,4,26–29 Consistent with previous studies, this study also observed increased atrial and ventricular susceptibility to arrhythmia induction, which was associated with prolonged APD, decreased threshold of APD ALT and slow conduction in the heart of mice with a hyperlipidemia condition (Fig. 2 and Table 1). In addition, our study further found that increased activation and expression of CaMKII was observed in HFD-induced heart, coupled with depressed protein levels of Cav1.2, K4.2/Kv4.3 and abnormal Ca2+ handling protein (Figs. 4, 5). Importantly, inhibition of CaMKII with KN93 remarkably improved these abnormalities in HFD-induced heart. Collectively, these results revealed an important role of CaMKII in mediating cardiac arrhythmias under obesity/hyperlipidemia conditions.
In this study, prolonged APD was observed in the HFD-induced heart, although this did not reflect on the surface ECG recording data as the QT and QTc showed no apparent changes between control and HFD group (Figs. 1, 2). This discrepancy may be raised from the different sensibility of the 2 detecting methods. APD prolongation may result from either an increased inward ionic flow or a decreased outward current, or a combination of both. Herein, we found that HFD-induced heart showed decreased protein level of Kv4.2/4.3 (Fig. 4), the potassium channels (Ito) that contribute to the phase 1 repolarization of AP both in atrial and ventricles.30,31 Thus, the reduced potassium channel protein level may cause prolonged repolarization and APD. Supporting our data, there is compelling evidence that intracellular lipid content can regulate potassium channels. In transgenic model of lipotoxic cardiomyopathy such as PPARγ or PPARα cardiac-overexpression mouse and MHC-FATP mouse, which has increased cardiomyocyte lipid content, reduced Kv expression and current have been demonstrated.29 These results suggested a decreased potassium channel function in the heart of obesity/hyperlipidemia conditions. In addition, decreased expression of Cav1.2, the pore-forming subunit of L-type calcium current (ICaL) present in all cardiomyocytes that contribute to phase 2 depolarization of AP, has also been observed in HFD-induced heart (Fig. 4), which is consistent with a previous study in the obese Zucker rat model (OZR).32 Interestingly, although the author observed decreased cardiac Cav1.2 expression in OZR, ICaL in OZR exhibited impaired gating properties, leading to increased Ca2+ influx.32 The reason for the decreased Cav1.2 level and conversely increased ICaL may be that not all channels expressed in the cell function at the same time. Collectively, these data suggested increased ICaL and decreased Ito may work together to contribute to the prolongation of APD in the heart under hyperlipidemia condition. However, inhibition of CaMKII with KN93 markedly improved the decline in these channel expressions and corrected the prolongation of APD, suggesting a role of CaMKII activation in regulating the channel turnover under hyperlipidemia conditions. In fact, activation of CaMKII has been reported to cause increased L-type Ca2+-current or impaired K+-channel function and thus prolong repolarization.33,34 Therefore, these data revealed a critical role of CaMKII activation in remodeling ion channel function under obesity/hyperlipidemia conditions.
In the HFD-induced heart, we observed a disturbed Ca2+ handling phenotype shaped by increased Ca2+-release evidenced by increased phosphorylation of Ca2+-release channel-RyR2 (both at S2808 and S2814 site), and decreased Ca2+ uptake as shown by decreased expression of Ca2+ pump-SERCA2, coupled with decreased activity of PLB, inhibitor of SERCA2, promoting intracellular (Ca2+) increasing (Fig. 5). Hyperphosphorylation of RyR2 can lead to a strong increase of spontaneous elementary Ca2+-release events from SR (“Ca2+-leak”), especially during diastole.35,36 Coupled with the decreased SERCA2 function, the diastolic (Ca2+) may further increase in the cytosol, which can activate the electrogenic NCX (Na+-Ca2+ exchanger), to produce an arrhythmogenic depolarizing current, causing delayed afterdepolarizations (DADs), which are a known trigger of atrial and VAs.37,38 These results indicate an arrhythmogenic role for cardiac abnormal calcium homestasis under hyperlipidemia. Phosphorylation of PLB at T17 site is a specific target of CaMKII and activated CaMKII is generally associated with increased phosphorylation of PLB at T17 site.39 However, we found that p-PLB (at T17 site) is significantly decreased in the HFD-induced heart, where activated CaMKII was observed, although no apparent change was in total PLB abundance (Fig. 5). In line with our data, Ellena et al also observed reduced protein expression of phosphorylated PLB at T17 site in rats on the HFD and sucrose diet compared with lean rats.40 Thus, increased occurrence of dephosphorylation of PLB may be involved in this process, which may need further investigation. Interestingly, all these alterations were corrected by KN93 treatment (Fig. 5). Collectively, these results suggested a critical role of Ca2+ handling abnormalities and its regulation by CaMKII in promoting cardiac arrhythmogenesis under obesity/hyperlipidemia conditions.
