

Abstract
β-Lactamases are enzymes produced by bacterial cells that provide resistance to β-lactam antibiotics. The CTX-M class of β-lactamases provides resistance against the antibiotic, cefotaxime, but not a related oxyimino-cephalosporin antibiotic, ceftazidime. β-lactamases that carry the P167S substitution, however, have been reported to provide ceftazidime resistance. The mechanism by which the P167S substitution expands the substrate profile of CTX-M enzymes is not known. In this study, CTX-M-14 was used as the model enzyme to study the structural changes caused by the P167S mutation that may accelerate ceftazidime turnover. X-ray crystallography was used to determine the structures of the CTX-M-14 P167S apo-enzyme along with the structures of the S70G/P167S, E166A/P167S and E166A mutant enzymes complexed with ceftazidime as well as the E166A/P167S apo-enzyme. The S70G and E166A mutations allow the capture of the enzyme-substrate complex and acylated forms of the ceftazidime molecule, respectively. The results showed a large conformational change in the Ω-loop of the CTX-M-14 ceftazidime acyl-enzyme complex of the P167S mutant but not in the enzyme-substrate complex suggesting the conformational change occurs upon acylation. The conformational change results in a larger active site cavity that prevents steric clash between the aminothiazole ring of ceftazidime and the Asn170 residue in the Ω-loop, allowing for accommodation of ceftazidime for hydrolysis. In addition, the conformational change in the Ω-loop was not observed in the E166A/P167S apo-enzyme, suggesting the presence of acylated ceftazidime influences the conformational change. Finally, the E166A acyl-enzyme structure with ceftazidime did not exhibit the altered Ω-loop conformation, indicating the P167S substitution is required for the change. Taken together, the results reveal that the P167S substitution and the presence of acylated ceftazidime both drive the structure towards a conformational change of the Ω-loop and that in CTX-M P167S enzymes found in drug-resistant bacteria this will lead to increased ceftazidime hydrolysis. This study demonstrates how a naturally occurring substitution can dramatically alter the active site to expand the substrate profile of an enzyme due to antibiotic selective pressure.
Graphical Abstract
INTRODUCTION
One of the most common modes of resistance to β-lactam antibiotics in Gram-negative bacteria is the production of β-lactamases.1 The CTX-M family of extended spectrum β-lactamases (ESBLs) was identified in the 1990’s shortly after the introduction of oxyimino-cephalosporin antibiotics such as cefotaxime and ceftazidime.2 The CTX-M enzymes belong to the class A family of β-lactamases. Class A β-lactamases are serine hydrolases that function by hydrolyzing the peptide bond within the β-lactam ring of β-lactam antibiotics through sequential acylation and deacylation steps. The catalytic Ser70 residue in the active site of the enzyme attacks the carbonyl carbon of the β-lactam leading to cleavage of the amide bond, protonation of the nitrogen atom in the β-lactam ring and formation of an acyl-enzyme intermediate with the antibiotic. Subsequently, Glu166 activates the catalytic water molecule which attacks the carbonyl carbon of the ester bond between the oxygen of the nucleophilic serine residue and the β-lactam ring leading to hydrolysis to regenerate the enzyme and release the inactivated antibiotic.2–4 This can be represented by the following reaction scheme:
In the reaction above, E represents the β-lactamase enzyme, ES represents the substrate complex, E-Ac represents the acyl-enzyme complex and P represents the product. The association and dissociation rate constants for the substrate complex are represented by k1 and k-1, respectively. The acylation and deacylation rate constants are represented by k2 and k3, respectively.
The CTX-M ESBLs are divided into 5 clusters based on amino acid sequence homology including CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-9 and CTX-M-25 with the names based on the prominent member of each subgroup. The subgroups differ from one another by ≥10% amino acid sequence divergence and each subgroup contains a number of variants that differ from one another by ≤5% sequence divergence.5,6 The CTX-M enzymes are named for their enhanced ability to hydrolyze cefotaxime over ceftazidime.7 In recent years, however, allelic variants have emerged that are able to hydrolyze ceftazidime. In particular, the D240G and P167S substitutions occur individually in multiple CTX-M subgroups where they enhance ceftazidime hydrolysis.8–11
The crystal structure of the D240G mutant enzyme has previously been determined and anisotropic B-factor analysis was used to demonstrate the increased flexibility of the B3 β-strand that forms the side of the catalytic cavity in CTX-M enzymes.10 The increased flexibility of the B3 β-strand in the D240G mutant enzyme is thought to expand its substrate profile by allowing access to the bulkier ceftazidime molecule.10,12
Similar to what is observed in the CTX-M enzymes carrying the D240G mutation, CTX-M enzymes that contain the P167S mutation exhibit increased rates of ceftazidime hydrolysis and thus higher resistance against the antibiotic. The increased catalytic efficiency for ceftazidime hydrolysis by CTX-M-14 P167S, comes at a cost of stability and decreased ability to provide resistance against older β-lactam antibiotics.8,11,13–15 The P167S mutation is located in the Ω-loop, which forms the bottom of the active site in CTX-M enzymes.16,17 It is located adjacent to the Glu166 residue that is critical in activating the catalytic water molecule for deacylation.4,17–19 Molecular dynamics simulations have previously been performed using Toho-1 (CTX-M-44) as the model enzyme to explain the increased hydrolysis of ceftazidime due to the P167S substitution.13 The results suggested that in the P167S mutant enzyme, the aminothiazole ring of ceftazidime is displaced to prevent steric clash with the hydroxyl group of Ser167 upon substrate binding. This displacement causes the C-4 carboxylate group of ceftazidime to form hydrogen bonds with Ser237 and Ser130 allowing for hydrolysis of ceftazidime.13 However, the crystal structure of the CTX-M P167S mutant enzyme has not been determined.
In this study, CTX-M-14 was used as the model enzyme to study the structural changes caused by the P167S mutation that may accelerate ceftazidime turnover. X-ray crystallography was used to determine the structures of the CTX-M-14 P167S apo-enzyme along with the structures of the S70G/P167S, E166A/P167S and E166A mutant enzymes complexed with ceftazidime as well as the E166A/P167S apo-enzyme. The S70G and E166A mutations allow the capture of the enzyme-substrate complex and acylated forms of the ceftazidime molecule, respectively. The results showed a large conformational change in the Ω-loop of the CTX-M-14 ceftazidime acyl-enzyme complex of the P167S mutant but not in the enzyme-substrate complex suggesting the conformational change occurs upon acylation. The conformational change results in a larger active site cavity that prevents steric clash between the aminothiazole ring of ceftazidime and the Asn170 residue in the Ω-loop, allowing for accommodation of ceftazidime for hydrolysis. In addition, the conformational change in the Ω-loop was not observed in the E166A/P167S apo-enzyme, suggesting the presence of acylated ceftazidime influences the conformational change. Finally, the E166A acyl-enzyme structure with ceftazidime did not exhibit the altered Ω-loop conformation, indicating the P167S substitution is required for the change.
