
Fig 2. Long terminal repeat (LTR) promoter toggling is sufficient to generate bimodality and control HIV fate.
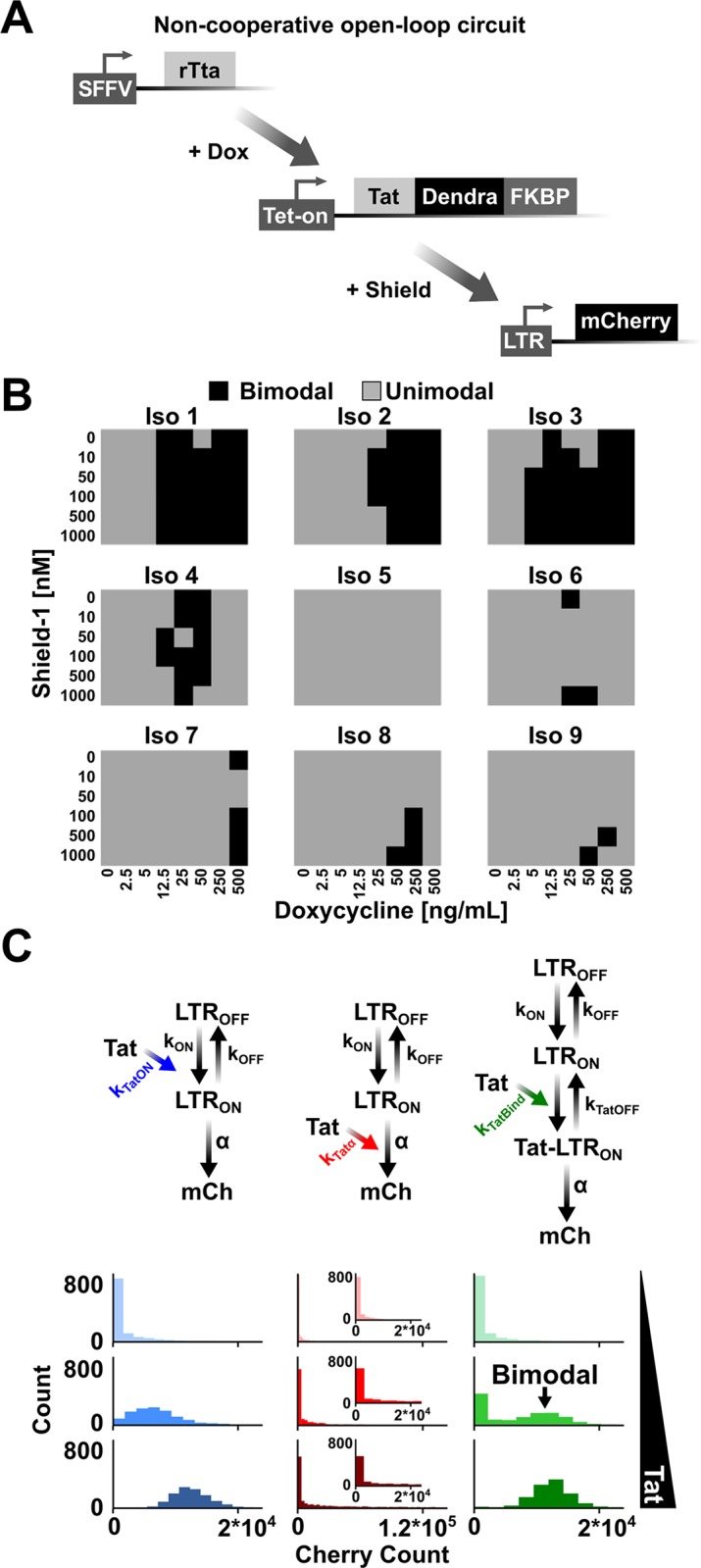
(A) Schematic of the open-loop HIV circuit. Doxycycline addition induces transcription from the Tet-ON promoter. Shield-1 addition controls the stability of the transactivator of transcription (Tat) fused to Dendra-FKBP fusion protein. Tat induces transcription from the HIV LTR. (B) The (Iso) term represents an independent isoclonal population; consequently, each cell within a clone has the same integration site for the LTR. Nine Iso populations were exposed to 48 different doxycycline and Shield-1 conditions (S2 and S3 Figs and S1–S23 Data), and bimodality was tested for by the Hartigan Dip Test [39] (the threshold for determining bimodality was p < 0.3, agreeing with an independent test, S3 Fig and S24 Data). Gray squares indicate populations that were determined to be unimodal, and black squares represent bimodal populations. (C) Open-loop stochastic model of Tat transactivation of the LTR by 1 of 3 mechanisms. Left column, increasing burst frequency by promoting transitions into the LTRON state (left, increasing kON, blue); middle column, increasing burst size by increasing transcriptional efficiency (middle, increasing α, red); and right column, increasing burst size through addition of a third promoter state (effectively inhibiting kOFF, green arrow). Note that for the model in which Tat effectively modulates kOFF (right), there is an additional production of mCh from the LTRON state (arrow not shown) at rate α so that changes in burst sizes can be generated without altering transcriptional efficiency. Model equations and details are presented in S1–S3 Tables. Plotted histograms are steady-state results of 1,000 simulations (at 1,000 hours) showing that slowing promoter toggling by inhibiting transitions into the active state is sufficient to generate bimodal distributions (i.e., right column, middle panel). Insets: Zoom of α modulation so the scale of the x-axis matches the kON (left column) and kOFF (right column) modulation graphs (S1 Data).