

Abstract
A series of six novel metallocenyl-7-ADCA (metallocenyl = ferrocenyl or ruthenocenyl; 7-ADCA = 7-aminodesacetoxycephalosporanic acid) conjugates were synthesized and their antibacterial properties evaluated by biochemical and microbiological assays. The ruthenocene derivatives showed a higher level of inhibition of DD-carboxypeptidase 64-575, a Penicillin Binding Protein (PBP), than the ferrocene derivatives and the reference compound penicillin G. Protein X-ray crystallographic analysis revealed a covalent acyl-enzyme complex of a ruthenocenyl compound with CTX-M β-lactamase E166A mutant, corresponding to a similar complex with PBPs responsible for the bactericidal activities of these compounds. Most interestingly, an intact compound was captured at the crystal-packing interface, elucidating for the first time the structure of a metallocenyl β-lactam compound that previously eluded small molecule crystallography. We propose that protein crystals, even from biologically unrelated molecules, can be utilized to determine structures of small molecules.
Graphical abstract
First used clinically in the 1940’s, the β-lactam antibiotics treat bacterial infections by inhibiting Penicillin Binding Proteins (PBPs), which are enzymes that catalyze the final steps of bacterial cell wall biosynthesis.1, 2 The β-lactams inhibit the PBPs by forming a covalently linked acyl-enzyme adduct with a catalytic serine in the active site.3–5 One of the main bacterial resistance mechanisms against β-lactam antibiotics, particularly in Gram-negative bacteria, is the production of β-lactamases.6–11 Among the four classes of β-lactamases, Classes A, C, and D all share a common active site serine residue, like the PBPs, whereas Class B are Zn2+-based metallo enzymes.12, 13 It is hypothesized that serine β-lactamases originated from PBPs by evolving the ability to hydrolyze the covalent acyl-enzyme linkage.9
Organometallic compounds have been explored within medicinal chemistry mainly as anticancer14–17 and antimalarial agents.18 However, they have recently attracted increased attention for their antibacterial properties.19–22 Antibacterial mechanisms of organometallic compounds are diversified, and in some cases they overcome antibiotic resistance in pathogenic bacteria.20, 21 The idea of metallocene to β-lactam antibiotic conjugation is not new and fits into the more general concept of organometallic drug derivatization often used in bioorganometallic chemistry.15 The first ferrocenyl (Fc) derivatives of 6-aminopenicillinic acid (6-APA) and 7-aminocephalosporanic acid (7-ACA) were reported in 1970s.23 Recently, novel ferrocenyl-penems with antibacterial activity against Gram-positive and Gram-negative bacteria have been reported, further substantiating the importance of metallocenyl conjugates of β-lactams as antibacterial agents.24
In continuing our program studying the biologically active organometallic compounds,25 we became interested in metallocenyl derivatives of 6-APA.26, 27 Our preceding results show noticeable activity of ferrocenyl and ruthenocenyl (Rc) 6-APA conjugates against Gram-positive bacteria, including clinically isolated S. aureus strains.26, 27 We have also determined a 1.18 Å resolution X-ray crystal structure of a hydrolyzed Rc-6-APA derivative in a complex with CTX-M-14 E166A β-lactamase.27 However, in all previous studies including our own, no experimental structure has been determined for any of the intact metallocenyl β-lactam compounds, mainly due to the difficulty in obtaining suitable small molecule crystals.
Here we report the synthesis, biological, and structural properties of ferrocenyl and ruthenocenyl conjugates of 7-aminodesacetoksycephalosporanic acid (7-ADCA; 1) (Scheme 1). The desired metallocenyl-β-lactams have been obtained by an active ester methodology.26 Compounds were characterized by 1H NMR, 13C NMR, FTIR, MS and elemental analysis, confirming the proposed structures. 1H-NMR spectra of 2–7 are shown in the SI.