MATERIALS AND METHODS
Bacterial Strains and Plasmids
The pET28a plasmid, which contains a kanamycin resistance marker, was used to express and purify the CTX-M-14 P167S, CTX-M-14 S70G/P167S, CTX-M-14 E166A/P167S and CTX-M-14 E166A mutant enzymes to obtain large amounts of protein for X-ray crystallography. The CTX-M-14-pET28a plasmid was created by inserting the gene encoding wild-type CTX-M-14 into the pET28a plasmid using the Gibson Assembly Kit (New England BioLabs, Ipswich, MA) as previously described.11 The CTX-M-14-pET28a plasmid was transformed into E. coli XL1-Blue [recA1, endA1, gyrA96, thi-1, hsdR17, supE44, relA1, lac, [F9 proAB lacIq lacZΔM15, Tn10 (tetr)]] (Stratagene, Inc., La Jolla, CA) for construction of the CTX-M-14 mutants by site-directed mutagenesis.20 The pET28a plasmids encoding the wild-type and mutant β-lactamases were transformed into the E. coli strain BL21 (DE3)(fhuA2[lon]ompTgal(λDE3) [dcm]ΔhsdSλDE3=λsBamHIoΔEcoRI-B int::(lacI::PlacUV5::T7gene1) i21 Δnin5) for protein expression and purification.21 Proteins expressed from the pET28a plasmid contained an N-terminal polyhistidine tag but lacked a signal sequence.
Site-Directed Mutagenesis
Phusion DNA Polymerase (New England BioLabs, Ipswich, MA) was used for site-directed mutagenesis to create all CTX-M-14 β-lactamase mutants in the CTX-M-14-pET28a plasmid. The primers listed in Table 1 were obtained from Integrated DNA Technologies (Coralville, IA) and used to introduce mutations into the CTX-M-14 gene. The sequences of all site-directed mutants were confirmed using DNA sequencing performed by Genewiz (South Plainfield, NJ).
Table 1.
Primers used for site-directed mutagenesis to create the mutant CTX-M enzymes
| Primer | Sequence |
|---|---|
| E166A | 5′-GATCGCACTGCTCCTACGCTGAAT-3′ |
| P167S-F | 5′-CTGGATCGCACTGAAAGCACGCTGAATACCGCC-3′ |
| P167S-R | 5′-GGCGGTATTCAGCGTGCTTTCAGTGCGATCCAG-3′ |
| P167S:E166A-F | 5′-CTGGATCGCACTGCTAGCACGCTGAATACC-3′ |
| S70G-F | 5′-TGAACGCTTTCCAATGTGCGG-3′ |
| S70G-R | 5′-ACTTGCGAAAGGTTACACGCC-3′ |
Protein Expression and Purification
The CTX-M-14-pET28a plasmid was introduced into E. coli BL21 (DE3) cells and the CTX-M-14 P167S, S70G/P167S, E166A/P167S, and E166A mutant enzymes were purified in this system as previously described.11 The protein lysate from the cell was isolated using a French press at 1250 psi. The protein was purified using a HisTrap FF column (GE Healthcare) and the N-terminal polyhistidine tag was cleaved using TEV protease as previously reported.11 An imidazole gradient was used to elute the protein from the HisTrap FF column. All β-lactamase enzymes were purified to >90% purity as determined by SDS-PAGE.
Crystallography
The PEGs, PACT and JCSG Core I Suites from Qiagen were used to screen conditions for crystal growth using the hanging drop vapor diffusion method. A Mosquito robot (TTP Labtech Ltd., Melbourn, UK) was used to set up crystallization screens with protein concentrated at 1100 μM. The CTX-M-14 P167S mutant enzyme crystallized in 0.2 M magnesium chloride, 0.1 M TRIS pH8, 20% (w/v) PEG 6000. The CTX-M-14 S70G/P167S mutant enzyme crystallized in 0.2 M ammonium chloride, 20% (w/v) PEG 3350. The CTX-M-14 E166A/P167S enzyme crystallized in 0.2 M sodium fluoride, 20% (w/v) PEG 3350. The CTX-M-14 E166A enzyme crystallized in 0.2 M ammonium nitrate, 20% (w/v) PEG 3350. The crystals of CTX-M-14 S70G/P167S, E166A/P167S, and E166A complexed with ceftazidime were obtained by soaking the protein crystals in 50 mM ceftazidime overnight, before collecting data. 25% glycerol was used as the cryoprotectant in all cases. The data was collected at the Berkeley Center for Structural Biology at the Advanced Light Source synchrotron beam line. All data was processed using iMosflm22 and the CCP4i Suite.23 Phaser24 was used for molecular replacement using CTX-M-14 (PDB ID: 1YLT)10 as the model enzyme for phasing. Coot25 was used to fit the model to the density and the phenix.refine26 and REFMAC527 programs were used for refinement. The data collection and refinement statistics for all structures are reported in Table 2. All figures were created using the UCSF Chimera program.28
Table 2.
X-ray crystallography data collection and refinement statistics for CTX-M-14 mutant enzymesa.
| Data collection | P167S 5TWDb |
E166A/P167S 5VTH |
S70G/P167S/CAZ 5TWE |
E166A/P167S/CAZ 5TW6 |
E166A/CAZ 5U53 |
|---|---|---|---|---|---|
| Space group | P 32 2 1 | P 32 2 1 | P 32 2 1 | P 32 2 1 | P 41 21 2 |
| a,b,c (Å) | 41.6, 41.6, 231.9 | 41.7, 41.7, 233.1 | 41.6, 41.6, 232.2 | 41.5, 41.5, 231.2 | 42.0, 42.0, 262.9 |
| α,β,γ (°) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 90.0 |
| Resolution Range (Å) | 30.60 - 1.70 (1.79-1.70) | 30.69 – 2.20 (2.28-2.20) | 30.62 -1.50 (1.58 -1.50) | 77.05 -1.70 (1.76 -1.70) | 30.33 -1.40 (1.48 -1.40) |
| R-merge (%) | 5.3 (12.0) | 13.3 (31.1) | 12.2 (26.6) | 9.2 (50.9) | 14.4 (98.4) |
| Rpim (%) | 1.6 (3.5) | 7.0 (15.1) | 4.3 (7.9) | 5.2 (28.1) | 3.8 (24.9) |
| I/sigma | 30.6 (16.5) | 6.1 (3.4) | 13.4 (7.9) | 7.2 (2.3) | 11.0 (2.9) |
| CC(1/2) | 1.0 (1.0) | 1.0 (1.0) | 1.0 (1.0) | 1.0 (0.8) | 1.0 (0.9) |
| Multiplicity | 11.9 (12.5) | 2.0 (2.0) | 2.0 (2.0) | 4.7 (5.0) | 2.0 (2.0) |
| Completeness (%) | 100 (100) | 95 (98) | 100 (100) | 100 (100) | 98 (99) |
| Wilson B-factor (Å2) | 12.93 | 23.31 | 14.2 | 17.5 | 11.3 |
| No. of unique reflections | 26936 (2641) | 12217 (1195) | 38791 (3790) | 26575 (2595) | 47186 (4664) |
| Refinement | |||||
| R-work/R-free (%) | 21.0/23.8 | 22.0/26.9 | 15.1/16.9 | 17.9/22.16 | 17.0/19.5 |
| No. of protein residues | 260 | 261 | 262 | 259 | 261 |
| No. of water molecules | 266 | 146 | 324 | 248 | 328 |
| Ramachandran | |||||
| Favored (%) | 98 | 97 | 98 | 98 | 98 |
| Outliers (%) | 0 | 0 | 0 | 0 | 0 |
| RMS deviations | |||||
| Bond length (Å) | 0.01 | 0.008 | 0.007 | 0.009 | 0.01 |
| Bond angles (°) | 0.98 | 0.82 | 1.49 | 0.96 | 1.12 |
| Average B-factor (Å2) | 24.8 | 36.1 | 22.2 | 27.4 | 16.5 |
| Protein | 24.1 | 36.2 | 19.9 | 26.1 | 14.0 |
| Ligand | - | 52.2 | 29.7 | 23.1 | |
| Solvent | 30.3 | 34.0 | 33.6 | 37.0 | 30.9 |
Values in parentheses in the body of the table indicate highest resolution shell.
