

Abstract
KIF23 was recently suggested to be a potential molecular target for the treatment of lung cancer. This proposal is based on elevated expression of KIF23 in several tumors affecting breast, lung, brain, and liver, and also on the presence of KIF23 mutations in melanoma and colorectal cancer. Recently, we identified a mutation in the KIF23 gene causing a rare hereditary form of dyserythropoietic anemia (CDA III) with predisposition to blood cancer. We suggested that KIF23 overexpression in tumors might be due to the presence of activating somatic mutations, and therefore, mutation screening of the KIF23 in 15 non-small-cell lung cancer (NSCLC) cases with elevated expression level of KIF23 was undertaken. Eight sequence variants were found in all samples. Furthermore, one variant was present in two cases, and one variant was case specific. Nine variants were previously reported while one variant lacks frequency information. Nine of ten cases available for single nucleotide polymorphism-array analysis demonstrated aberrant karyotypes with additional copy of entire chromosome 15. Thus, no activating somatic mutations in coding regions of the KIF23 were found. Furthermore, no mutations were detected in cell cycle genes homology region in KIF23 promoter responsible for p53-dependent repression of KIF23 expression. We showed that the elevated level of KIF23 could be due to additional copy of chromosome 15 demonstrated in 90% of NSCLC cases analyzed in this study. Considering the crucial role of KIF23 in the final step of mitosis, the gene is a potential molecular marker, and for better understanding of its role in cancer development, more tumors should be analyzed.
Keywords: KIF23, overexpression, mutation, copy number alteration, CNA, lung cancer
Introduction
Currently, multiple studies aim on screening for novel diagnostic and prognostic biomarkers in different types of tumors. Molecular markers in combination with clinical features allow better understanding of cancer development and progression compared to only histological examination.1
A key regulator of cytokinesis, kinesin motor protein KIF23 also known as MKLP1, was proposed to be a potential marker for several tumors.2 KIF23 overexpression assessed by cDNA microarray or by quantitative reverse transcription-polymerase chain reaction (PCR) was recently shown in lung cancer, breast cancer, gliomas, paclitaxel-resistant gastric cancer, and hepatocellular carcinomas.3–8 Notably, the poor survival in patients with several cancer types was associated with KIF23 overexpression.4,6,7
Dysfunctional KIF23 causes a cytokinesis failure leading to appearance of bi-and multinucleated cells.9,10 Recently, we reported on a KIF23 mutation causative of congenital dyserythropoietic anemia type III (CDA III), a rare hereditary disease, characterized by large multinuclear erythroblasts in patients’ bone marrow.11 Knockdown experiments using KIF23 siRNA and following rescue with mutant P916R KIF23 in HeLa cells resulted in cytokinesis failure with bi-nucleated cells present.11 Moreover, cancer diagnosis was overrepresented among CDA III patients in the Swedish family. It might be due to the effect of mutated KIF23 on cytokinesis resulting in polyploid cells, a feature associated with a risk for carcinogenesis.12
In this study, we hypothesized that overexpression of KIF23 might be explained by the presence of activating somatic mutations or genomic amplifications. For this purpose, we conducted direct sequencing of KIF23 and copy number analysis by genome-wide genotyping in non-small-cell lung cancer (NSCLC) samples exhibiting high level of KIF23. Along with our experiments, we browsed the cBioPortal for Cancer Genomics and Catalogue of Somatic Mutations in Cancer (COSMIC) in order to retrieve KIF23 mutations spectrum in cancer analyzed by whole exome sequencing (WES).
Materials and methods
NSCLC patients were part of the study3 on identification of new NSCLC biomarkers by whole-genome gene expression microarray (Illumina, San Diego, CA, USA). Fifteen samples with elevated expression of KIF23 were selected for the current study (Table 1). Tumor samples were examined by a pathologist, and the samples rich in content of tumor cells were selected for DNA extraction using a NucleoSpin® Tissue kit (Macherey–Nagel, Düren, Germany). Corresponding DNA extracted from peripheral blood was available for some of the patients. An informed written consent for genetic analyses of biological samples was obtained from all patients, and the Ethics Review Committee on Human Research of the University of Tartu, Estonia approved the project. All samples analyzed at Umeå University were anonymized and did not contain any personal identification information.
Table 1.
Characteristics of 15 NSCLC patients with elevated level of KIF23
| Sample | Histology | Gender | Age (years) | Smoking (packs/year) | TNM | Stage |
|---|---|---|---|---|---|---|
| 15 | SCC | Male | 69 | 50 | pT2N0M1pul | IV |
| 16 | SCC | Male | 64 | 50 | pT2N0M0 | Ib |
| 52 | SCC | Male | 72 | 30 | pT1N0M0 | Ia |
| 66 | SCC | Male | 69 | 30 | pT3N0M0 | IIb |
| 67 | SCC | Male | 61 | 40 | pT2N0M0 | Ib |
| 68 | SCC | Male | 80 | 62 | pT2N0M0 | Ib |
| 73 | SCC | Male | 44 | 18 | pT2N0M0 | Ib |
| 84 | SCC | Male | 72 | 55 | pT2N2M0 | IIIa |
| 87 | SCC | Male | 77 | 40 | pT2N0M0 | Ib |
| 103 | SCC | Male | 44 | 60 | pT2N1M0 | IIb |
| 104 | In situ adenocarcinoma | Female | 62 | 30 | pT1N0M0 | Ib |
| 105 | SCC | Male | 57 | 15 | pT1N0M0 | Ia |
| 108 | SCC | Male | 75 | 50 | pT3N2M0 | IIIa |
| 109 | SCC | Male | 68 | 50 | pT2N0M0 | Ib |
| 114 | In situ adenocarcinoma | Male | 64 | 30 | pT1N0M0 | Ia |
Abbreviations: SCC, squamous cell carcinoma; NSCLC, non-small-cell lung cancer.