Inhibitory activity of compounds 2–7 was evaluated against several bacterial enzymes: a D-alanyl-D-alanine carboxypeptidase/transpeptidase from Saccharopolyspora erythraea 64-575 (DD-carboxypeptidase 64-575), CTX-M-14 Class A β-lactamase,28–31 and Bacillus cereus 569/H9 β-lactamase (Table 1).32–34 DD-carboxypeptidase 64-575 is an exocellular serine peptidase. Similar to high molecular mass (HMM) PBPs, DD-carboxypeptidase 64-575 shows affinity to a plethora of common β-lactam antibiotics, including cephalosporin C, cefamandole, and cefotaxime. These antibiotics cause 50% inhibition of the DD-carboxypeptidase enzyme in the low concentration range from 5.8×10−9 to 7.5×10−6 M.35 Therefore, DD-carboxypeptidase 64-575 can be utilized as a model enzyme in the study of β-lactam-PBP interactions and inhibition. CTX-M-14 β-lactamase belongs to the class A β-lactamase family and its active site shares many key catalytic features with that of the PBP’s.9, 36, 37 Moreover, high-resolution X-ray crystal structures of CTX-M β-lactamases in their apo, as well as substrate-bound, states were solved, providing detailed insights into the mode of action of these enzymes.27, 30, 31, 38, 39 The third enzyme of interest, a B. cereus 569/H9 β-lactamase, belongs to the class B metallo-β-lactamases (MBLs).40 In our biochemical assay, compounds 2–7 were treated as ‘competitive inhibitors’ to compare their interactions with the enzyme active site, although they function as covalent ligands for the DD-carboxypeptidase and substrates for β-lactamases. Compared with the 7-ADCA starting scaffold and the benzyl side chain of penicillin G, the ferrocenyl and ruthenocenyl moieties have enhanced the interactions with the protein and consequently improved the inhibitory activity as demonstrated by the IC50 values (Table 1). In particular, for the DD-carboxypeptidase, the ruthenocenyl group appears to enhance binding more than the ferrocenyl group.
Table 1.
Inhibition of compounds 2–7 on PBP and β-lactamases.
| IC50 μM | ||||||||
|---|---|---|---|---|---|---|---|---|
| Enzyme | 1 | 2 | 3 | 4 | 5 | 6 | 7 | penicillin G |
| DD-carboxypeptidase 64-575 | 176 ± 2.38 | 0.158 ± 0.002 | 0.052 ± 0.001 | 0.133 ± 0.002 | 0.027 ± 0.000 | 0.083 ± 0.003 | 0.081 0.003 ± | 0.153 ± 0.002 |
| CTX-M-14 | 1620 ± 144 | 31 ± 5 | 44 ± 4 | 716 ± 108 | 7 ± 1 | 36 ± 4 | 363 ± 49 | nd* |
| B. cereus 569/H9 | 4661 ± 422 | 142 ± 28 | 200 ± 25 | 82 ± 12 | 117 10 ± | 126 ± 27 | 65 ± 5 | nd |
nd, not determined.
The antibacterial activity of compounds 2–7 was tested against a panel of Gram-positive strains, namely: methicillin-sensitive Staphylococcus aureus (MSSA), methicillin-resistant S. aureus (MRSA), vancomycin-intermediate S. aureus (VISA), and S. epidermidis (Table S3). In contrast to the biochemical results, the compounds displayed overall less activity in the cell-based assays compared with the reference compounds penicillin G and ampicillin. This suggests the metallocenyl moieties may have affected the compounds’ ability to reach the PBPs or may have varied effects on PBPs from different bacterial species.