Protein data bank code for the deposited structure (PDB ID).
RESULTS
Crystal Structure of the CTX-M-14 P167S Apo-enzyme
The structure of the P167S mutant enzyme was determined to 1.7 Å resolution to study the effects of the P167S mutation on the structure of the CTX-M-14 β-lactamase. The Ser167 residue is observed to adopt two conformations in the P167S mutant enzyme (Fig. 1). In one conformation, the hydroxyl group of Ser167 forms a hydrogen bond with Asn104, which is located in the 103-VNYN-106 loop that forms the side of the active site in CTX-M enzymes (Fig. 1). In the alternate conformation, the hydroxyl group of Ser167 faces the solvent. The overall structures of the wild-type CTX-M-14 enzyme (PDB ID: 1YLT)10 and the P167S mutant enzyme are very similar as evidenced by an RMSD of 0.32 Å between the Cα atoms of the two enzymes. In addition, key interactions that have previously been reported between residues in the Ω-loop of class A ESBLs are observed in the P167S mutant and wild-type enzymes.10,13,14,29,30 For example, the hydrogen bond between Arg164 and Thr171 is preserved, as is the hydrogen bonding network between Glu166 and Asn170 and the deacylating water molecule. In addition, the salt bridges formed between Arg164 and Asp179, Lys73 and Glu166, Arg161 and Asp163, and Asp176 and Arg178 that maintain the Ω-loop’s structural integrity are also present. In the wild-type CTX-M-14, as well as most class A β-lactamases, the peptide unit preceding Pro167 is in cis conformation. Ser167 in the mutant also exhibits a cis peptide despite a non-proline cis conformation being energetically unfavorable.31,32 In fact, the presence of the cis peptide would be expected to be destabilizing and could contribute to the observed decreased stability of the P167S enzyme relative to wild type.11 Overall, there is significant similarity between the structures of the wild-type and P167S apo-enzyme.
Figure 1.

Alternate conformations of Ser167 in the CTX-M-14 P167S apo-enzyme structure. A ribbon diagram of CTX-M-14 P167S is shown on the left. The right panel shows residues Glu166, Ser167, and Asn170 in the Ω-loop and the Ser70, Asn104, and the Asn132 residues represented as gray sticks. It depicts the two different conformations adopted by Ser167 in the apo-enzyme. The catalytic water molecule that is coordinated by Ser70, Glu166 and Asn170 is represented as a red sphere and dashed lines depict hydrogen bonds. Oxygen atoms are represented in red and nitrogen atoms are represented in blue.
Crystal Structure of the CTX-M-14 S70G/P167S Enzyme with Ceftazidime
Because there were only minor structural changes due to the P167S mutation in the apo-enzyme, the effects of the P167S mutation on the structure of the non-covalent complex with ceftazidime was determined. The mutation of serine 70 to glycine was used to prevent the formation of the acyl-enzyme and ceftazidime hydrolysis. The structure of CTX-M-14 S70G/P167S/CAZ was determined to 1.5 Å resolution. The overall structure was very similar to that of the CTX-M-14 P167S apo-enzyme including the cis peptide bond preceding Ser167 (Fig. 2A,B,E). The hydrogen bonds and ionic interactions observed in the Ω-loop of the CTX-M-14 P167S apo-enzyme were also found when ceftazidime non-covalently binds to the active site (Fig. 2B,E).
Figure 2.

Electron density and conformation of Ω-loop residues 166-172 in the CTX-M-14 mutant enzymes crystallized with and without ceftazidime. A) Structural alignment of the P167S apo-enzyme (gray), S70G/P167S/CAZ (olive green), E166A/P167S/CAZ (orange), E166A/P167S apo-enzyme (cyan) and E166A/CAZ (blue) β-lactamase structures. Note the ceftazidime molecules are not shown for clarity. Conformational changes in the Ω-loop between the crystal structures following the same color scheme (inset). In the following panels, for each of the crystal structures, simulated annealing Fo-Fc map for the Ω-loop (contoured at 2.5 σ) alongside with the stick model using the same color scheme as in A) is shown. B) P167S apo-enzyme, C) E166S/P167S apo-enzyme, D) E166A/P167S/CAZ E) S70G/P167S/CAZ, and F) E166A/CAZ. Oxygen and nitrogen atoms in the stick model are represented in red and blue, respectively.
Currently, there are no reported crystal structures of β-lactamases in complex with an intact ceftazidime molecule. The structure of CTX-M-14 S70G/P167S/CAZ reveals the placement of the intact ceftazidime molecule within the active site of the enzyme (Fig. 3A). Electron density is clearly present for the β-lactam ring, dihydrothiazine ring and the side chain aminothiazole ring as well as the amide of the acyl side chain of ceftazidime. However, electron density is lacking for the pyridine ring and the dimethyl and carboxyl groups on the acyl side chain of ceftazidime. The ceftazidime molecule has an average occupancy of 0.8 in the CTX-M-14 S70G/P167S/CAZ crystal structure. The partial density for ceftazidime may be due to high mobility of these groups and could be related to the high Km observed for ceftazidime hydrolysis in the CTX-M-14 P167S enzyme.11
Figure 3.

Electron density for ceftazidime in the crystal structures of A) CTX-M-14 S70G/P167S/CAZ, B) E166A/P167S/CAZ and C) E166A/CAZ mutant enzymes. The alignment of the CTX-M-14 S70G/P167S/CAZ, E166A/P167S/CAZ and E166A/CAZ mutant enzymes is shown at the top. A) The intact ceftazidime molecule displayed in the active site of S70G/P167S/CAZ. Partial density is present for ceftazidime. B) The acylated ceftazidime molecule displayed in the active site of E166A/P167S/CAZ. C) The acylated ceftazidime molecule displayed in the active site of E166A/CAZ. For each of the crystal structure Fo-Fc (2.5 σ) and 2Fo-Fc (1.0 σ) maps are shown in green (top) and blue (bottom), respectively. In all panels, ceftazidime is shown as a stick model with oxygen atoms represented in red, nitrogen atoms in blue and sulfur atoms in yellow.