Sanger sequencing
For bidirectional sequencing of KIF23 (NM_138555.3) coding exons and adjacent intronic sequences were amplified from genomic tumor DNA. Primers were designed with Primer3 software and are available upon request. Sequence primers for cell cycle genes homology region (CHR) in KIF23 promoter were 5′-ACCTCAAGATCTCCCTCCAG-forward and 5′-CCGGCAGCAAGAACTAGAAC-reverse. PCR amplification was carried out using AmpliTaq® DNA polymerase enzyme system (Applied Biosystems, Waltham, MA, USA). An amount of 30 ng of tumor DNA was amplified in a 15 µL PCR mix containing 3 nmol dNTPs, 1× PCR Buffer II, 4.5 nmol MgCl2, 2% dimethyl sulfoxide, 0.45 U AmpliTaq Gold, and 3 pmol of each M13 tagged primer. PCR was performed using Veriti®96 Well Thermal Cycler (Applied Biosystems) with ramp time set to 50%. PCR included denaturation at 95°C for 10 minutes, followed by a touchdown step for 7 cycles (denaturation at 95°C for 30 seconds, annealing at 64°C to 1°C/cycle for 30 seconds, elongation at 72°C for 45 seconds), cycling step for 40 cycles (denaturation at 95°C for 30 seconds, annealing at 58°C for 30 seconds, elongation at 72°C for 45 seconds), followed by a final elongation at 72°C for an additional 10 minutes. Amplicons for exon 7 and 17 were amplified without touchdown step with elongation at 60°C. Any remaining primers were removed from the PCR products by treatment with 0.06 U of exonuclease I, ExoI (Thermo Scientific, Waltham, MA, USA) at 37°C for 15 minutes, followed by enzyme inactivation at 85°C for 15 minutes. Further, PCR clean up was done by dilution 1:6 with H2O. Sequencing was performed using BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). PCR product of 1.0 µL was used in a 10 µL sequencing reaction as well as 0.5 µL BigDye Terminator reaction mix, 1× sequencing buffer, and 3.2 pmol of M13 tagged primer. Reactions were cycled according to manufacturer’s instructions. Sequencing reactions were separated using ABI 3730xl DNA Analyzer (Applied Biosystems). Raw data were analyzed using DNA Sequencing Analysis Software v5.1 (Applied Biosystems) and then aligned and evaluated using Sequencher software version 4.9 (Gene Codes Corporation, Ann Arbor, MI, USA). All sequence variants were described according to the nomenclature recommended by Human Genome Variation Society by using GRCh37/hg19 as reference. To predict the impact of sequence variants, the Alamut software version 2.9 (Interactive Biosoftware, Rouen, France) was applied. Variants detected in intronic sequences were analyzed with the splice site prediction programs: GeneSplicer, Splice Site Finder, MaxEntScan, Human Splicing Finder, and NNSPLICE, all included in Alamut package. Population frequency of KIF23 sequence variants was obtained from NHLBI (National Heart, Lung, and Blood Institute) Exome Sequencing Project (ESP) or Exome Aggregation Consortium.
Mutations in KIF23 gene in tumors were retrieved from cBioPortal for Cancer Genomics13,14 and from COSMIC.15
SNP array genome-wide genotyping
High-resolution genome-wide single nucleotide polymorphism (SNP)-array genotyping was applied for identification of DNA copy number alterations (CNAs) in 10 NSCLC cases. DNA was genotyped according to manufacturer’s instructions on a SNP microarray (HumanOmniExpress-24 BeadChip, Illumina) containing more than 715,000 SNPs. The data were analyzed using GenomeStudio software (Illumina) and cnvPartition 3.2.0 was applied for CNAs detection by retrieving log R ratio (LRR) and the B allele frequency (BAF). LRR is the ratio between the observed and the expected probe intensity. A region without evidence of CNA shows an LRR around zero, and BAF 0, 0.5, or 1 correspond to the genotypes AA, AB, and BB. Human genome build 19 was used for assigning chromosome positions.
Results
Mutation screening
Tumor DNA from 15 NSCLC patients with elevated level of KIF23 was available for mutation screening (Figure 1). Detailed characteristics of these cases are shown in Table 1. In short, the age of the NSCLC patients (14 males and 1 female) ranged from 44 to 80 years (mean 65 years). Histologically, the tumors were presented in situ adenocarcinoma (n=2) and squamous cell carcinoma (n=13).
Figure 1.
