

Abstract
Multidrug resistance (MDR) resulting from overexpression of P-glycoprotein (Pgp) transporters increases the drug efflux and thereby limits the chemotherapeutic efficacy. It is desirable to administer both an MDR1 gene silencer and a chemotherapeutic agent in a sequential way to generate a synergistic therapeutic effect in multidrug-resistant cancer cells. Herein, we rationally designed an anti-MDR1 molecular beacon (MB)-based micelle (a-MBM) nanosystem, which is composed of a diacyllipid core densely packed with an MB corona. One of Pgp-transportable agents, doxorubicin (DOX), was encapsulated in the hydrophobic core of the micelle and in the stem sequence of MB. The a-MBM-DOX nanosystem showed an efficient self-delivery, enhanced enzymatic stability, excellent target selectivity, and high drug-loading capacity. With its relatively high enzymatic stability, a-MBM-DOX initially facilitated intracellular MDR1 mRNA imaging to distinguish multidrug-resistant and non-multidrug-resistant cells and subsequently downregulated the MDR1 gene expression owing to an antisense effect. After that, the MB corona was degraded, destroying the micellar nanostructure and releasing DOX, which resulted in a high accumulation of DOX in OVCAR8/ADR cells and a high chemotherapeutic efficacy owing to successful restoration of drug sensitivity. This micelle approach has the potential for both visualizing MDR1 mRNA and overcoming MDR in a sequential and synergistic way.
Keywords: micelles, gene regulation, drug delivery, mRNA imaging, multidrug resistance
Graphical abstract
Molecular beacon-based micelle system (a-MBM) presents its capability to combat multidrug resistance (MDR) in a sequential and synergistic way. With enhanced enzymatic stability, excellent target selectivity, and high drug-loading capacity, a-MBMs can not only visualize MDR1 mRNA and specifically inhibit MDR1 gene expression but also result in a high chemotherapeutic efficacy owing to successful restoration of drug sensitivity.
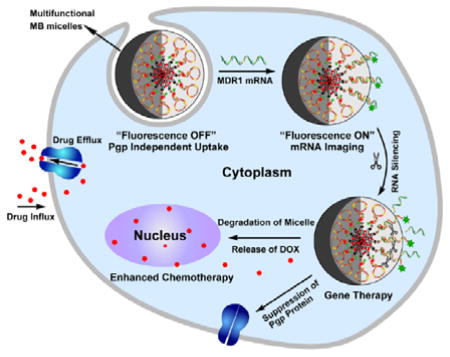
1. Introduction
Chemotherapy is the most widely used approach to cancer treatment. However, development of multidrug resistance (MDR) is one of the most challenging impediments leading to the failure of chemotherapy and a high cancer recurrence rate.[1–3] A major mechanism of MDR is the active efflux of drugs by multidrug transporters,[2,3] which involves overexpression of the drug efflux transporter, i.e. P-glycoprotein (Pgp, also known as ABCB1), multidrug resistance associated-protein 1 (MRP1, also known as ABCC1), breast cancer resistance protein (BCRP, also known as ABCG2), and so on. Of these transporters, Pgp, encoded by the MDR1 gene, is the most commonly encountered in the clinic; and it is overexpressed in many cancer cells.[2–4] Overexpressed Pgp pumps a variety of foreign drugs out of cells, thereby reducing the accumulation of therapeutic agents in MDR cancer cells and resulting in a low chemotherapeutic efficacy.