To investigate the molecular interactions between these compounds and the target protein, we used CTX-M-14 E166A mutant to prepare complex crystals with compound 3. Class A β-lactamases such as CTX-M are hypothesized to have evolved from PBPs, acquiring Glu166 to activate a water molecule that breaks the acyl-enzyme linkage with the β-lactam compound.9, 36, 37 CTX-M-14 E166A mutant behaves like a PBP, forming a stable acyl-enzyme intermediate with β-lactam antibiotics. This is indeed what we observed in a 1.35 Å resolution crystal structure of CTX-M-14 E166A complexed with 3 (Fig. 1). The electron density maps clearly identified the binding pose of the compound in the protein active site (Fig. 1A), as well as a covalent bond between the β-lactam ring carbon and Ser70Oγ (Fig. 1B). The β-lactam ring carbonyl oxygen is nestled in the oxyanion hole formed by the backbone –NH groups of Ser70 and Ser237. The structure also illustrates other favorable interactions between the compound and the protein, including hydrogen bonds (HBs) between the C4 carboxylate group and Thr235/Ser130, and between the ligand amide group and Asn104/Asn132/Ser237. Interestingly, the ruthenocenyl group appears to display two conformations: conformation 1 with the base ring of the cyclopentadienyl group stacked against the protein backbone of Gly238-Asp240 (note: there is a gap in the residue numbering due to class A β-lactamase convention); conformation 2 has the base ring rotated away from the β-strand and the distal ring establishing more contact with Thr171. Together with our previous structure, these observations demonstrate the versatility of metallocenyl groups in interacting with protein.
Figure 1.
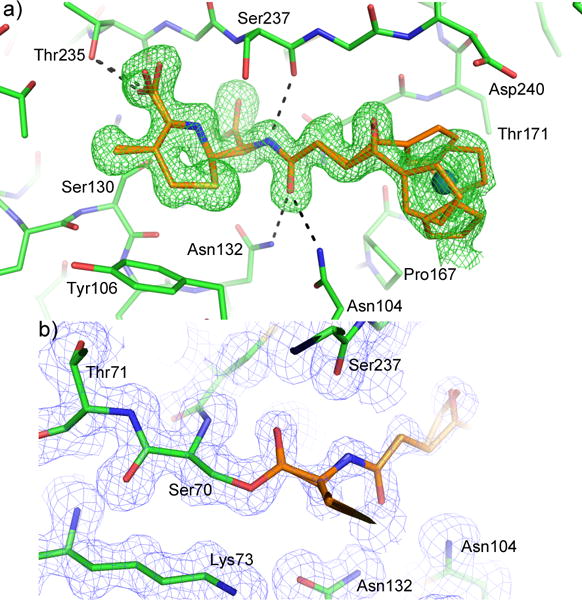
Acyl-enzyme complex of CTX-M-14 E166A mutant with compound 3. The protein and compound are shown in green and orange respectively. (A) The unbiased Fo-Fc density map is shown in green at 2σ. (B) 2Fo-Fc map (0.5σ, blue) shows the covalent acyl-enzyme linkage between Ser70 and compound 3.
The CTX-M crystal also allowed us to capture an intact compound 3 at the crystal-packing interface. This is particularly worth noting because none of the metallocenyl β-lactam antibiotics have been structurally characterized by small molecule X-ray crystallography due to the challenges in obtaining suitable single crystals. We initially resorted to computational modeling using the B3LYP hybrid functional as described in the SI. The conformation of the compound is identified unambiguously in the unbiased Fo-Fc electron density map (Fig. 2A). The intact compound resides outside the active site and establishes multiple interactions with three monomers at the crystal-packing interface (Fig. 2B). The six-membered ring is sandwiched between Arg65 and Gly175 of one monomer (monomer 1), forming extensive non-polar and stacking interactions with these residues. The C4-carboxylate group forms a HB with the Gly175 backbone, while establishing favorable electrostatic interactions with nearby Arg43 and Lys88 from two different monomers (monomers 1 and 2 respectively). The β-lactam ring contacts both monomer 1 and 2, forming a HB with Gln89. The ruthenocenyl moiety and the linker also interact with two monomers (1 and 3), including non-polar interactions with Pro177 (monomer 1) and Ile173/Pro174 from monomer 3, as well as stacking interactions with the Arg65 side chain. There is also close proximity (4.5 Å) between the ruthenium atom and the hydroxyl group of Tyr241, suggesting a possible HB. This contact highlights a unique feature of Ru in interacting with –OH,41, 42 particularly in the context of protein-ligand interactions.21
Figure 2.