A key feature of β-lactam hydrolysis is the presence of the oxyanion hole. In class A β-lactamases, the main chain nitrogen of residues 70 and 237 form hydrogen bonds with the carbonyl oxygen of the β-lactam and this is thought to stabilize the developing negative charge on the tetrahedral intermediate during acylation.33 In addition, the carboxylate group at the C3/C4 position of β-lactam substrates forms hydrogen bonds to Ser/Thr235 in class A enzymes including wild-type CTX-M-14.34–37 The structure of the non-covalent complex (S70G/P167S/CAZ) shows the carbonyl oxygen of the β-lactam ring present in the oxyanion hole forming hydrogen bonds with the main chain NH of residues 70 and 237. In addition, the carboxylate group on the dihydrothiazine ring of ceftazidime forms hydrogen bonds with the side chain hydroxyl groups of Ser130 and Thr235 (Fig. 4A). Thus, the β-lactam and dihydrothiazine rings of ceftazidime are positioned similarly to previously observed non-covalent complexes with the good substrate, ceftotaxime.20
Figure 4.

The hydrogen bond network between key active site residues and ceftazidime in the CTX-M-14 S70G/P167S/CAZ, E166A/P167S/CAZ and E166A/CAZ mutant enzymes. A) Hydrogen bonds between the carbonyl and C3 carboxylate groups of ceftazidime and key active site residues represented as green stick models in S70G/P167S/CAZ. The β-lactam carbonyl oxygen is present in the oxyanion hole. B) Hydrogen bonds between the carbonyl and C3 carboxylate groups of ceftazidime and active site residues depicted as orange stick models in E166A/P167S/CAZ. The carbonyl group is positioned within the oxyanion hole for optimal hydrolysis in the enlarged active site. C) The acyl side chain forms hydrogen bonds with Asn104, Asn132 and Ser237 and is well accommodated in the enlarged active site of the E166A/P167S/CAZ enzyme. C) Hydrogen bond interactions between the carbonyl and C3 carboxylate groups of ceftazidime and active site residues depicted as blue stick models in E166A/CAZ. The interactions are similar to those observed in the E166A/P167S/CAZ enzyme. Hydrogen bond contacts are shown between the acyl side chain of ceftazidime and the narrow active site of the E166A/CAZ enzyme. Ceftazidime is represented as gray sticks in all panels. Hydrogen bonds are represented by dotted lines. Oxygen, nitrogen, and sulfur atoms are shown in red, blue, and yellow, respectively.
Crystal Structure of the CTX-M-14 Acyl-enzyme (E166A/P167S) with Ceftazidime
The mutation, E166A, removes the catalytic base for activating the deacylating water molecule allowing the formation of a stable acyl-enzyme with β-lactams. The structure of the acyl-enzyme of CTX-M-14 E166A/P167S and ceftazidime was determined to 1.7 Å resolution. The overall structure of CTX-M-14 E166A/P167S/CAZ was similar to the structure of the P167S apoenzyme except within the Ω-loop (Fig. 2D). All of the hydrogen bonds and ionic interactions that were observed in the P167S apo-enzyme outside of the Ω-loop were also observed in the E166A/P167S/CAZ mutant enzyme, with the exception of those involving Glu166.
The Ω-loop of the E166A/P167S acyl-enzyme with ceftazidime exhibited a large conformational change that altered the positioning of residues 168-TLN-170 (Fig. 2D). Specifically, the unraveling of the Ω-loop in the E166A/P167S/CAZ mutant enzyme resulted in the rotation of the Leu169 side chain out of the active site to face the solvent (Fig. 2D). It also caused a 4.2 Å shift between the Cα atoms of Asn170 in the E166A/P167S/CAZ acyl-enzyme when compared to the P167S apo-enzyme (Fig. 5A). The increased flexibility of the Ω-loop in the E166A/P167S/CAZ acyl-enzyme structure is further supported by an increase in B-factors for residues 166–172 (Fig. 5B). The change in conformation is also associated with a change in the peptide bond preceding Ser167 to trans. The conformational change in the Ω-loop results in the formation of a more open, larger active-site cavity that is better able to accommodate ceftazidime for hydrolysis. Finally, note that the 168-TLN-170 region of the Ω-loop does not interact with any symmetry-related neighboring molecule for this or any of the structures reported here.
Figure 5.

Position of residue Asn170 and temperature factors for residues in the Ω-loop of the CTX-M-14 mutant enzymes crystallized with and without ceftazidime. A) The Asn170 residue in the Ω-loop is displayed as a gray model in the P167S apo-enzyme, as a green model in S70G/P167S/CAZ, as an orange model in E166A/P167S/CAZ, as a blue model in E166A/CAZ and as a cyan model in E166A/P167S apo-enzyme. The conformational change in the Ω-loop of the E166A/P167S/CAZ mutant enzyme shifts the Asn170 residue resulting in the formation of a wider active site cavity to accommodate ceftazidime. Oxygen atoms are represented in red and nitrogen atoms are represented in blue. B) Graph representing the temperature factors of residues 160-180 in the Ω-loop. The temperature factors are normalized by dividing by the Wilson B factor for each structure. The P167S apo-enzyme is represented as gray triangles, the S70G/P167S/CAZ mutant enzyme is represented as green circles, the E166A/P167S/CAZ mutant enzyme is represented as orange squares, the E166A/CAZ mutant enzyme is represented as blue triangles and the E166A/P167S apo-enzyme is shown with cyan diamonds. The flexibility in the Ω-loop of the E166A/P167S/CAZ mutant enzyme is supported by an increase in B-factors.