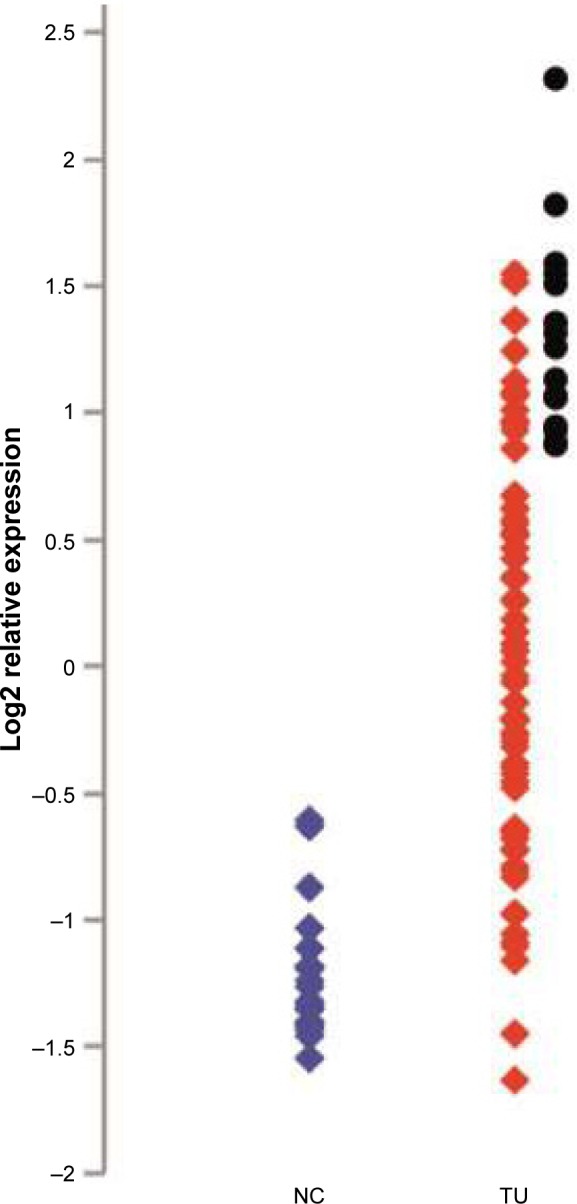
Relative expression of KIF23 in NSCLC and noncancerous lung samples from the study.
Notes: NSCLC samples with the highest KIF23 expression analyzed in the present work are indicated with black circles. Expression level was detected using Illumina, Human-6 microarrays, quantile-normalized, and log2-transformed as described.3 Differential gene expression analysis comparing NSCLC samples to noncancerous lung tissue was performed using t-tests with an empirical Bayes correction from the Bioconductor Limma package. On average, the KIF23 expression in tumor samples was 2.79 times higher than in noncancerous samples. Matched samples (tumor and noncancerous tissue from the same individual) were available only for a minority of the cases. Data from Valk et al.3
Abbreviations: TU, tumor; NC, non cancerous; NSCLC, non-small-cell lung cancer.
Somatic KIF23 mutations
DNA was extracted from the tumors with high content of cancer cells; therefore, we undertook Sanger sequencing as a method with detection level high enough for mutation detection in these samples. DNA from 15 NSCLC cases was available for Sanger sequencing.
All samples were found to harbor eight sequence alterations, six intronic (introns 2, 5, 6, and 14) and two exonic (exons 14 and 17). Genomic locations, frequencies, and bioinformatics prediction are shown in Table 2. Three of the changes (c.454–14delT, c.564–3delT, and c.1555+50delT) present in all NSCLC cases were deletions of a single T in the chain of 12–13 T; therefore, five control-DNA from healthy individuals were sequenced. Control sequences were identical to tumor sequences; thus, these three variants represent either a common sequence variant or a result of problematic sequencing of long-stretch repetitive DNA and, therefore, considered unable to cause overexpression of KIF23 in these samples.
Table 2.