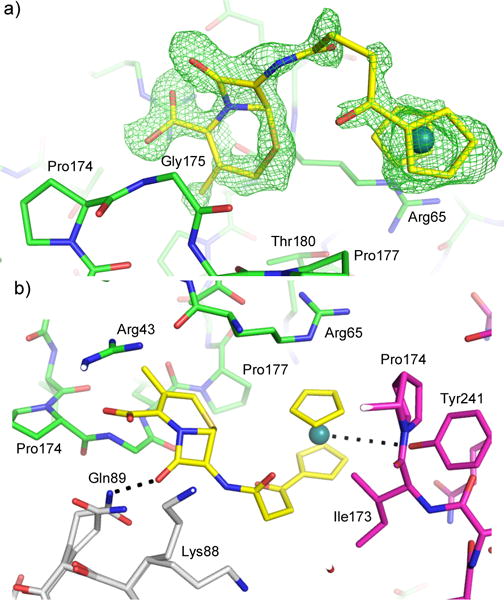
Intact compound 3 (yellow) observed at the crystal-packing interface. (A) The unbiased Fo-Fc density map is shown in green at 3σ. (B) Interactions between compound 3 and three protein monomers. Monomers 1, 2, and 3 are colored in green, white, and magenta respectively. Potential hydrogen bonds are shown as black dashed lines.
The observation of an intact compound 3 at the crystal-packing interface, unrelated to CTX-M’s biological activity, highlights the value of protein X-ray crystallography in determining novel structures for small molecules. Although this is certainly not an entirely new concept because protein-small molecule complex structures are determined routinely, including those in the intended binding pockets, as well as some accidental discoveries at the crystal packing interface. But in most cases, the structure of the ligand has already been characterized by small molecule crystallography, or the ligand is bound by a biologically related macromolecule. However, based on previous studies, we propose that protein crystals can be utilized as ‘crystalline sponges’ in a fashion similar to those formed by certain framework metallo compounds as previously described.43 First, protein crystals provide an anisotropic binding environment allowing the capture of specific and ordered ligand conformations. Second, although the vast majority of protein crystals are filled with water, it has been shown that crosslinking can stabilize protein crystals in organic solvent in multiple-solvent crystal structure (MSCS) experiments, and such crosslinking does not block the access of small molecules to the protein surface.44–47 Lastly, there is a wide range of protein crystals described in the Protein Data Bank, including many capable of diffracting to sub-Angstrom resolution, like those of CTX-M. These crystals can provide a diverse set of interfaces to capture different small molecules and the high diffracting quality of some of these crystals can permit accurate structure determination even for ligands with low occupancies. It is therefore conceivable that a series of high quality and diverse protein crystals can be analyzed and prepared to screen and capture small molecule ligands for structure determination, independent of the biological relationship between the proteins and the small molecules.
In conclusion, our synthesis and characterization of metallocenyl-7-ADCA conjugates has demonstrated how metallocenyl groups can be utilized to enhance the interactions between β-lactam compounds and the target proteins. These results provide valuable information for designing future metallocenyl compounds with biological activities, and offer insights into how protein crystals can facilitate the structural analysis of novel small molecules.
Supplementary Material
Scheme 1.
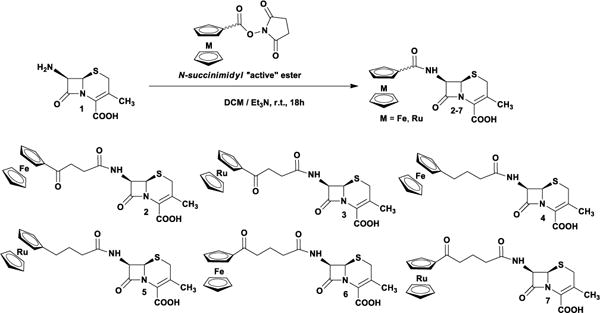
Synthesis of metallo-7-ADCA compounds 2–7.
Acknowledgments
Y.C. has been supported by the NIH (AI103158). K.K., L.S., J.S., and J.S. thank the National Science Centre (Krakow, Poland) for financial support (Grant no. DEC-2013/11/B/ST5/00997)
Footnotes
Supporting Information
Compound synthesis, conformational analysis, computational data, microbiological data, computed Cartesian coordinates of all of the molecules reported in this study, and x-ray crystallographic statistics.