It has been demonstrated that Pgp-mediated drug resistance can be reversed by downregulating the MDR1 gene with antisense oligonucleotides or small interfering RNAs (siRNAs)[5–9] or by increasing the circulation time of drugs in cells and bypassing the drug efflux through various nanocarriers such as liposomes, micelles, and inorganic nanoparticles.[10–14] However, any single approach has its intrinsic limitations. For example, the efficacy of anti-MDR1 approaches using small nucleic acids has been compromised by bioavailability problems such as poor enzymatic stability and low cellular uptake, thus requiring an additional delivery reagent to improve the stability and transfection efficiency.[15, 16] Nanocarriers can counteract the efflux mechanisms through a local release of high concentrations of drugs inside cells, but they cannot prevent released drugs from being pumped out because the MDR efflux pump still actively works. In addition, long-term cytotoxicity of nanomaterials cannot be ignored, even for gold nanoparticles, considering their intracellular use at relatively high concentrations.[17,18] Therefore, a novel, all-in-one nanosystem is highly desired not only to circumvent the aforementioned limitations of the current strategies but also to administer both an MDR1 gene silencer and a chemotherapeutic agent in a sequential way to produce a synergistic therapeutic effect, since chemotherapy cannot be effective until drug sensitivity is restored.[19–22]
In this study, we report an anti-MDR1 molecular beacon (MB)-based micelle (a-MBM-DOX) nanosystem obtained through self-assembly of diacyllipid–MB (L–MB) conjugates and doxorubicin (DOX) molecules, as shown in Scheme 1A. This a-MBM-DOX nanostructure is composed of a diacyllipid core and an MB corona, as well as DOX in the hydrophobic core of the micelle and in the stem sequence of MB. We hypothesized that this design might show a great potential to effectively overcome MDR based on the following considerations: 1) unlike naked MBs, the micellar nanostructure would enable the self-delivery and enhance the resistance to enzymatic cleavage owing to a densely packed MB corona and also show superior biocompatibility since it does not require any biohazardous materials;[23–26] 2) with a high target specificity and a high sensitivity of MB,[27] a-MBM-DOXs could effectively inhibit the expression of specific target genes through an antisense effect[28, 29] and could also be applied to monitor the MDR1 mRNA expression, which may be an indicator of increased MDR progression, through fluorescence enhancement upon target binding;[24, 30–33] 3) the hydrophobic core of the micelle provides more space;[34,35] this will not only solve the problem of the low DOX-loading capacity of MBs, because the number of GC/CG intercalation sites are limited within one MB, but will also offer the possibility to investigate MDR induced by other Pgp-transportable cytotoxic agents, which cannot be intercalated into the GC/CG site.[25, 36] It is worth mentioning that the enhanced enzymatic stability of a-MBM plays a vital role in our design. As shown in Scheme 1B, initially, stable a-MBM specifically binds to MDR1 mRNA within a very short time, enabling fast mRNA imaging to monitor the MDR1 mRNA activity. Subsequently, this hybridization event induces gene silencing, leading to the suppression of Pgp expression. After that, the MB corona is degraded, which destroys the micellar nanostructure and releases DOX for effective chemotherapy. Therefore, it is their relative stability that makes a-MBMs a promising tool for visualizing MDR1 mRNA and overcoming MDR in a sequential way, while avoiding MB’s drawbacks.
Scheme 1.

The design and function of multifunctional DNA Micelles. (A) Self-assembly of diacyllipid-molecular-beacon conjugates (L-MBs) and doxorubicin molecules (DOXs) into a-MBM-DOXs. (B) Intracellular functions of a-MBM-DOXs, including self-delivery, MDR1 mRNA imaging, MDR1 gene silencing, and reversal of drug resistance. C) Sequences of anti-MDR1 MB and control MB.
2. Result and Discussion
2.1. Preparation and Characterization of a-MBM-DOXs
L–MBs were readily synthesized by directly coupling a hydrophobic diacyllipid tail with the 5′-end of MBs through a solid-phase phosphoramidite chemistry on an automatic DNA/RNA synthesizer (described in the synthetic part of the Supporting Information).[37] DOX was chosen as a Pgp-transportable cytotoxic agent[7] because it not only has been widely used to treat various cancers but is also able to be intercalated into GC/CG base pairs[25] and encapsulated into a hydrophobic domain.[34] a-MBM-DOXs were easily prepared by self-assembly of anti-MDR1 L–MBs and DOX in an aqueous solution owing to hydrophobic interactions, as shown in Scheme 1A. In a-MBM-DOXs, the MB part is a DNA sequence composed of a 21-basepair (bp) target recognition loop flanked by two short complementary stem sequences (5bp). The recognition loop of MBs perfectly complements a region (nucleotides 572–592) in the MDR1 mRNA (Scheme 1C). A fluorophore, Alexa 488, is conjugated to the 3′-end of MB, and 4-(4-dimethylaminophenylazo)-benzoic acid (DABYCL) is included as a quencher between the 5′-end and the diacyllipid tail (Table S1). As shown in Scheme 1B, in the absence of MDR1 mRNA, a-MBM-DOXs maintain a stem–loop structure, and the fluorescence of the fluorophore is quenched by the closely located quencher (OFF state). Upon hybridization to the target mRNA, MBs in a-MBM-DOXs undergo a conformational change, which opens the hairpin structure, causing the fluorophore to move away from the quencher to restore the fluorescence signal (ON state). Subsequently, hybridization of a-MBM-DOXs to the target mRNA can specifically inhibit the gene expression either by blocking translation of the target mRNA or by forming a DNA/RNA hybrid with the target mRNA, which can be degraded by RNase H, leading to the inhibition of Pgp expression.[28,38] Highly loaded DOX provides chemotherapy after the MDR1 gene expression is suppressed and drug sensitivity of multidrug-resistant cancer cells is restored. To confirm the importance of gene silencing in synergistic therapy, non-complementary MBM-DOX (denoted as ctrl-MBM-DOX) was prepared as a control using DOX and mutant L–MBs with eight mismatches (Scheme 1C). In addition, a-MBM and ctrl-MBM were also prepared by self-assembly of anti-MDR1 L–MBs and ctrl L–MBs, respectively (Figure S1).