KIF23 mutations in cancer
| Position GRCh37 | DNA, amino acid change | rs ID | MAFa | Prediction | Genotype | Cancer type | References |
|---|---|---|---|---|---|---|---|
| 69,709,610, intron 2 | c.82–112C>G | rs150343049 | G=0.0004 (1000G) | No predictable effect | C/G, case 68 | NSCLC | This study |
| 69,709,686, intron 2 | c.82–36G>A | rs3759825 | G=0.0749 (ExAC) | Splicing changeb | A/A, all cases | NSCLC | This study |
| 69,714,651, intron 5 | c.454–14delT | rs769142360 | −=0.0186 (ExAC) | No predictable effect | −/T, all cases | NSCLC | This study |
| 69,714,707, exon 6 | c.496G>A, p.E166K | – | NA | Possibly damagingc Splicing changed |
Melanoma | Cifola et al 201324 | |
| 69,715,488, intron 6 | c.564–10C>T | rs7180624 | C=0.2610 (ExAC) | No predictable effect | T/T, all cases | NSCLC | This study van de Wetering et al 201525 |
| 69,715,488, intron 6 | c.564 10delC | rs199725995 | −=0.2552 (ExAC) | Splicing changee | −/−, all cases | NSCLC | This study |
| 69,715,495, intron 6 | c.564 3delT | – | NA | Splicing changef | −/T, all cases | NSCLC | This study |
| 69,728,530, exon 13 | c.1326A>C, p.(V442=) | rs11852675 | C=0.0481 (ExAC) | Splicing changeg | A/C, cases 16, 108 | NSCLC | This study |
| 69,728,949, exon 14 | c.1443A>G, p.(P481=) | rs3825858 | A=0.1097 (ExAC) | No predictable effect | G/G, all cases | NSCLC | This study |
| 69,729,001, exon 14 | c.1495G>A, p.E499K | – | NA | Benignc Splicing changeh |
Melanoma | Cifola et al 201324 | |
| 69,729,111, intron 14 | c.1555+50delT | rs72161188 | −=0.0044 (ExAC) | No predictable effect | −/T, all cases | NSCLC | This study |
| 69,732,736, exon 17 | c.1977C>T, p.(T659=) | rs937724 | C=0.1095 (ExAC) | Splicing changei | T/T, all cases | NSCLC | This study |
| 69,732,770, exon 17 | c.2011C>T, p.R671W | rs377162847 | T=7.7e-5 (EVS) | Probably damagingc Splicing changej |
Colorectal cancer | DeRycke et al 201323 |
Notes:
Allele frequencies for all variants were retrieved from http://www.ncbi.nlm.nih.gov/variation/view using data from Exome Variant Server (http://evs.gs.washington.edu), Exome Aggregation Consortium (http://exac.broadinstitute.org) and a Deep Catalog of Human Genetic Variation (http://browser.1000genomes.org).
Created splicing signal at branch point according to SpliceSiteFinder-like (from 0/100 to 72.3/100), and increased splicing signal according to Human Splicing Finder (from 71.2/100 to 79.5/100).
According to PolyPhen2.
Created splicing signal at intron 5 acceptor site according to NNSPLICE (from 0/1.0 to 1.0/1.0). Increased splicing signal in exon 6 according to MaxEntScan (from 2.9/12 to 5.2/12), and increased splicing signal nearby according to MaxEntScan (from 1.5/12 to 4.2/12). Abolished splicing binding site for splicing factor SF2/ASF in exon 6 according to ESE finder. Abolished splicing signal at intron 6 donor site according to NNSPLICE (from 1.0/1.0 to 0/1.0).
Decreased splicing signal at acceptor site according to MaxEntScan (from 9.0/16 to 6.4/16).
Decreased splicing signal at acceptor site according to MaxEntScan (from 9.0/16 to 7.6/16) and increased splicing signal according to GeneSplicer (from 7.8/15 to 8.5/15).
Splicing binding sites for SC35 and SRp55 are changed to SF2/ASF and SF2/ASF (IgM-BrCa1) according to ESE finder.
Created splicing binding sites for splicing binding factor SC35 and SRp40 in exon 14.
Created splicing signal according to Human Splicing Finder (from 0/100 to 66.4/100). Binding sites for splicing factor SF2/ASF (IgM-BrCa1) were changed to a binding site for SRp40.
Two splicing binding sites for each of splicing binding factor SF2/ASF, SF2/ASF (IgM-BrCa1), and SRp40 were abolished.
Abbreviation: NSCLC, non-small-cell lung cancer.
Two case-specific sequence changes were detected in cases 16, 68, and 108 (Table 2). Variant c.82–112C>G in intron 2 (case 68) and synonymous change c.1326A>C, p.(V442=) (cases 16 and 108) have been previously detected by WES and reported in NHLBI ESP. Case-specific sequence variants are shown in Figure 2. We also tested those two case-specific variants in the corresponding DNA extracted from peripheral blood of the patients. c.82–112C>G was detected only in tumor tissue. c.1326A>C was present in both tumor and peripheral blood in the patient 108. Notably, patient 16 was heterozygous for this variant in peripheral blood but, probably, hemizygous for an A allele in the tumor.
Figure 2.

Sequences of case-specific heterozygous DNA variants present in 3 of 15 NSCLC samples.
Notes: (A) c.1326A>C in exon 13, case 16. The same change was seen in case 108 (C). (B) c.82–112C>G in intron 2, case 68. Upper panels show sequence of DNA from peripheral blood, and lower panels show sequence of tumor DNA.
Abbreviation: NSCLC, non-small-cell lung cancer.
CHR sequencing
Recently, it was shown that reduction of KIF23 expression of the mRNA and the protein level in different cell types is regulated by p53.16 The introduction of the mutations into the highly conserved CHR in the KIF23 promoter (TTTGAA) resulted in a significant increase of KIF23 promoter activity.16–18 Thus, we speculated whether the patients included in our study with increased KIF23 level carry any CHR mutations.
Nine NSCLC samples (15, 52, 73, 87, 104, 105, 108,109, and 114; Table S1) were subjected to Sanger sequencing of the CHR in KIF23 promoter. Wild-type sequence was detected in all tumors and corresponding peripheral blood samples (Figure S1A).