Notes
The coordinates and structure factors have been deposited in the Protein Data Bank (PDB), www.rcsb.org, with the accession code PDB ID: 5UJO. The authors declare no competing financial interest.
References
- 1.Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. FEMS Microbiol Rev. 2006;30:673–691. doi: 10.1111/j.1574-6976.2006.00024.x. [DOI] [PubMed] [Google Scholar]
- 2.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. FEMS Microbiol Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 3.Llarrull LI, Testero SA, Fisher JF, Mobashery S. Curr Opin Microbiol. 2010;13:551–557. doi: 10.1016/j.mib.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Testero SA, Fisher JF, Mobashery S, Abraham DJ. Burger’s Medicinal Chemistry and Drug Discovery. John Wiley & Sons Inc; Hoboken: 2003. [Google Scholar]
- 5.Yao Z, Kahne D, Kishony R. Mol Cell. 2012;48:705–712. doi: 10.1016/j.molcel.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bush K, Fisher JF. Annu Rev Microbiol. 2011;65:455–478. doi: 10.1146/annurev-micro-090110-102911. [DOI] [PubMed] [Google Scholar]
- 7.Drawz SM, Bonomo RA. Clin Microbiol Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikolaidis I, Favini-Stabile S, Dessen A. Prot Sci. 2014;23:243–259. doi: 10.1002/pro.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith JD, Kumarasiri M, Zhang W, Hesek D, Lee M, Toth M, Vakulenko S, Fisher JF, Mobashery S, Chen Y. Antimicrob Agents Chemother. 2013;57:3137–3146. doi: 10.1128/AAC.00505-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taubes G. Science. 2008;321:356–361. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- 11.Therrien C, Levesque RC. FEMS Microbiol Rev. 2000;24:251–262. doi: 10.1111/j.1574-6976.2000.tb00541.x. [DOI] [PubMed] [Google Scholar]
- 12.Bush K, Jacoby GA, Medeiros AA. Antimicrob Agents Chemother. 1995;39:1211–1233. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livermore DM. Clin Microbiol Rev. 1995;8:557–584. doi: 10.1128/cmr.8.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ornelas C. New J Chem. 2011;35:1973–1985. [Google Scholar]
- 15.Jaouen G, Vessieres A, Top S. Chem Soc Rev. 2015;44:8802–8817. doi: 10.1039/c5cs00486a. [DOI] [PubMed] [Google Scholar]
- 16.Gasser G, Ott I, Metzler-Nolte N. J Med Chem. 2011;54:3–25. doi: 10.1021/jm100020w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartinger CG, Metzler-Nolte N, Dyson PJ. Organometallics. 2012;31:5677–5685. [Google Scholar]
- 18.Biot C, Dive D. In: Medicinal Organometallic Chemistry. Jaouen G, Metzler-Nolte N, editors. Springer Berlin Heidelberg; Berlin, Heidelberg: 2010. pp. 155–193. [Google Scholar]
- 19.Patra M, Gasser G, Metzler-Nolte N. Dalton Trans. 2012;41:6350–6358. doi: 10.1039/c2dt12460b. [DOI] [PubMed] [Google Scholar]
- 20.Wenzel M, Patra M, Senges CHR, Ott I, Stepanek JJ, Pinto A, Prochnow P, Vuong C, Langklotz S, Metzler-Nolte N, Bandow JE. ACS Chem Biol. 2013;8:1442–1450. doi: 10.1021/cb4000844. [DOI] [PubMed] [Google Scholar]
- 21.Albada HB, Prochnow P, Bobersky S, Bandow JE, Metzler-Nolte N. Chem Sci. 2014;5:4453–4459. [Google Scholar]
- 22.Patra M, Gasser G, Wenzel M, Merz K, Bandow JE, Metzler-Nolte N. Organometallics. 2010;29:4312–4319. [Google Scholar]
- 23.Edwards EI, Epton R, Marr G. J Organomet Chem. 1976;107:351–357. [Google Scholar]
- 24.Long B, He C, Yang Y, Xiang J. Eur J Med Chem. 2010;45:1181–1188. doi: 10.1016/j.ejmech.2009.12.045. [DOI] [PubMed] [Google Scholar]