To examine the formation of the micellar nanostructure, agarose gel electrophoresis, dynamic light scattering (DLS), and transmission electron microscopy (TEM) were carried out. As shown in Figure 1A, a non-lipid-conjugated MB migrated much faster than the assembled a-MBM, which has a higher molecular weight and larger micellar nanostructure, suggesting that a-MBMs were successfully formed. The average hydrodynamic diameter of a-MBMs, measured by DLS, was about 24 nm (Figure 1C and Figure S2A). The TEM analysis, as shown in Figure 1B, directly confirmed that a-MBMs were spherical in shape, with a size of about 20 nm. The incorporation of DOX did not have a significant effect on the a-MBM nanostructure. After adding a synthetic complementary DNA target (designated as cDNA), the migration of a-MBM became much slower (Figure 1A) and the hydrodynamic diameter of a-MBM increased to 33 nm, while incubation of a-MBM with a random DNA control (denoted as rDNA) or DOX resulted in a negligible size increase (Figure 1C). The results demonstrated that a-MBM maintained the target recognition capability and neither the binding event nor DOX loading caused a disruption of the micellar nanostructure. In addition, zeta potential data for a-MBM only, a-MBM treated with cDNA, a-MBM treated with rDNA, and a-MBM loaded with DOX were found to be −7.6, −10.8, −7.9, and −5.0 mV, respectively (Figure 1D and Table S2). These results further illustrated successful binding of the negatively charged cDNA and loading of the positively charged DOX. Because fluorescence of DOX could be quenched after intercalation into the GC/CG site or encapsulation into the hydrophobic domain, it was calculated that a-MBM-DOXs had a high encapsulation efficiency (96.8% vs. 23.6%) and a high drug-loading capacity (22.8w/w% vs. 4.3w/w%), i.e., approximately five-fold higher than those of anti-MDR1 MB (Table S3). The detailed sequence information for all experiments can be found in Table S1.
Figure 1.

Characterization of a-MBMs. (A) Agarose gel electrophoresis of MBs (Lane 1), MBs with synthetic complementary target (cDNA; Lane 2), a-MBMs (Lane 3), and a-MBMs with cDNA (Lane 4). (B) TEM images of a-MBMs (top) and a-MBM-DOXs (down) after negative staining by 2% aqueous uranyl acetate. (C) DLS and (D) Zeta-potential measurements of a-MBMs, a-MBMs with cDNA, a-MBMs with non-complementary target (rDNA), and a-MBM-DOXs.