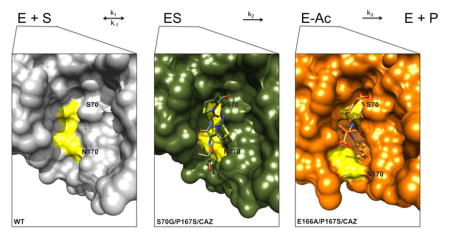
The crystal structure of CTX-M-14 E166A/P167S/CAZ also revealed a clearly defined electron density due to the presence of the acylated ceftazidime molecule within the active site of the enzyme (Fig. 3B). The acylated ceftazidime molecule has an average occupancy of 1.0 in the CTX-M-14 E166A/P167S/CAZ crystal structure and clearly shows a covalent bond between the oxygen of serine 70 and the carbonyl group of the β-lactam carbonyl. The ceftazidime molecule itself does not contain the pyridine ring, which is consistent with cephalosporin antibiotic fragmentation upon acylation.38–40 The hydrogen bond interactions between ceftazidime and key residues in the active site are highlighted in Figure 4B.34 The entire ceftazidime molecule is accommodated in the active site of E166A/P167S/CAZ (Fig. 3B & 4B). The carboxylate group on the dihydrothiazine ring forms hydrogen bonds with Ser130, Ser237 and Thr235, but not with Ser70. This allows the carbonyl group of the β-lactam ring to be placed within the oxyanion hole, composed of the amide nitrogens Ser70 and Ser23733, for optimal hydrolysis (Fig. 4B). A major difference from the S70G/P167S/CAZ structure is the orientation of the acyl side chain where in the non-covalent complex the aminothiazole ring is oriented up away from the active site while in the E166A/P167S/CAZ complex it is buried deeply within the active site, which is possible due to the movement of the omega loop (Fig. 6). The acyl side chain of ceftazidime in the E166A/P167S/CAZ complex forms hydrogen bonds with Cys69, Asn104, Asn132 and Thr171 (Fig. 4B). The ceftazidime molecule does not adopt this conformation in the narrow active site of the S70G/P167S/CAZ mutant enzyme because it would cause a steric clash between the aminothiazole ring and residue Asn170 in the Ω-loop (Fig. 6). Overall, these results demonstrate that the P167S substitution causes a conformational change in the Ω-loop upon substrate acylation and that the larger active site cavity is essential to alleviate steric clash in accommodating ceftazidime for hydrolysis.
Figure 6.

Protein surface representation of wild-type CTX-M-14 (gray), S70G/P167S with bound ceftazidime (green), E166A/P167S with acylated ceftazidime (orange) and E166A with acylated ceftazidime (blue). The positions of the Ser70 and Asn170 residues are highlighted in yellow. Ceftazidime is shown as green sticks and acylated ceftazidime as orange sticks for E166A/P167S and blue sticks for E166A. Oxygen and nitrogen atoms are colored red and blue respectively.
Additional electron density was observed in the active site adjacent to the ceftazidime molecule in the 2Fo-Fc map contoured at 1σ. Ethylene glycol, which was a part of the initial crystallization condition, was fit to the density and has an average B-factor of 28.8. The ethylene glycol molecule does not interact with the Ω-loop of the enzyme, and is unlikely to cause the conformational change observed. However, it does hydrogen bond to the aminothiazole ring of ceftazidime, potentially stabilizing the substrate in the active site.
Crystal Structure of the CTX-M-14 E166A/P167S Apo-Enzyme
In order to examine if the conformational change of the Ω-loop observed in the E166A/P167S/CAZ acyl-enzyme structure is triggered by the presence of acylated ceftazidime, the structure of the E166A/P167S apo-enzyme was determined to 2.2 Å resolution. Interestingly, the conformation of the Ω-loop is closed, resembling the P167S apo structure (Fig. 2C). The Leu169 and Asn170 residues are within the active site in a similar position as that in the P167S apo-enzyme structure. The side chain of Asn170, however, assumes a slightly altered conformation compared to that in the P167S apo-enzyme structure (Fig. 2B, C). However, the closed conformation of the Ω-loop in the E166A/P167S apo-enzyme and the open conformation in the E166A/P167S/CAZ acyl-enzyme structure indicate the presence of the acylated ceftazidime facilitates the change to the open Ω-loop conformation.
It is also noteworthy that, in contrast to the P167S apo-enzyme structure, the peptide bond preceding Ser167 is trans in the E166A/P167S apo-enzyme structure despite the Ω-loop being in a closed conformation (Fig. 2C). Thus, the presence of the trans peptide bond preceding residue Ser167 is not sufficient to convert the Ω-loop to the open conformation.
A comparison of the P167S apo-enzyme structure with the E166A/P167S apo-enzyme structure suggests the E166A substitution, which was included in this study to trap the acylated version of ceftazidime, also affects the peptide bond preceding Ser167 in that the bond is cis in the P167S apo-enzyme and trans in the E166A/P167S apo-enzyme structure (Fig. 2B,C). The Glu166 side chain forms a hydrogen bond with the Asn170 side chain in the wild type and P167S enzymes, which may act to stabilize the cis peptide bond at residue 167. However, as noted below, the E166A substitution is not sufficient and the P167S substitution is also required for conversion of the peptide bond from cis to trans.
Crystal Structure of the CTX-M-14 Acyl-enzyme (E166A) with Ceftazidime
The crystal structure of the CTX-M-14 E166A mutant enzyme with ceftazidime (CTX-M-14 E166A/CAZ) was determined to examine if the conformational change observed in the Ω-loop of the E166A/P167S/CAZ acyl-enzyme resulted from the P167S mutation or the E166A mutation. The crystal structure of E166A/CAZ acyl-enzyme was determined to 1.4 Å resolution. The Ω-loop does not display a conformational change as observed in the E166A/P167S/CAZ crystal structure (Fig. 2F). This is confirmed by the electron density and low B-factors of the residues in the Ω-loop in E166A/CAZ acyl-enzyme structure (Fig. 2F & 5B). In addition, the peptide bond preceding Pro167 is cis in the E166A/CAZ acyl-enzyme. Since the E166A/P167S/CAZ and E166A/P167S apo-enzyme structures have a trans peptide bond at residue 167, this result indicates the P167S substitution contributes to the presence of a trans bond. This is consistent with the unfavorable energetics of a non-prolyl cis peptide bond.31,32 The lack of a conformation change in the Ω-loop of the acyl-enzyme structure of E166A/CAZ also indicates that the P167S mutation, rather than E166A, is responsible for the conformational change observed in the E166A/P167S/CAZ mutant enzyme.
The position of the ceftazidime molecule in the active site of the E166A/CAZ mutant enzyme is clearly defined by electron density (Fig. 3C). The acylated ceftazidime molecule has an average occupancy of 0.88 in the E166A/CAZ crystal structure. The hydrogen bond interactions between ceftazidime and key residues in the active site are highlighted in Figure 4C. The region near the acylation site of the ceftazidime molecule, including the oxyanion hole, makes similar interactions to those found in the E166A/P167S/CAZ structure, however, the catalytic efficiency (kcat/KM) of the wild-type enzyme is 10-fold lower than that of the CTX-M-14 P167S mutant enzyme (Fig. 4B).10–11 This difference may be explained by changes in the conformation of the aminothiazole ring, which is not situated as deep in the active site as in the E166A/P167S enzyme (Fig. 6). The shallower binding observed in the narrow active site of the E166A/CAZ acyl-enzyme results in fewer hydrogen bond and van der Waals contacts between the acyl side chain of ceftazidime and the enzyme, which may lead to an increased Km and decreased catalytic efficiency (Fig. 4B). The aminothiazole ring of ceftazidime does not take on the same conformation in the E166A/CAZ acyl-enzyme as it does in the E166A/P167S/CAZ enzyme because it would clash with the Asn170 residue. Overall, this suggests that the active site expansion observed in the E166A/P167S/CAZ structure is necessary for proper placement and efficient hydrolysis of ceftazidime.