- 25.Kowalski K, Skiba J, Oehninger L, Ott I, Solecka J, Rajnisz A, Therrien B. Organometallics. 2013;32:5766–5773. [Google Scholar]
- 26.Skiba J, Rajnisz A, de Oliveira KN, Ott I, Solecka J, Kowalski K. Eur J Med Chem. 2012;57:234–239. doi: 10.1016/j.ejmech.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 27.Lewandowski EM, Skiba J, Torelli NJ, Rajnisz A, Solecka J, Kowalski K, Chen Y. Chem Commun. 2015;51:6186–6189. doi: 10.1039/c5cc00904a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonnet R. Antimicrob Agents Chemother. 2004;48:1–14. doi: 10.1128/AAC.48.1.1-14.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradford PA. Clin Microbiol Rev. 2001;14:933–951. doi: 10.1128/CMR.14.4.933-951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Bonnet R, Shoichet BK. J Am Chem Soc. 2007;129:5378–5380. doi: 10.1021/ja0712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, Delmas J, Sirot J, Shoichet B, Bonnet R. J Mol Biol. 2005;348:349–362. doi: 10.1016/j.jmb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Sabath LD, Abraham EP. Biochem J. 1966;98:11C–13C. doi: 10.1042/bj0980011c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bebrone C. Biochem Pharmacol. 2007;74:1686–1701. doi: 10.1016/j.bcp.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 34.Davies RB, Abraham EP. Biochem J. 1974;143:115–127. doi: 10.1042/bj1430115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurzątkowski W, Solecka J, Filipek J, Kurzątkowski JD, Kuryłowicz W. Appl Microbiol Biotechnol. 1990;33:452–454. doi: 10.1007/BF00170067. [DOI] [PubMed] [Google Scholar]
- 36.Adamski CJ, Cardenas AM, Brown NG, Horton LB, Sankaran B, Prasad BVV, Gilbert HF, Palzkill T. Biochemistry. 2015;54:447–457. doi: 10.1021/bi501195g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massova I, Mobashery S. Antimicrob Agents Chemother. 1998;42:1–17. doi: 10.1128/aac.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y, Shoichet B, Bonnet R. J Am Chem Soc. 2005;127:5423–5434. doi: 10.1021/ja042850a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delmas J, Leyssene D, Dubois D, Birck C, Vazeille E, Robin F, Bonnet R. J Mol Biol. 2010;400:108–120. doi: 10.1016/j.jmb.2010.04.062. [DOI] [PubMed] [Google Scholar]
- 40.Palzkill T. Ann N Y Acad Sci. 2013;1277:91–104. doi: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shubina ES, Krylov AN, Kreindlin AZ, Rybinskaya MI, Epstein LM. J Mol Struct. 1993;301:1–5. [Google Scholar]
- 42.Shubina ES, Krylov AN, Kreindlin AZ, Rybinskaya MI, Epstein LM. J Organomet Chem. 1994;465:259–262. [Google Scholar]
- 43.Inokuma Y, Yoshioka S, Ariyoshi J, Arai T, Hitora Y, Takada K, Matsunaga S, Rissanen K, Fujita M. Nature. 2013;495:461–466. doi: 10.1038/nature11990. [DOI] [PubMed] [Google Scholar]
- 44.English AC, Done SH, Caves LS, Groom CR, Hubbard RE. Proteins: Struct, Funct Bioinf. 1999;37:628–640. [PubMed] [Google Scholar]
- 45.Quiocho FA, Richards FM. Proc Natl Acad Sci US A. 1964;52:833–839. doi: 10.1073/pnas.52.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattos C, Bellamacina CR, Peisach E, Pereira A, Vitkup D, Petsko GA, Ringe D. J Mol Biol. 2006;357:1471–1482. doi: 10.1016/j.jmb.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 47.Fitzpatrick PA, Steinmetz AC, Ringe D, Klibanov AM. Proc Natl Acad Sci US A. 1993;90:8653–8657. doi: 10.1073/pnas.90.18.8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.