2.2. Specificity and Stability of a-MBMs in Vitro
We first assessed the binding specificity of a-MBM by adding the cDNA target to the buffer solution and using fluorescence spectroscopy. It could be clearly seen that the fluorescence of a-MBM significantly increased, with approximately nine-fold signal enhancement, upon the addition of excess amount of cDNA, which was much higher than the increase in fluorescence reported in the literature for gold–nucleic acid conjugates,[30] while only minimal fluorescence enhancement was observed when rDNA was added (Figure 2A). More importantly, a-MBMs could discriminate between a completely complementary target and a mismatched target with a single mutation. These results suggested that a-MBMs exhibited a remarkable target selectivity and high sensitivity. The specificity of a-MBMs was further confirmed with the cDNA target and off-targets, including survivin, β-actin, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). As shown in Figure 2B, the off-targets did not induce a significant fluorescence enhancement. In addition, ctrl-MBM also showed little fluorescence enhancement, even at a high concentration of cDNA or off-targets because of a low binding affinity (Figure S3). Figure S4 showed that the fluorescence signal of a-MBM increased with the increasing cDNA concentrations in the range from 0 to 1 μM, with a wide linear range from 0 to 200 nM. These results demonstrated that the fluorescence intensity was highly associated with the concentration of the cDNA target, indicating that a-MBMs could be used for quantitative analysis of intracellular mRNA expression. We then evaluated the hybridization kinetics of a-MBM to the cDNA target. As shown in Figure 2C, a-MBMs responded to the addition of the cDNA sequence in less than 20 s. This fast response makes a-MBMs suitable for live-cell mRNA imaging, essentially because this extremely short delay can help avoid unnecessary interferences with data analysis of real-time mRNA imaging in cells. To compare the stability of a-MBMs and MBs against enzymatic digestion, we incubated each complex with the DNase I endonuclease or serum, and monitored the fluorescence change as a function of time. As shown in Figure 2D and Figure S5, MBs demonstrated a substantial increase in fluorescence. In contrast, the equivalent amount of a-MBMs showed no significant change in the fluorescence signal in the initial 5 h and a much slower subsequent increase in fluorescence, indicating an enhanced stability of a-MBM to enzymatic digestion.
Figure 2.

Performance evaluation of a-MBMs. (A) Fluorescence spectra of a-MBMs in the presence of no target (black line), cDNA (red line), cDNA with one mismatch (blue line), and rDNA (green line). (B) Specificity of a-MBMs to several DNA targets. (C) Hybridization kinetics of a-MBMs associated with the cDNA target. a-MBMs (200 nM) were mixed with 300 nM of cDNA target. (D) Enzymatic stability study of anti-MDR1 MB (black) and a-MBMs (red) treated with DNase I.
2.3. Internalization, Distribution, and Cytotoxicity of a-MBMs
We investigated the cellular uptake of micelles by OVCAR8/ADR cells using confocal laser scanning microscopy (CLSM). An “always-on” a-MBM probe (denoted as aa-MBM) was prepared by self-assembly of anti-MDR1 L–MBs without the quencher. We found that aa-MBMs were initially distributed on the cell membrane and showed a weak fluorescence signal at 1 h (Figure S6A). Afterwards, the increase in the fluorescence signal from 1 to 4 h showed gradual accumulation of aa-MBMs in the cytoplasm but not the nucleus (Figure S6A) because the nanoparticles with the size of 20 nm could not penetrate the nuclear membrane. In addition, OVCAR8 cells incubated with aa-MBMs for 4 h displayed a similar fluorescence signal to that of OVCAR8/ADR cells (Figure S7A). On the contrary, MBs showed no obvious fluorescence signal in cells, even after 12 h of incubation (Figure S7B). These results indicated that the introduction of the diacyllipid moiety did enable efficient self-delivery, and the kinetics of cellular uptake was consistent with that of spherical nucleic acid nanoparticles reported in the literature.[23,36] Subcellular distribution of aa-MBMs after 4-h treatment was examined by CLSM, with Lysotracker red used to identify endosomes/lysosomes. The results showed that few aa-MBMs were located in endosomes/lysosomes, while most of aa-MBMs were distributed in the cytoplasm (Figure S8), indicating that aa-MBM might flip and diffuse from the cell membrane to the cytoplasm rather than permeate the cell through the endocytosis pathway.[23] Next, cytotoxicity of a-MBMs was evaluated against OVCAR8/ADR cells by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. As shown in Figure S9, no measurable cytotoxicity was observed at concentrations of 0–1,500 nM of a-MBMs after 48 h of incubation. (Note: the concentration of micelles represents that of MBs in all experiments.) This indicated that a-MBMs exhibited excellent biocompatibility with living cells, which can probably be attributed to the structural similarity of the diacyllipid to phospholipids, the basic components of the cell membrane. Collectively, our results clearly confirmed the ease of synthesis, efficient self-delivery, enhanced enzymatic stability, remarkable target specificity, high sensitivity, high drug-loading capacity, and superior biocompatibility of a-MBMs, which were the prerequisites for mRNA imaging, gene silencing, and chemotherapy in subsequent experiments.