DISCUSSION
The P167S mutation has been identified in both the CTX-M-1 and CTX-M-9 subfamilies but not in other subfamilies.6 The fact that this mutation has arisen independently in two different subfamilies of CTX-M β-lactamases suggests that it plays an important role in the evolution of these enzymes. Escherichia coli cells that express the P167S variant have an increased MIC for ceftazidime but a decreased MIC for ampicillin, cephalothin and cefotaxime, which are good substrates for the wild-type enzyme.8,11,13–15,41 In addition, the catalytic efficiency (kcat/KM) of the P167S mutant enzyme is increased 10-fold for ceftazidime hydrolysis in comparison to the wild-type enzyme.11 However, the increased activity against ceftazidime has been associated with decreased stability and protein expression of the mutant enzyme.11
Molecular dynamics simulations in Toho-1 P167S have previously suggested that the Ser167 side chain would sterically block binding of the aminothiazole ring of ceftazidime leading to an altered placement of the substrate and formation of new interactions for hydrolysis in the narrow active site of CTX-M ESBLs.13 However, the findings of this study suggest that active site expansion is necessary for CTX-M enzymes to accommodate ceftazidime for hydrolysis.
The crystal structure of CTX-M-14 E166A/P167S/CAZ acyl-enzyme reveals a conformational change leading to enlargement of the active site (Figs. 2,6). A comparison of X-ray structures determined here suggests both the P167S substitution and the presence of acylated ceftazidime contribute to the conformational change. The P167S apo-enzyme and the S70G/P167S/CAZ structure with the ceftazidime substrate bound do not exhibit a conformational change in the Ω-loop. In addition, in both of these structures there is a cis peptide bond preceding Ser167. The addition of the E166A substitution to P167S to create the E166A/P167S enzyme results in a trans peptide bond preceding Ser167 but no conformational change in the Ω-loop in the apo-enzyme. The addition of ceftazidime to E166A/P167S crystals results in the trapped acyl-enzyme, which displays a trans bond preceding Ser167 and the conformational change opening the Ω-loop. In contrast, the structure of the E166A enzyme with acylated ceftazidime shows a cis peptide bond preceding Pro167 and no conformational change in the Ω-loop. Taken together, these results suggest P167S and acylated ceftazidime contribute to a trans peptide bond preceding residue 167 and the formation of the open conformation of the Ω-loop. However, the E166A substitution may also contribute in that the E166A/P167S apo-enzyme displays a trans peptide bond at residue 167. Nevertheless, P167S and acylated ceftazidime are also necessary in that the E166A/CAZ structure exhibits a cis peptide bond and no conformational change of the Ω-loop. The E166A substitution was included in the study in order to trap the acylated ceftazidime but this substitution will not be found in enzymes from drug resistant bacterial isolates because it is catalytically impaired. Our interpretation of the results is that the P167S substitution and the presence of acylated ceftazidime both drive the structure towards a trans peptide bond at residue 167 and the conformational change of the Ω-loop and that in CTX-M P167S enzymes found in drug-resistant bacteria this will lead to the trans peptide bond and conformational change leading to increased ceftazidime hydrolysis. It is not possible to determine the structure of the P167S enzyme with acylated ceftazidime without the E166A substitution as the reaction proceeds to completion.
Active site expansion for the accommodation of oxyimino cephalosporin antibiotics has previously been observed in TEM β-lactamases.29,42 A TEM-1 triple mutant (W165Y/E166Y/P167G) and the natural mutant TEM-64 (E104K/R164S/M182T) both contain substitutions within the Ω-loop that increase their activity against ceftazidime.29,42 The P167G substitution in TEM-1 W165Y/E166Y/P167G functions similarly to the P167S substitution in CTX-M-14, by allowing the peptide bond between Glu166 and Pro167 to shift from cis to trans and allow an altered conformation of the Ω-loop.29,43 In TEM-64, the R164S substitution abolishes the ionic interaction between the conserved Arg164 and Asp179 residues on either end of the Ω-loop of class A β-lactamases leading to a conformational change, although the change is less pronounced than that observed for TEM-1 W165Y/E166Y/P167G or the CTX-M-14 E166A/P167S/CAZ structures (Fig. 7).17,42–44 In contrast to the TEM-1 W165Y/E166Y/P167G and CTX-M-14 E166A/P167S/CAZ structures, the peptide bond preceding Pro167 in TEM-64 is cis. The crystal structures of the TEM-1 W165Y/E166Y/P167G apo-enzyme and TEM-64 with a boronic acid inhibitor, both reveal a conformational change in the Ω-loop similar to the CTX-M-14 E166A/P167S/CAZ structure (Fig. 7). In contrast to TEM-1 W165Y/E166Y/P167G, the conformational change in the Ω-loop is not observed in the CTX-M-14 P167S apo-enzyme or the CTX-M-14 S70G/P167S/CAZ structure, suggesting the change occurs in the CTX-M enzymes upon substrate acylation (Fig. 2 & 5A).
Figure 7.

Position of the Ω-loop in the structures of TEM-1 W165Y/E166Y/P167G, TEM-64 crystallized with boronic acid inhibitor, CTX-M-14 E166A/P167S/CAZ and wild-type CTX-M-14. The conformation of the Ω-loop is represented in dark gray for TEM-1 W165Y/E166Y/P167G (PDB ID: 4RVA), in white for TEM-64 crystallized with boronic acid inhibitor (PDB ID: 1JWZ), in orange for CTX-M-14 P167S/E166A/CAZ, and in pink for wild-type CTX-M-14 (PDB ID: 1YLT). The catalytic Ser70 residue and the Asn170 residue are represented as stick models in all enzymes. The Ω-loop is unraveled and takes on different conformations in the three mutant enzymes. All conformations of the Ω-loop in the mutant enzymes result in an active site cavity that is wider than that of the wild-type enzyme. The substrates have been omitted for clarity. Oxygen atoms are shown in red and nitrogen atoms are shown in blue.
Molecular docking studies with ceftazidime in TEM-1 W165Y/E166Y/P167G as well as the structure of TEM-64 crystallized with boronic acid inhibitor and the structure of CTX-M-14 E166A/P167S/CAZ all show that enlargement of the active site cavity is necessary to relieve steric clash between the substrate and the Asn170 residue in the Ω-loop.29,42 The crystal structures of all three mutant enzymes reveal unwinding of the short helix in the Ω-loop, where the Asn170 residue resides (Fig. 7). The loss of α-helical content in TEM-1 mutants has previously been confirmed through CD and molecular dynamics studies.42,45,46 The unwinding of the Ω-loop results in a shift in the Asn170 residue which leads to a larger active site cavity that relieves the steric clash with the substrate in these enzymes (Fig. 5A & 7). The crystal structures of TEM-1 W165Y/E166Y/P167G and TEM-64 with boronic acid inhibitor revealed an 8.4 Å and 4.5Å shift, respectively, in the Cα atoms of the Asn170 residue compared to wild-type TEM-1. Similarly, a 4.2Å shift was observed in the Cα atoms of Asn170 in CTX-M-14 E166A/P167S/CAZ when compared to the CTX-M-14 P167S apo-enzyme. These studies suggest that enlargement of the active site, which results in a shift in the Asn170 residue, is necessary for the enzyme to accommodate the bulky ceftazidime molecule for efficient hydrolysis.