DNA copy number alterations
CNAs were analyzed by using high-resolution genome-wide genotyping of SNP-array technology (Illumina). All NSCLC cases tested showed extensively aberrant karyotypes, with large gains and losses involving even entire chromosomes (Tables S1 and S2; Figure S2). Average number of CNAs detected by cnvPartition frequency was 112 (n=10) ranging from 43 to 172 excluding case 87 with 11 CNAs detected (Table S1). Additional copy of chromosome 15 where KIF23 resides was detected in nine NSCLC cases examined (Figure 3). B-allele indicated presence of a third copy of chromosome 15. No CNAs were detected specifically at KIF23 locus (15q23).
Figure 3.

DNA CNA of chromosome 15 profiles in NSCLC. Whole-genome genotyping was performed using Illumina, SNP-array. Analysis was done using GenomeStudio and cnvPartition v. 3.2.0 with manual revision of the SNP-array profiles. BAFs for SNPs along the chromosome 15 show additional copy of chromosome 15 in 9 NSCLC cases. Only case 87 demonstrates normal profile with BAF =0.5 indicating absence of CNA on chromosome 15.
Abbreviations: CNA, copy number alteration; NSCLC, non-small-cell lung cancer; SNP, single nucleotide polymorphism; BAF, B-allele frequency.
Discussion
In this study, we investigated whether the somatic mutations in the KIF23 gene potentially present in NSCLC might explain protein overexpression. Fifteen cases of NSCLCs with high level of KIF23 expression were examined in regard to sequence changes and DNA CNAs. We considered that high percentage of cancer cells in our samples would allow mutation detection by Sanger sequencing, despite limited sensitivity for mutation detection in tumor samples representing heterogeneous mixture of normal and cancer cells. By assessment of CNAs by high-resolution genome-wide genotyping, we aimed to investigate whether KIF23 locus (15q23) was possibly amplified.
Recent publications highlighted role of KIF23 in cancer development and proposed KIF23 to be a potential biomarker for cancers with poor outcome.3–7 KIF23, also known as MKLP1, is a member of the kinesin-like protein family, which interacts with Aurora family protein kinases, regulates cell division, and ensures proper cytokinesis.19 We reported KIF23 mutation, p.P916R, as a genetic cause of a rare hereditary form of anemia, CDA III. The disease characterized by giant multinuclear erythroblasts in patients’ bone marrow is due to cytokinesis failure proven in knockdown experiments in HeLa cells.11 Correct performance of cell division is crucial for stable maintenance of the genome while a failure of cytokinesis might lead to aneuploidy and genomic instability, often associated with cancer, and probably representing initial step in cancer development.12 Notably, the incidence of blood cancer such as myeloma is higher in a large Swedish family with CDA III than a general population risk.11,20,21
By means of next generation sequencing, many tumors have been investigated in order to understand molecular mechanisms of cancer, and identify diagnostic and prognostic biomarkers. A mutation analysis of 3281 tumors from 12 cancer types revealed mutations in seven cell cycle genes among 127 significantly mutated,22 though KIF23 was not one of them. Nevertheless, in the study of 16 families with colorectal cancer among 375 missense single nucleotide variants, a rare KIF23 variant, c.2011C>T, p.R671W segregated with the disease in one family23 (Table 2). Furthermore, mutations in KIF23 were also found in melanoma cell lines derived from metastatic lesions patients24 (Table 2). Currently, by WES, 136 mutations in KIF23 have been detected in a range of tumours13 available via cBioPortal for Cancer Genomics. Majority of the mutations scattered all over the gene are missense mutations with five nonsense mutations and two mutations affecting splicing. A total of 123 mutations were submitted to COSMIC. KIF23 mutations were detected in a half out of 38 cancer types tested with minimum frequency 0.07% in hematopoietic and lymphoid tissues, and maximum mutation frequency of 2.03% in stomach. All tested tumor types (20 out of 38) confirmed upregulation of KIF23. Three out of ten tumors analyzed for CNAs showed gain, two confirmed loss, and five had both gain and loss.
In our study, we found in total ten sequence variants, eight were present in all NSCLC samples while two additional variants were found in three cases. One of those variants was present only in tumor DNA and not in blood. The bioinformatics tools retrieved predicted effect of all variants detected in this study. Three variants in exons were synonymous and did not cause amino-acid change although two of them (c.1326A>C and c.1977C>T) were predicted to have some effect on splicing. Variant c.564–3delT might affect splicing resulting in skipping of exon 7 though located in the chain of 12 Ts, it is difficult to assign this change to exact sequence position, and to exclude sequencing error. Bioinformatics tools such as Human Splicing Finder, SpliceSiteFinder, and ESE predict some changes in splicing factors binding sites for two other intronic variants found in this study (Table 2). Despite the prediction of subtle changes in splicing signals, the possible effect of those two variants cannot be completely neglected without further functional studies.
Intronic variant c.564–10C>T (rs7180624) was reported as somatic mutation in a living organoid of a patient with colorectal cancer;25 however, this variant was present in all NSCLC samples and healthy control cases in this study. None of the mutations reported in lung cancer on BioPortal, E203Q, D211Y, L525V, X188splice, G869W, and P415H were present in any of our samples.13
Recently, it was shown that p53 indirectly represses transcription of the cell cycle genes including KIF23 via p53–p21-DREAM-CDE/CHR pathway.17 p53 wild-type downregulates KIF23 expression at mRNA and protein levels by regulation of KIF23 promoter activity, and the CHR is the crucial element mediating p53-dependent repression of KIF23 expression.16 The increased KIF23 levels in this study could be explained by p53 mutations or deletions or mutations in the CHR in KIF23 promoter. We observed a slight but not significant correlation between KIF23 and TP53 expression (Figure S1B) and investigated the presence of CHR mutations. None of the examined samples were positive for the CHR mutations.