2.4. Evaluation of a-MBMs as mRNA Imaging Probes in OVCAR8/ADR Cancer Cells
We then studied the ability of a-MBMs to specifically detect MDR1 mRNA in cancer cells by restoring the quenched fluorescence of a-MBM. The OVCAR8/ADR cell line, which overexpresses the MDR1 gene (Figure S10), was chosen. As shown in Figure S6B, the fluorescence intensity of Alexa 488 in the cytoplasm gradually increased with the incubation time, and the time profile was similar to that of the cellular uptake of aa-MBMs (Figure S6A). To obtain the best results for intracellular detection of MDR1 mRNA, we optimized both the incubation time and a-MBM concentration. Based on the data, the incubation time of 4 h and the a-MBM concentration of 400 nM were determined to be the optimal assay conditions for fluorescence imaging (Figure S6B and Figure S11).
Under the optimal conditions, fluorescence signals were acquired from OVCAR8/ADR and OVCAR8 cells incubated with the same amount of non-diacyllipid-conjugated MBs, a-MBMs, and ctrl-MBMs. As shown in Figure 3A, OVCAR8/ADR cells treated with a-MBMs displayed an intense fluorescence, while cells treated with MBs alone (Figure 3B) did not show any noticeable fluorescence because naked MBs cannot be internalized by cells. To confirm that the fluorescence in the cancer cells was indeed triggered by MDR1 mRNA, OVCAR8/ADR cells were pretreated with siRNA (15 nM) for 48 h to downregulate the MDR1 mRNA expression before incubating the cells with a-MBMs (Figure S12). It is noteworthy that the siRNA-treated cells showed weaker fluorescence signals than cells without siRNA treatment (Figure 3C). This result confirmed that the strong fluorescence observed in Figure 3A did come from specific binding of a-MBMs to MDR1 mRNA. To further exclude other factors (i.e. degradation of a-MBM or hairpin opening by off-targets or protein binding) that are likely to contribute to fluorescence enhancement, OVCAR8/ADR and OVCAR8 cells were treated with ctrl-MBMs and a-MBMs, respectively. Little fluorescence was observed in both cases (Figures 3D and 3E), suggesting a strong target specificity and high stability of a-MBMs at the current stage. The flow cytometry results (Figure S13) were very consistent with the confocal imaging: 3.5-times signal enhancement of a-MBMs was observed compared to those of MBs or ctrl-MBMs, and siRNA treatment of OVCAR8/ADR cells decreased the signal of a-MBMs by 1.4 times. Thus, using a-MBMs provides a promising strategy for monitoring the MDR1 mRNA progression and distinguishing multidrug-resistant and non-multidrug-resistant cells.
Figure 3.

Confocal microscopy fluorescence images of MDR1 mRNA targets with 400 nM of a-MBMs (A), MBs with non-diacyllipid conjugation (B), a-MBMs after siRNA treatment of cells (C), ctrl-MBMs (D) in OVCAR/ADR cells, and (E) a-MBMs in OVCAR cells. Scale bar: 20 μm.
2.4. Suppression of MDR1 mRNA in a-MBM-treated OVCAR8/ADR cells
Substantial evidence has suggested that antisense oligonucleotides can specifically and efficiently inhibit mRNA transcription when properly delivered.[5,6,28,29] Thus, we investigated the intracellular activity of a-MBMs in OVCAR8/ADR cells by quantitative real-time polymerase chain reaction (qRT-PCR) and Western blotting. As shown in Figure 4A, ctrl-MBMs at concentrations of 200, 600, and 1,200 nM showed no measurable effects on MDR1 mRNA levels after 48 h. In contrast, a-MBMs at identical concentrations caused a marked decrease in MDR1 mRNA transcription in a dose-dependent manner, which further supports an excellent intracellular target specificity of a-MBMs. Western blot analysis displayed consistent results with those of qRT-PCR at the protein level (Figure 4B). These results unambiguously demonstrated that a-MBMs were able to selectively and efficiently induce the MDR1 gene silencing and consequently inhibit the expression of the Pgp protein in multidrug-resistant cells. This effective inhibition was attributed to a significant RNase H-dependent antisense effect, which was further confirmed as a gene inhibition mechanism of a-MBMs (Figure S14). Therefore, our a-MBM system is useful for the intracellular mRNA detection and imaging-guided gene therapy in living cells.