In this study, decreased ventricular CV was found in the HFD-induced heart, which was associated with increased expression of CX43 and cardiac fibrosis (Fig. 3). Similarly, decreased CV in atria has also been reported in HFD-fed mice.28 CX43, the predominant ventricular gap junction protein, is critical for maintaining normal cardiac electrical conduction. Dynamic changes of CX43 expression have been reported during the progression from compensated cardiac hypertrophy to heart failure with upregulated CX43 in compensated hypertrophy and diminished CX43 in decompensated hypertrophy.41 Consistent with this concept, in our mouse model, 16-week HFD feeding did not impair cardiac function but induced cardiac remodeling (cardiac fibrosis), coupled with CX43 upregulation in the heart, which is consistent with a previous study using mice fed HFD for 8 weeks.4 However, in 34-week HFD feeding, apparent cardiac dysfunction was observed, which was associated with decreased expression of CX43 in the heart of mouse model.42 In addition to its abundance, aberrant distribution of cardiac CX43 such as lateralization was also demonstrated in HFD-fed mice.42 Thus, alterations in the amount and distribution of the CX43 may lead to anisotropic conduction and altered patterns of conduction, promoting arrhythmogenesis.41 However, treatment with KN93 significantly attenuated the slow conduction cardiac fibrosis and recovered of CX43 level in the HFD-induced heart, implicating a role for CaMKII activation in regulating conduction abnormalities under hyperlipidemia conditions.
It should be noted that, in this study, peritoneal injection of KN93 for 8 weeks greatly improved metabolic parameters in the HFD-fed mice (Table 1), suggesting a metabolic protective role of KN93, which may be attributed to the concomitant inhibition of liver CaMKII that has been demonstrated to play an important role in insulin resistance in obesity.43 The metabolic improvement by KN93 may relieve some metabolic stress on heart and improve electrical abnormalities. So it is not clear whether the effects of KN93 in vivo is due to an effect on the heart or is secondary to the drug action on the liver. In addition, although KN93 has been widely used to inhibit CaMKII activity in cultured cells, in isolated muscles, and in live animals, some off-target effects of KN93 have also been reported that it can inhibit voltage-dependent K+ currents and L-type calcium channel activity.44,45 Overall, KN93 is not suitable to be used to test the role of cardiac CaMKII in obesity/hyperlipidemia-induced arrhythmias, because of the numerous other explanations. Furthermore, the arrhythmogenic effect of hyperlipidaemia evaluated through induction in ex vivo condition is still tenuous, weak, and probably of little relevance to real cardiac arrhythmias. Thus, a deeper mechanistic understanding of the role of CaMKII would be derived from cardiac-targeted CaMKII KO mice, and the rhythm effects of obesity/hyperlipidaemia would be markedly improved with in vivo arrhythmia induction and an assessment of arrhythmias without induction through telemetry.
IN CONCLUSION
Taken together, our results suggested that HFD or hyperlipidemia has strongly adverse effects on cardiac electrophysiology, promoting arrhythmogenesis. Ion channel dysfunction, disturbed Ca2+ handling, conduction abnormalities, and cardiac fibrosis may participate in this process and CaMKII activation may be the upstream signaling mediating these downstream pathological electrical events under hyperlipidemia conditions.
Footnotes
Supported by grants from the key Project of Hubei Science and Technology Support Program (No. 2013BCB013), and the National Natural Science Foundation of China (No. 81270249 and No. 81570306).
The authors report no conflicts of interest.