Since the sequence variants in our NSCLC cases were absent, previously reported, had high frequency in general population, or lacked convincing predicted impact on splicing or protein function, we investigated CNAs in these patients. Ninety percent of NSCLC were found to contain an additional copy of entire chromosome 15 without amplification of KIF23 locus at 15q23. Therefore, overexpression of KIF23 might be due to extra chromosome 15 in nine out of ten NSCLC patients examined here.
Conclusion
Importance of KIF23 as a key player in cytokinesis and chromosome segregation during cell cycle for cancer development is undeniable. Overexpression of KIF23 in several different cancer types promotes tumor cell proliferation, and also down-regulation of KIF23 decreases proliferation of glioma and lung cancer cells,4,6 indicating its potential as a prognostic bio-marker and therapeutic target. Sequence alterations in KIF23 detected in this study might represent “passenger” mutations with no obviously causative role for cancer progression, though further investigations in larger data sets are required, and more data mining should be done in the future.
Supplementary materials
(A) Sequencing of cell cycle homology region (CHR) in the promoter of KIF23 in 9 NSCLC tumors. The consensus wild-type sequence TTTGAAA was present in all samples. (B) Correlation between TP53 and KIF23. Scatter plot of log2 mean-centered expression values of KIF23 and TP53, TP53: Pearson’s correlation 0.256, uncorrected p-value =0.008. Pearson product-moment correlation (r) and uncorrected p-value were calculated using R software package.
Abbreviation: NSCLC, non-small-cell lung cancer.
CNV region display from GenomeStudio performed by cnvPartition 3.2.
Table S1.
Sanger sequencing and CNAs detection in NSCLC
| Sample | Sex | Histology | Sanger sequencing | CNAs number detected by SNP-array |
|---|---|---|---|---|
| 15 | Male | SCC | + | 153 |
| 16 | Male | SCC | + | NAa |
| 52 | Male | SCC | + | 148 |
| 66 | Male | SCC | + | NA |
| 67 | Male | SCC | + | 171 |
| 68 | Male | SCC | + | 172 |
| 73 | Male | SCC | + | 92 |
| 84 | Male | SCC | + | 97 |
| 87 | Male | SCC | + | 11 |
| 103 | Male | SCC | + | NA |
| 104 | Female | In situ adenocarcinoma | + | 68 |
| 105 | Male | SCC | + | 43 |
| 108 | Male | SCC | + | NA |
| 109 | Male | SCC | + | NA |
| 114 | Male | In situ adenocarcinoma | + | 164 |
Note:
Data not available, SNP-array was not performed due to a lack of DNA.
Abbreviations: SSC, squamous cell carcinoma; CNA, copy number alterations; NSCLC, non-small-cell lung cancer.
Table S2.
Chromosomal copy number alterations in NSCLC analyzed by genome wide genotyping using Illumina SNP-array
| Chromosome case | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15 | +1 | +2 | +3 | +4 | +5 | +6 | +7 | +8 | +9 | +10 | +11p | +12p, q | +13 | +15 | +16 | +17 | +18 | +19 | +20p | +21 | +22 | |
| 52 | +1p | +2 | +3 | +4 | +5q | +6 | +9p | +13 | +14 | +15 | +17p | +21 | ||||||||||
| 67 | +1 | +2 | +3 | +4 | +5 | +6 | +7 | +8 | +9p | +10 | +11 | +12q | +13 | +14 | +15 | +16 | +17p, q | +18 | +19 | +20p | +21 | +22q |
| 68 | +3p | +4 | +8q | +9p, q | +11q | +12 | +13 | +15 | +16 | +17p | +18 | +19 | +20p | +22 | ||||||||
| 73 | +1 | +2 | +3p | +4q | +5p, q | +6 | +7 | +9p | +10 | +11q | +12p | +13 | +15 | +16q | +17p | +18 | +19 | +21q | +22 | |||
| 84 | +1q | +2 | +3p, q | +4q | +5 | +6 | +7p | +8 | +9 | +10p | +11 | +12 | +13q | +14 | +15 | +16 | +17 | +18q | +19 | +20 | +21 | +22 |
| 87 | +1p | +3 | +4q | +5q | +6p | +7p | +8p | +9 | +10 | +11p | +13 | +14 | +16p | +17p, q | +22 | |||||||
| 104 | +1q | +2 | +3 | +4 | +5 | +6q | +7q | +8 | +9 | +11 | +12p, q | +13 | +14 | +15 | +17 | +18 | +19q | +20 | +21 | +22 | ||
| 105 | +1q | +2q | +3q | +4 | +5 | +6p | +8 | +9 | +10 | +12p | +13q | +14q | +15 | +16p | +17p | +18 | +19q | +20q | +21 | +22q | ||
| 114 | +1 | +2q | +3q | +4 | +5 | +6 | +7q | +8 | +9q | +10 | +11 | +12 | +13 | +14q | +15 | +16 | +17 | +18q | +19 | +21q | +22 |
Notes: Chromosome gains are marked with “+”. Partial gain of a short (p) or a long arm (q) of chromosome is indicated with +p or +q.
