
Figure 3. Pharmacologic methods to disrupt core spliceosome function.
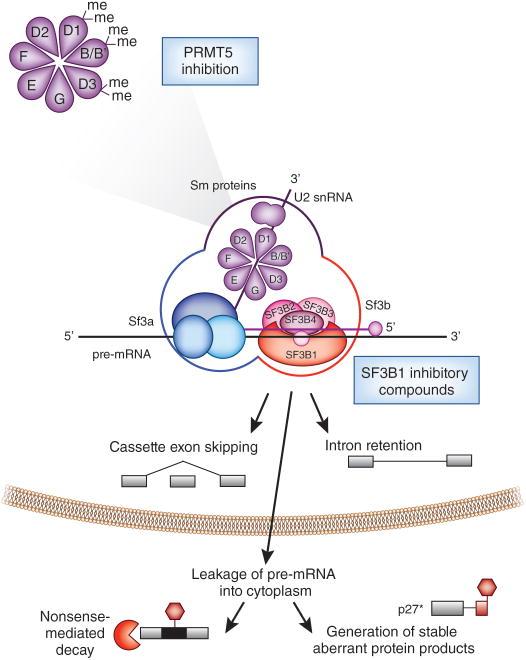
Current methods to directly inhibit spliceosome function include a series of compounds that bind to the SF3B component of U2 snRNP and inhibit early spliceosome assembly. While the precise biochemical interactions between SF3B1 inhibitors and U2 snRNP are not well understood, the effects of each of these drugs on cell toxicity is nearly completely abrogated by mutation of a single residue of SF3B1 (SF3B1 R1074H), suggesting that each of these compounds specifically functions through interactions with SF3B1. Inhibition of U2 snRNP function has been shown to result in widespread intron retention and cassette exon skipping in a time- and dose-dependent manner in a variety of cell types. While this inhibition of splicing results in an accumulation of pre-mRNAs in the nucleus, pharmacologic inhibition of U2 snRNP is also associated with leakage of pre-mRNA into the cytoplasm. Although most unspliced mRNAs are expected to become substrates for nonsense-mediated decay, a portion of these mRNAs may undergo translation to generate aberrant protein products which themselves may have cellular toxicity. For example, a functionally active form of the cell cycle inhibitory protein p27 (termed “p27*”) which lacks the C-terminal domains required for degradation is generated following exposure to several SF3B1 inhibitory compounds.
In addition to SF3B1 inhibitory compounds, recent data suggests that inhibition of Sm protein methylation through downregulation of PRMT5 (protein arginine methyltransferase-5) may also inhibit splicing. PRMT5 symmetrically methylates arginine residues of SmB/B′, SmD1, and SmD3.