P. Zhong and D. Quan have contributed equally.
REFERENCES
- 1.Tsang TS, Petty GW, Barnes ME, et al. The prevalence of atrial fibrillation in incident stroke cases and matched population controls in Rochester, Minnesota: changes over three decades. J Am Coll Cardiol. 2003;42:93–100. [DOI] [PubMed] [Google Scholar]
- 2.Wang TJ, Parise H, Levy D, et al. Obesity and the risk of new-onset atrial fibrillation. JAMA. 2004;292:2471–2477. [DOI] [PubMed] [Google Scholar]
- 3.McCully BH, Hasan W, Streiff CT, et al. Sympathetic cardiac hyperinnervation and atrial autonomic imbalance in diet-induced obesity promote cardiac arrhythmias. Am J Physiol Heart Circ Physiol. 2013;305:H1530–H1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aubin M-C, Cardin S, Comtois P, et al. A high-fat diet increases risk of ventricular arrhythmia in female rats: enhanced arrhythmic risk in the absence of obesity or hyperlipidemia. J Appl Physiol. 2010;108:933–940. [DOI] [PubMed] [Google Scholar]
- 5.Savio-Galimberti E, Kannankeril P, Wasserman D, et al. Weight loss reduces atrial fibrillation inducibility and burden in severe obesity induced by either high-fat diet or genetic hyperphagia in mice. Circulation. 2015;132(suppl 3):A14729–A. [Google Scholar]
- 6.Lin YK, Chen YC, Chen JH, et al. Adipocytes modulate the electrophysiology of atrial myocytes: implications in obesity-induced atrial fibrillation. Basic Res Cardiol. 2012;107:293. [DOI] [PubMed] [Google Scholar]
- 7.Kang JX, Xiao Y-F, Leaf A. Free, long-chain, polyunsaturated fatty acids reduce membrane electrical excitability in neonatal rat cardiac myocytes. Proc Natl Acad Sci. 1995;92:3997–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y-B, Wu C-C, Lu L-S, et al. Sympathetic nerve sprouting, electrical remodeling, and increased vulnerability to ventricular fibrillation in hypercholesterolemic rabbits. Circ Res. 2003;92:1145–1152. [DOI] [PubMed] [Google Scholar]
- 9.Erickson JR, Pereira L, Wang L, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rokita AG, Anderson ME. New therapeutic targets in cardiology arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation. 2012;126:2125–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swaminathan PD, Purohit A, Hund TJ, et al. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res. 2012;110:1661–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herren AW, Bers DM, Grandi E. Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am J Physiol Heart Circ Physiol. 2013;305:H431–H445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer TH, Neef S, Maier LS. The Ca-calmodulin dependent kinase II: a promising target for future antiarrhythmic therapies? J Mol Cell Cardiol. 2013;58:182–187. [DOI] [PubMed] [Google Scholar]
- 15.Qin M, Huang H, Wang T, et al. Absence of Rgs5 prolongs cardiac repolarization and predisposes to ventricular tachyarrhythmia in mice. J Mol Cell Cardiol. 2012;53:880–890. [DOI] [PubMed] [Google Scholar]
- 16.Wang D, Liu T, Shi S, et al. Chronic administration of catestatin improves autonomic function and exerts cardioprotective effects in myocardial infarction rats. J Cardiovasc Pharmacol Ther. 2016;21:1074248416628676. [DOI] [PubMed] [Google Scholar]
- 17.Verheule S, Sato T, Everett T, et al. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ Res. 2004;94:1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong P, Wu L, Qian Y, et al. Blockage of ROS and NF-kappaB-mediated inflammation by a new chalcone L6H9 protects cardiomyocytes from hyperglycemia-induced injuries. Biochim Biophys Acta. 2015;1852:1230–1241. [DOI] [PubMed] [Google Scholar]
- 19.Smith JA, Kohn TA, Chetty AK, et al. CaMK activation during exercise is required for histone hyperacetylation and MEF2A binding at the MEF2 site on the Glut4 gene. Am J Physiology Endocrinol Metab. 2008;295:E698–E704. [DOI] [PubMed] [Google Scholar]
- 20.Benter IF, Yousif MH, Canatan H, et al. Inhibition of Ca 2+/calmodulin-dependent protein kinase II, RAS-GTPase and 20-hydroxyeicosatetraenoic acid attenuates the development of diabetes-induced vascular dysfunction in the rat carotid artery. Pharmacol Res. 2005;52:252–257. [DOI] [PubMed] [Google Scholar]
- 21.Yousif MH. Signal transduction through Ras-GTPase and Ca2+/calmodulin-dependent protein kinase II contributes to development of diabetes-induced renal vascular dysfunction. Cell Biochem Funct. 2006;24:299–305. [DOI] [PubMed] [Google Scholar]