Abbreviations: NSCLC, non-small-cell lung cancer; SNP, single nucleotide polymorphism.
Acknowledgments
We acknowledge the TCGA Research Network for using the TCGA data set. This investigation was supported by a regional agreement between Umeå University and Västerbotten County Council in cooperation in the fields of Medicine, Odontology, and Health and Cancerforskningsfonden Norrland.
Footnotes
Author contributions
ALV and IG contributed to design of this study. TV, KL, and TA contributed to collection of patients’ data and expression studies. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Golub TR. Genome-wide views of cancer. N Engl J Med. 2001;344(8):601–602. doi: 10.1056/NEJM200102223440809. [DOI] [PubMed] [Google Scholar]
- 2.Nislow C, Lombillo VA, Kuriyama R, McIntosh JR. A plus-end-directed motor enzyme that moves antiparallel microtubules in vitro localizes to the interzone of mitotic spindles. Nature. 1992;359(6395):543–547. doi: 10.1038/359543a0. [DOI] [PubMed] [Google Scholar]
- 3.Valk K, Vooder T, Kolde R, et al. Gene expression profiles of non-small cell lung cancer: survival prediction and new biomarkers. Oncology. 2010;79(3–4):283–292. doi: 10.1159/000322116. [DOI] [PubMed] [Google Scholar]
- 4.Takahashi S, Fusaki N, Ohta S, et al. Downregulation of KIF23 suppresses glioma proliferation. J Neurooncol. 2012;106(3):519–529. doi: 10.1007/s11060-011-0706-2. [DOI] [PubMed] [Google Scholar]
- 5.Sun X, Jin Z, Song X, et al. Evaluation of KIF23 variant 1 expression and relevance as a novel prognostic factor in patients with hepatocellular carcinoma. BMC Cancer. 2015;15(1):961. doi: 10.1186/s12885-015-1987-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato T, Wada H, Patel P, et al. Overexpression of KIF23 predicts clinical outcome in primary lung cancer patients. Lung Cancer. 2016;92:53–61. doi: 10.1016/j.lungcan.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 7.Zou JX, Duan Z, Wang J, et al. Kinesin family deregulation coordinated by bromodomain protein ANCCA and histone methyltransferase MLL for breast cancer cell growth, survival, and tamoxifen resistance. Mol Cancer Res. 2014;12(4):539–549. doi: 10.1158/1541-7786.MCR-13-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murakami H, Ito S, Tanaka H, Kondo E, Kodera Y, Nakanishi H. Establishment of new intraperitoneal paclitaxel-resistant gastric cancer cell lines and comprehensive gene expression analysis. Anticancer Res. 2013;33(10):4299–4307. [PubMed] [Google Scholar]
- 9.Zhu C, Bossy-Wetzel E, Jiang W. Recruitment of MKLP1 to the spindle midzone/midbody by INCENP is essential for midbody formation and completion of cytokinesis in human cells. Biochem J. 2005;389(Pt 2):373–381. doi: 10.1042/BJ20050097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X, Zhou T, Kuriyama R, Erikson RL. Molecular interactions of Polo-like-kinase 1 with the mitotic kinesin-like protein CHO1/MKLP-1. J Cell Sci. 2004;117(Pt 15):3233–3246. doi: 10.1242/jcs.01173. [DOI] [PubMed] [Google Scholar]
- 11.Liljeholm M, Irvine AF, Vikberg AL, et al. Congenital dyserythropoietic anemia type III (CDA III) is caused by a mutation in kinesin family member, KIF23. Blood. 2013;121(23):4791–4799. doi: 10.1182/blood-2012-10-461392. [DOI] [PubMed] [Google Scholar]
- 12.Steigemann P, Wurzenberger C, Schmitz MH, et al. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136(3):473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 13.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(database issue):D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer M, Grundke I, Sohr S, et al. p53 and cell cycle dependent transcription of kinesin family member 23 (KIF23) is controlled via a CHR promoter element bound by DREAM and MMB complexes. PLoS One. 2013;8(5):e63187. doi: 10.1371/journal.pone.0063187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer M, Quaas M, Steiner L, Engeland K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016;44(1):164–174. doi: 10.1093/nar/gkv927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seguin L, Liot C, Mzali R, et al. CUX1 and E2F1 regulate coordinated expression of the mitotic complex genes Ect2, MgcRacGAP, and MKLP1 in S phase. Mol Cell Biol. 2009;29(2):570–581. doi: 10.1128/MCB.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neef R, Klein UR, Kopajtich R, Barr FA. Cooperation between mitotic kinesins controls the late stages of cytokinesis. Curr Biol. 2006;16(3):301–307. doi: 10.1016/j.cub.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 20.Haraldsdottir J, Sandstrom B. Detection of underestimated energy intake in young adults. Int J Epidemiol. 1994;23(3):577–582. doi: 10.1093/ije/23.3.577. [DOI] [PubMed] [Google Scholar]
- 21.Sandstrom H, Wahlin A, Eriksson M, Bergstrom I, Wickramasinghe SN. Intravascular haemolysis and increased prevalence of myeloma and monoclonal gammopathy in congenital dyserythropoietic anaemia, type III. Eur J Haematol. 1994;52(1):42–46. doi: 10.1111/j.1600-0609.1994.tb01283.x. [DOI] [PubMed] [Google Scholar]
- 22.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeRycke MS, Gunawardena SR, Middha S, et al. Identification of novel variants in colorectal cancer families by high-throughput exome sequencing. Cancer Epidemiol Biomarkers Prev. 2013;22(7):1239–1251. doi: 10.1158/1055-9965.EPI-12-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cifola I, Pietrelli A, Consolandi C, et al. Comprehensive genomic characterization of cutaneous malignant melanoma cell lines derived from metastatic lesions by whole-exome sequencing and SNP array profiling. PLoS One. 2013;8(5):e63597. doi: 10.1371/journal.pone.0063597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van de Wetering M, Francies HE, Francis JM, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161(4):933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Sequencing of cell cycle homology region (CHR) in the promoter of KIF23 in 9 NSCLC tumors. The consensus wild-type sequence TTTGAAA was present in all samples. (B) Correlation between TP53 and KIF23. Scatter plot of log2 mean-centered expression values of KIF23 and TP53, TP53: Pearson’s correlation 0.256, uncorrected p-value =0.008. Pearson product-moment correlation (r) and uncorrected p-value were calculated using R software package.