Figure 4.

Anti-MDR1 effects of a-MBMs in OVCAR8/ADR cells. (A) qRT-PCR analyses of MDR1 mRNA transcription in the OVCAR8/ADR cells treated with different concentrations of a-MBMs (blue) and ctrl-MBMs (red). Each bar presents the mean and standard deviation derived from three independent experiments. (B) Western blot analyses of Pgp expression in the OVCAR8/ADR cells after treatment with a-MBMs at different concentrations, which was probed with anti-Pgp antibody and anti-β-actin antibody, respectively. Fluorescence images of OVCAR/ADR cells treated with free DOXs (C), ctrl-MBM-DOXs of 1,200 nM (D), a-MBM-DOXs of 200 nM (E) and 1,200 nM (F). Scale bar: 20μm.
2.5. DOX Accumulation in a-MBM-DOX Treated OVCAR8/ADR Cells
As we mentioned before, elevated Pgp expression in cancer cells is one of the most prevalent mechanisms that cause anticancer drug efflux, leading to a low accumulation of drugs in cells and a decreased therapeutic efficacy. Since a-MBMs induced strong inhibition of Pgp both at the mRNA and protein levels, a higher accumulation of DOX is expected to occur in multidrug-resistant cancer cells. Thus, we further evaluated the retention of DOX in cancer cells using CLSM because DOX fluorescence should be restored upon DOX release from a-MBM-DOXs. According to our strategy, the relatively stable a-MBM-DOXs are supposed to be degraded at this stage to ensure a high DOX accumulation for effective chemotherapy. As depicted in Figures 4C and 4D, there was an obvious fluorescence signal in the cytoplasm of the Pgp-overexpressing OVCAR8/ADR cells treated with ctrl-MBM-DOXs, while only weak fluorescence was observed in the cells exposed to free DOX. This result indicated that the micellar nanostructure was favorable for drug accumulation in cells; however, the released drug could unavoidably be pumped out by the Pgp efflux pump. Nevertheless, a-MBM-DOXs significantly enhanced the accumulation of DOX compared to ctrl-MBM-DOXs (Figures 4E and 4F), even at a relatively lower concentration (200 nM a-MBM-DOXs vs. 1,200 nM ctrl-MBM-DOXs). More importantly, the released DOX was directed, to a significant extent, to the nucleus with the increased concentrations of a-MBM-DOXs (Figures 4E and 4F), suggesting that the efficient suppression of the Pgp pump by a-MBM-DOXs indeed resulted in higher DOX retention in the multidrug-resistant cancer cells. The flow cytometry results (Figure S15) were in agreement with the confocal imaging. Compared to fluorescence signal of free DOX, fluorescence intensity of a-MBM-DOXs of 1,200 nM, a-MBM-DOXs of 200 nM, ctrl-MBM-DOXs of 1,200 nM increased by 3.2, 2.7, 1.8 times, respectively. This higher DOX retention capability was probably due to the synergistic effect caused by efficient suppression of the Pgp pump by a-MBM-DOXs and the ability of the micellar nanostructure to bypass the drug efflux.