- 22.Sato K, Suematsu A, Nakashima T, et al. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat Med. 2006;12:1410–1416. [DOI] [PubMed] [Google Scholar]
- 23.Mahajan R, Lau DH, Sanders P. Impact of obesity on cardiac metabolism, fibrosis, and function. Trends Cardiovasc Med. 2015;25:119–126. [DOI] [PubMed] [Google Scholar]
- 24.Pietrasik G, Goldenberg I, McNitt S, et al. Obesity as a risk factor for sustained ventricular tachyarrhythmias in MADIT II patients. J Cardiovasc Electrophysiol. 2007;18:181–184. [DOI] [PubMed] [Google Scholar]
- 25.Morrow JP. High-fat diet, obesity and sudden cardiac death. Acta Physiol (Oxf). 2014;211:13–16. [DOI] [PubMed] [Google Scholar]
- 26.Ashrafi R, Yon M, Pickavance L, et al. Altered left ventricular ion channel transcriptome in a high-fat-fed rat model of obesity: insight into obesity-induced arrhythmogenesis. J Obes. 2016;2016:7127898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang F, Hartnett S, Sample A, et al. High fat diet induced alterations of atrial electrical activities in mice. Am J Cardiovasc Dis. 2016;6:1–9. [PMC free article] [PubMed] [Google Scholar]
- 28.Takahashi K, Sasano T, Sugiyama K, et al. High-fat diet increases vulnerability to atrial arrhythmia by conduction disturbance via miR-27b. J Mol Cell Cardiol. 2016;90:38–46. [DOI] [PubMed] [Google Scholar]
- 29.Huang H, Amin V, Gurin M, et al. Diet-induced obesity causes long QT and reduces transcription of voltage-gated potassium channels. J Mol Cell Cardiol. 2013;59:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant AO. Cardiac ion channels. Circ Arrhythmia Electrophysiol. 2009;2:185–194. [DOI] [PubMed] [Google Scholar]
- 31.Aomine M, Yamato T. Electrophysiological properties of ventricular muscle obtained from spontaneously diabetic mice. Exp Anim. 2000;49:23–33. [DOI] [PubMed] [Google Scholar]
- 32.Lin Y-C, Huang J, Kan H, et al. Defective calcium inactivation causes long QT in obese insulin-resistant rat. Am J Physiology Heart Circ Physiol. 2012;302:H1013–H1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antzelevitch C, Burashnikov A. Overview of basic mechanisms of cardiac arrhythmia. Card Electrophysiol Clin. 2011;3:23–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mustroph J, Maier LS, Wagner S. CaMKII regulation of cardiac K channels. Front Pharmacol. 2014;5:20.24600393 [Google Scholar]
- 35.Chelu MG, Sarma S, Sood S, et al. Calmodulin kinase II–mediated sarcoplasmic reticulum Ca 2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neef S, Dybkova N, Sossalla S, et al. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. 2010;106:1134–1144. [DOI] [PubMed] [Google Scholar]
- 37.Voigt N, Li N, Wang Q, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mustroph J, Neef S, Maier LS. CaMKII as a target for arrhythmia suppression. Pharmacol Ther. 2016. 10.1016/j.pharmthera.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Tzimas C, Terrovitis J, Lehnart SE, et al. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition ameliorates arrhythmias elicited by junctin ablation under stress conditions. Heart Rhythm. 2015;12:1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paulino EC, Ferreira JCB, Bechara LR, et al. Exercise training and caloric restriction prevent reduction in cardiac Ca2+-handling protein profile in obese rats. Hypertension. 2010;56:629–635. [DOI] [PubMed] [Google Scholar]
- 41.Kostin S, Dammer S, Hein S, et al. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res. 2004;62:426–436. [DOI] [PubMed] [Google Scholar]
- 42.Noyan-Ashraf MH, Shikatani EA, Schuiki I, et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation. 2013;127:74–85. [DOI] [PubMed] [Google Scholar]
- 43.Ozcan L, de Souza JC, Harari AA, et al. Activation of calcium/calmodulin-dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell Metab. 2013;18:803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rezazadeh S, Claydon TW, Fedida D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)] amino-N-(4-chlorocinnamyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther. 2006;317:292–299. [DOI] [PubMed] [Google Scholar]
- 45.Gao L, Blair LA, Marshall J. CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem Biophys Res Commun. 2006;345:1606–1610. [DOI] [PubMed] [Google Scholar]