The crystal structures of CTX-M-14 S70G/P167S/CAZ and E166A/P167S/CAZ capture the enzyme in two different steps of ceftazidime hydrolysis. The high Km for ceftazidime hydrolysis in the CTX-M-14 P167S mutant enzyme may be related to the fact that the aminothiazole ring of the ceftazidime side chain is in a shallow position in the S70G/P167S/CAZ structure compared to that in the E166A/P167S/CAZ acyl-enzyme structure (Fig. 4A,C; Fig. 6). In addition, the aminothiazole ring forms more hydrogen bond contacts with active site residues in the E166A/P167S/CAZ acyl-enzyme compared to the E166A/CAZ acyl-enzyme, which could explain the lower catalytic efficiency for ceftazidime hydrolysis by wild-type CTX-M-14 (Fig. 4B & C). Overall, this suggests that the ceftazidime molecule is placed optimally for deacylation in enzymes containing the P167S substitution.
Lastly, the ability of the CTX-M-14 P167S mutant enzyme to hydrolyze the bulkier cephalosporin antibiotic ceftazidime comes at a cost of decreased stability and a decreased ability to provide resistance against smaller β-lactam antibiotics.8,11,15,41 It has been hypothesized that a larger active site cavity would not make optimal contacts with smaller antibiotics making it less effective for catalysis.42 Enlarging an active site could decrease the stability of the enzyme by increasing the strain on the active site.42 Activity-stability trade-offs have previously been reported in β-lactamase enzymes. Decreased stability resulting from functional mutations that expand the substrate profile of the β-lactamase enzyme can be overcome by secondary stabilizing mutations.11,47–49 For example, the A77V mutation has been identified alongside P167S and serves to stabilize the enzyme without affecting the catalytic efficiency of the enzyme.11 The data presented here shows how a single point mutation can result in a large conformational change in the active site of CTX-M β-lactamase enzymes to expand their substrate profile while still maintaining the overall structure of the enzyme. It demonstrates the extraordinary ability of the CTX-M enzymes to adapt and evolve through amino acid substitutions in the presence of antibiotic selective pressure.
Acknowledgments
This work was supported by NIH grant R01 AI32956 to T.P. M.P.P. was supported in part by Award Number T32 GM088129 from the National Institute of General Medical Sciences. This work was also supported by the Robert Welch Foundation Grant (Q1279) to BVVP and the Berkeley Center for Structural Biology, supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH1123. We thank Hiram Gilbert for discussions and comments on the manuscript.
References
- 1.Pfeifer Y, Cullik A, Witte W. Resistance to cephalosporins and carbapenems in Gram-negative bacterial pathogens. Int J Med Microbiol. 2010;300:371–379. doi: 10.1016/j.ijmm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Drawz SM, Bonomo R. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ambler RP. The structure of β-lactamases. Philos Trans R Soc Lond B Biol Sci. 1980;289:321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 4.Hata M, Fujii Y, Tanaka Y, Ishikawa H, Ishii M, Neya SN, Tsuda M, Hoshino T. Substrate deacylation mechanisms of serine β-lactamases. Biol Pharm Bull. 2006;29:2151–2159. doi: 10.1248/bpb.29.2151. [DOI] [PubMed] [Google Scholar]
- 5.Cantón R, González-Alba JM, Galán JC. CTX-M enzymes: origin and diffusion. Front Microbiol. 2012;3:1–19. doi: 10.3389/fmicb.2012.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Andrea MM, Arena F, Pallecchi L, Rossolini GM. CTX-M-type β-lactamases: a successful story of antibiotic resistance. Int J Med Microbiol. 2013;303:305–317. doi: 10.1016/j.ijmm.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 7.Bonnet R. Growing group of extended-spectrum β-lactamases: the CTX-M enzymes. Antimicrob Agents Chemother. 2004;48:1–14. doi: 10.1128/AAC.48.1.1-14.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novais A, Cantón R, Coque TM, Moya A, Baquero F, Galán JC. Mutational events in cefotaximase extended-spectrum beta-lactamases of the CTX-M-1 cluster involved in ceftazidime resistance. Antimicrob Agents Chemother. 2008;52:2377–2382. doi: 10.1128/AAC.01658-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonnet R, Recule C, Baraduc R, Chanal C, Sirot D, De Champs C, Sirot J. Effect of D240G substitution in a novel ESBL CTX-M-27. Antimicrob Agents Chemother. 2003;52:29–35. doi: 10.1093/jac/dkg256. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Delmas J, Sirot J, Shoichet B, Bonnet R. Atomic resolution structures of CTX-M beta-lactamases: extended spectrum activities from increased mobility and decreased stability. J Mol Biol. 2005;348:349–362. doi: 10.1016/j.jmb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 11.Patel MP, Fryszczyn BG, Palzkill T. Characterization of the global stabilizing substitution A77V and its role in the evolution of CTX-M β-lactamases. Antimicrob Agents Chemother. 2015;59:6741–6748. doi: 10.1128/AAC.00618-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delmas J, Chen Y, Prati F, Robin F, Shoichet BK, Bonnet R. Structure and dynamics of CTX-M enzymes reveal insights into substrate accommodation by extended-spectrum beta-lactamases. J Mol Biol. 2008;375:192–201. doi: 10.1016/j.jmb.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 13.Kimura S, Ishiguro M, Ishii Y, Alba J. Role of a mutation at position 167 of CTX-M-19 in ceftazidime hydrolysis. Antimicrob Agents Chemother. 2004;48:1454–1460. doi: 10.1128/AAC.48.5.1454-1460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poirel L, Naas T, Thomas ILE, Karim A, Bingen E, Nordmann P, Debre R. CTX-M-type extended-spectrum β-lactamase that hydrolyzes ceftazidime through a single amino acid substitution in the omega loop. Antimicrob Agents Chemother. 2001;45:3355–3361. doi: 10.1128/AAC.45.12.3355-3361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welsh KJ, Barlow M, Tenover FC, Biddle JW, Rasheed JK, Clark LA, Mcgowan JE. Experimental prediction of the evolution of ceftazidime resistance in the CTX-M-2 extended-spectrum beta-lactamase. Antimicrob Agents Chemother. 2005;49:1242–1244. doi: 10.1128/AAC.49.3.1242-1244.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knox JR. Extended-spectrum and inhibitor-resistant TEM-type beta-lactamases: mutations, specificity, and three-dimensional structure. Antimicrob Agents Chemother. 1995;39:2593–601. doi: 10.1128/aac.39.12.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strynadka N, Adachi H, Jensen S, Johns K, Sielecki A, Betzel C, Sutoh K, James M. Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7A resolution. Nature. 1992;359:700–705. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 18.Guillaume G, Vanhove M, Lamotte-Brasseur J, Lendent P, Jamin M, Joris B, Frere J. Site-directed mutagenesis of glutamate 166 in two β-lactamases: kinetic and molecular modeling studies. J Biol Chem. 1997;272:5438–5444. doi: 10.1074/jbc.272.9.5438. [DOI] [PubMed] [Google Scholar]