2.6. Reversal of Drug Resistance of OVCAR8/ADR Cells by Treatment with a-MBM-DOXs
To assess whether the a-MBM-DOX-induced inhibition of Pgp could sensitize OVCAR8/ADR cells to DOX as we expected, we compared cell viabilities of a-MBM-DOX-treated, untreated, and ctrl-MBM-DOX-treated cells using the MTT assay (Figure 5A). A significant decrease in the cell survival was observed after a-MBM-DOX treatment. On the contrary, a less significant loss of viability was shown by the cells treated with free DOX and ctrl-MBM-DOX. Measuring the concentration of a drug that provides a half-maximal response (IC50) is the best way to evaluate the restoration of drug sensitivity. As shown in Figure 5B and Table S4, treatment with 1,200 nM of a-MBM-DOXs induced a significant decrease of IC50, which was 15-fold lower than that for untreated OVCAR8/ADR cells, indicating higher inhibition of multidrug-resistant cell proliferation by a-MBM-DOXs. In summary, the synergistic effect of the efficient suppression of the Pgp pump by a-MBM-DOXs and the ability of the micellar nanostructure to bypass drug efflux makes multidrug-resistant cancer cells more sensitive to the subsequent release of DOX, resulting in a high intracellular accumulation of DOX and thus providing a significantly high antitumor efficacy. Altogether, these results suggest that our system has the capabilities of supporting intracellular mRNA imaging and synergistic therapy in multidrug-resistant cancer cells in a specific and sequential way.
Figure 5.

(A) Cell viability of OVCAR8/ADR cells treated with free DOX, ctrl-MBM-DOXs of 1,200 nM, a-MBM-DOXs of 200 nM and 1,200 nM, and OVCAR8 treated with free DOX. The concentration of DOX is identical to be 10 μM. (B) Effect of a-MBM-DOXs on the sensitivity of OVCAR8/ADR cells to doxorubicin. OVCAR8 cells treated with free DOX (green), OVCAR8/ADR cells with free DOX (black) and a-MBM-DOXs of 1,200 nM (blue). Each point represents the average value from three independent experiments.
3. Conclusion
In summary, the present study demonstrates the capability of a-MBM-DOXs to specifically and efficiently combat MDR. A simple modification of MBs with a diacyllipid group resulted in an efficient self-delivery, enhanced enzymatic stability, excellent target selectivity, and high drug-loading capacityof a-MBMs. All these properties make a-MBMs a successful platform to overpower the limitations of current drug/gene delivery systems for the treatment of multidrug-resistant cancer cells. Our a-MBM system not only integrates a Pgp repression component and the anticancer drug DOX but also makes those working in a controlled, programmable manner. The study confirmed that a-MBMs are capable of entering cells and releasing the drug only after they bypass the MDR barrier, thus showing great promise for constructing an all-in-one micelle probe to carry out, sequentially and synergistically, mRNA imaging and image-guided therapy.
4. Experimental Section
Synthesis of a-MBM-DOX
L–MBs were synthesized by directly coupling a hydrophobic diacyllipid tail with the 5′-end of MBs through solid-phase phosphoramidite chemistry on an automatic DNA/RNA synthesizer (described in the synthetic part of the Supporting Information). a-MBM–DOXs were easily prepared by self-assembly of anti-MDR1 L–MBs and DOX in an aqueous solution owing to hydrophobic interactions. In a typical experiment, DOX were dissolved in 0.1 mL methanol at room temperature (molar ratio of doxorubicin: triethylamine = 1:2) and L–MBs was dissolved in 1 mL distilled water, respectively. The DOX solution was dropwise added into the L–MBs solution with constant stirring. The mixed solution was subject to dialysis against 2 L deionized water for 12 h. The micelle solution was lyophilized by a freeze dryer system to obtain the dried powder of a-MBM-DOXs. The dried powder was hydrated with phosphate buffer (pH 7.4). Particles were sterilized with a 0.2 μm acetate syringe filter. The DOX-encapsulation efficiency (EE%) of was calculated by Equation (1), and the DOX-loading efficiency (LE%) was calculated by Equation (2)
| (1) |
| (2) |
Agarose gel electrophoresis
DNA samples were analyzed by gel electrophoresis, which was performed at 90 V for about 45 min through a 4% agarose gel in 1x TBE buffer (89 mM tris(hydroxymethyl)aminomethane, 2 mM ethylenediamine tetraacetic acid and 89 mM boric acid, pH 8.0). The DNA bands were stained with ethidium Bromide (EB), visualized by UV illumination.
Characterization of micelles Using DLS, TEM, UV-Visand FL Spectra
The particle sizes and zeta potential were measured using a Zetasizer5000 (Malvern Instruments, U.K.) at 25 °C. TEM measurements were performed on a JEOL 200X. UV-Vis spectra were measured using a Shimadzu UV1701. FL spectra were measured using Hitachi F-7000 fluorescence spectrophotometer.