Abbreviation: NSCLC, non-small-cell lung cancer.
CNV region display from GenomeStudio performed by cnvPartition 3.2.
Table S1.
Sanger sequencing and CNAs detection in NSCLC
| Sample | Sex | Histology | Sanger sequencing | CNAs number detected by SNP-array |
|---|---|---|---|---|
| 15 | Male | SCC | + | 153 |
| 16 | Male | SCC | + | NAa |
| 52 | Male | SCC | + | 148 |
| 66 | Male | SCC | + | NA |
| 67 | Male | SCC | + | 171 |
| 68 | Male | SCC | + | 172 |
| 73 | Male | SCC | + | 92 |
| 84 | Male | SCC | + | 97 |
| 87 | Male | SCC | + | 11 |
| 103 | Male | SCC | + | NA |
| 104 | Female | In situ adenocarcinoma | + | 68 |
| 105 | Male | SCC | + | 43 |
| 108 | Male | SCC | + | NA |
| 109 | Male | SCC | + | NA |
| 114 | Male | In situ adenocarcinoma | + | 164 |
Note:
Data not available, SNP-array was not performed due to a lack of DNA.
Abbreviations: SSC, squamous cell carcinoma; CNA, copy number alterations; NSCLC, non-small-cell lung cancer.
Table S2.
Chromosomal copy number alterations in NSCLC analyzed by genome wide genotyping using Illumina SNP-array
| Chromosome case | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15 | +1 | +2 | +3 | +4 | +5 | +6 | +7 | +8 | +9 | +10 | +11p | +12p, q | +13 | +15 | +16 | +17 | +18 | +19 | +20p | +21 | +22 | |
| 52 | +1p | +2 | +3 | +4 | +5q | +6 | +9p | +13 | +14 | +15 | +17p | +21 | ||||||||||
| 67 | +1 | +2 | +3 | +4 | +5 | +6 | +7 | +8 | +9p | +10 | +11 | +12q | +13 | +14 | +15 | +16 | +17p, q | +18 | +19 | +20p | +21 | +22q |
| 68 | +3p | +4 | +8q | +9p, q | +11q | +12 | +13 | +15 | +16 | +17p | +18 | +19 | +20p | +22 | ||||||||
| 73 | +1 | +2 | +3p | +4q | +5p, q | +6 | +7 | +9p | +10 | +11q | +12p | +13 | +15 | +16q | +17p | +18 | +19 | +21q | +22 | |||
| 84 | +1q | +2 | +3p, q | +4q | +5 | +6 | +7p | +8 | +9 | +10p | +11 | +12 | +13q | +14 | +15 | +16 | +17 | +18q | +19 | +20 | +21 | +22 |
| 87 | +1p | +3 | +4q | +5q | +6p | +7p | +8p | +9 | +10 | +11p | +13 | +14 | +16p | +17p, q | +22 | |||||||
| 104 | +1q | +2 | +3 | +4 | +5 | +6q | +7q | +8 | +9 | +11 | +12p, q | +13 | +14 | +15 | +17 | +18 | +19q | +20 | +21 | +22 | ||
| 105 | +1q | +2q | +3q | +4 | +5 | +6p | +8 | +9 | +10 | +12p | +13q | +14q | +15 | +16p | +17p | +18 | +19q | +20q | +21 | +22q | ||
| 114 | +1 | +2q | +3q | +4 | +5 | +6 | +7q | +8 | +9q | +10 | +11 | +12 | +13 | +14q | +15 | +16 | +17 | +18q | +19 | +21q | +22 |
Notes: Chromosome gains are marked with “+”. Partial gain of a short (p) or a long arm (q) of chromosome is indicated with +p or +q.
Abbreviations: NSCLC, non-small-cell lung cancer; SNP, single nucleotide polymorphism.