- 19.Matagne A, Misselyn-Bauduin A, Joris B, Erpicum T, Granier B, Frère J. The diversity of the catalytic properties of class A beta-lactamases. The Biochem J. 1990;265:131–46. doi: 10.1042/bj2650131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adamski CJ, Cardenas AM, Brown NG, Horton LB, Sankaran B, Prasad BVV, Gilbert HF, Palzkill T. Molecular basis for the catalytic specificity of the CTX-M extended-spectrum β-lactamases. Biochemistry. 2015;54:447–457. doi: 10.1021/bi501195g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 22.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton Ea, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 28.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 29.Stojanoski V, Chow DC, Hu L, Sankaran B, Gilbert HF, Prasad BVV, Palzkill T. A triple mutant in the Ω-loop of TEM-1 β-lactamase changes the substrate profile via a large conformational change and an altered general base for catalysis. J Biol Chem. 2015;290:10382–10394. doi: 10.1074/jbc.M114.633438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ibuka A, Taguchi A, Ishiguro M, Fushinobu S, Ishii Y, Kamitori S, Okuyama K, Yamaguchi K, Konno M, Matsuzawa H. Crystal structure of the E166A mutant of extended-spectrum β-lactamase Toho-1 at 1.8 A resolution. J Mol Biol. 1999;285:2079–2087. doi: 10.1006/jmbi.1998.2432. [DOI] [PubMed] [Google Scholar]
- 31.Odefey C, Mayr LM, Schmid FX. Non-prolyl cis-trans peptide bond isomerization as a rate-determining step in protein unfolding and refolding. J Mol Biol. 1995;245:69–78. doi: 10.1016/s0022-2836(95)80039-5. [DOI] [PubMed] [Google Scholar]
- 32.Jorgensen WL, Gao J. Cis-trans energy difference for the peptide bond in the gas phase and in aqueous solution. J Am Chem Soc. 1988;110:4212–4216. [Google Scholar]
- 33.Matagne A, Lamotte-Brasseur J, Frère J. Catalytic properties of class A β-lactamases:efficiency and diversity. Biochemistry. 1998;330:581–598. doi: 10.1042/bj3300581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pérez-Llarena FJ, Kerff F, Abián O, Mallo S, Fernández MC, Galleni M, Sancho J, Bou G. Distant and new mutations in CTX-M-1 beta-lactamase affect cefotaxime hydrolysis. Antimicrob Agents Chemother. 2011;55:4361–4368. doi: 10.1128/AAC.00298-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delmas J, Leyssene D, Dubois D, Birck C, Vazeille E, Robin F, Bonnet R. Structural insights into substrate recognition and product expulsion in CTX-M enzymes. J Mol Biol. 2010;400:108–120. doi: 10.1016/j.jmb.2010.04.062. [DOI] [PubMed] [Google Scholar]
- 36.Dubus A, Wilkin J, Raquet X, Normark S, Frère J. Catalytic mechanism of active-site serine β-lactamases: role of the conserved hydroxy group of the Lys-Thr(Ser)-Gly triad. Biochem J. 1994;301:485–494. doi: 10.1042/bj3010485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imtiaz U, Manavathu EK, Lerner SA, Mobashery S. Critical hydrogen bonding by serine 235 for cephalosporinase activity of TEM-1 β-lactamase. Antimicrob Agents Chemother. 1993;37:2438–2442. doi: 10.1128/aac.37.11.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen CCH, Herzberg O. Structures of the acyl-enzyme complexes of the Staphylococcus aureus β-Lactamase mutant Glu166Asp:Asn170Gln with benzylpenicillin and cephaloridine. Biochemistry. 2001;40:2351–2358. doi: 10.1021/bi002277h. [DOI] [PubMed] [Google Scholar]
- 39.Shimamura T, Ibuka A, Fushinobu S, Wakagi T, Ishiguro M, Ishii Y, Matsuzawa H. Acyl-intermediate structures of the extended-spectrum class A beta-lactamase, Toho-1, in complex with cefotaxime, cephalothin, and benzylpenicillin. J Biol Chem. 2002;277:46601–46608. doi: 10.1074/jbc.M207884200. [DOI] [PubMed] [Google Scholar]
- 40.Powers RA, Caselli E, Focia PJ, Prati F, Shoichet BK, Campi V. Structures of ceftazidime and its transition-state analogue in complex with AmpC β-lactamase: implications for resistance mutations and inhibitor design. Biochemistry. 2001;40:9207–9214. doi: 10.1021/bi0109358. [DOI] [PubMed] [Google Scholar]
- 41.Kimura S, Ishii Y, Tateda K, Yamaguchi K. Predictive analysis of ceftazidime hydrolysis in CTX-M-type β-lactamase family members with a mutational substitution at position 167. Int J Antimicrob Agents. 2007;29:326–331. doi: 10.1016/j.ijantimicag.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol. 2002;320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 43.Palzkill T, Botstein D. Identification of amino acid substitutions that alter the substrate specificity of TEM-1 beta-lactamase. J Bacteriol. 1992;174:5237–5243. doi: 10.1128/jb.174.16.5237-5243.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vakulenko SB. Effects of Asp-179 mutations in TEMpUC19 β-lactamase on susceptibility to β-lactams. Antimicrob Agents Chemother. 1995;39:1878–1880. doi: 10.1128/aac.39.8.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mobashery S, Lerner SA. Reversal of clavulanate resistance conferred by a Ser-244 mutant of TEM-1 β-lactamase as a result of a second mutation (Arg to Ser at position 164) that enhances activity against ceftazidime. Antimicrob Agents Chemother. 1994;38:1134–1139. doi: 10.1128/aac.38.5.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taibi-tronche P, Massova I, Vakulenko SB, Lerner SA, Mobashery S, August RV. Evidence for structural elasticity of class A β-lactamases in the course of catalytic turnover of the novel cephalosporin cefepime. J Am Chem Soc. 1996;1996:7441–7448. [Google Scholar]
- 47.Marciano DC, Pennington JM, Wang X, Wang J, Chen Y, Thomas VL, Shoichet BK, Palzkill T. Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 beta-lactamase. J Mol Biol. 2008;384:151–164. doi: 10.1016/j.jmb.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang W, Palzkill T. A natural polymorphism in beta-lactamase is a global suppressor. Proc Natl Acad Sci USA. 1997;94:8801–8806. doi: 10.1073/pnas.94.16.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas VL, McReynolds AC, Shoichet BK. Structural bases for stability-function tradeoffs in antibiotic resistance. J Mol Biol. 2009;396:47–59. doi: 10.1016/j.jmb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]