Hybridization Experiments
a-MBMs of 200 nM in PBS buffer (10 mM PBS, pH 7.4, 150 mM NaCl, and 2 mM MgCl2) were treated with increasing concentrations of the cDNA targets (0, 10, 20, 50, 100, 200, 400, 700, 1000 nM). After 1-h incubation at 37 °C, the fluorescence of Alexa 488 was excited at 490 nm and measured at 530 nm (range from 500 to 700 nm).
Specificity experiments
a-MBMs or ctrl-MBMs of 200 nM in PBS buffer were treated with different DNA targets of 300 nM, including cDNA, cDNA with single mismatch, rDNA, survivin, β-actin, and GAPDH. After 1-h incubation at 37 °C, the fluorescence of Alexa 488 was excited at 490 nm and measured at 530 nm (range from 500 to 700 nm). All experiments were repeated at least three times.
Nuclease assay
DNase I (Invitrogen) with a concentration of 50 U/L was added to 200 nM of MB or a-MBM buffer solution. The fluorescence of two samples was then monitored at intervals for 20 h.
Cytotoxicity of a-MBMs on OVCAR8/ADR cells
OVCAR8/ADR cells were dispensed into 96-well plates at a final concentration of 1 × 104 cells/well in a culture medium (100 μL), and incubated overnight before treatment. The culture medium was then removed and replaced with new medium containing a-MBM at varying concentrations of 200, 400, 700, 1,000, and 1,500 nM. After 48-h incubation at 37 °C, cell viability was measured using a MTT assay.
Confocal laser scanning microscopy experiments
Cells were plated in a 35 mm coverglass-bottom dish (MatTek Corp., USA) and incubated overnight before the experiment. Cells were washed twice with 1x PBS and then incubated with the proper probes at the desired concentration for 1, 2 or 4 h at 37 °C in 5% CO2. After incubation, cells were washed twice with 1x PBS, dispersed in 1x PBS and then subjected to confocal imaging.
Anti-MDR1 Effects of a-MBM-DOXsonOVCAR8/ADR Cells
OVCAR8/ADR cells were grown in RPMI 1640 media supplemented with 10% fetal bovine serum and antibiotics at 37 °C in 5% CO2. Cells were seeded onto 35-mm wells of a 6-well plate and cultured overnight. The cells were treated with fresh media containing a-MBMs or ctrl-MBMsusing different concentrations of 200, 600 and1,200 nM for 48 h. Control cells were incubated only with the culture medium. After that, cells were lysed and assayed by qRT-PCR analyses and western blot.
RNA Extraction and qRT-PCR Analysis for the RNA Levels of MDR1, TNF-α, IFN-β and GAPDH
RNA samples were extracted from OVCAR8/ADR cells using aTriPure isolation reagent (Roche). To prevent DNA contamination, total RNA was treated with RNase-free DNase II (Invitrogen). Total RNA samples (1 μg per reaction) were reversely transcribed into cDNAs by SuperScriptTMFirst-StrandSynthesis System (Invitrogen). Then, cDNAs were used as templates in qRT-PCR. The amplification reactions were performed using Fast SYBRGreen Master Mix on a StepOne Plus RT-PCR system (Applied Biosystems, CA) according to the manufacturer’s protocol. The Primers for MDR1 gene were 5′-AAATTGGCTTGACAAGTT-GTATATGG-3′ (Forward) and 5′-CACCAGCATCATGAGAGGAAGTC-3′ (Reverse). GAPDH was used as an internal control in PCR amplification, and its primers were 5′-GGTCTCCTCTGACTTCAACA-3′ (Forward) and 5′-AGCCAAATTCGTTGTCATAC-3′ (Reverse).
Western blot analysis of Pgp proteins
Cell lysates were prepared by treating samples with ice-cold RIPA buffer (PIERCE) with a protease inhibitor cocktail (Roche) for 5 min on ice followed by centrifugation at 4 °C for 15 min to sediment particulate materials. Proteins then were separated by SDS-PAGE, transferred to nitrocellulose membranes. The membranes were incubated in blocking solution containing 5% skim milk for 1 h at room temperature, and then immunoblotted with monoclonal anti-Pgp mouse (C219) antibody (EMD Millipore) and β-actin (13E5) rabbit mAb (Cell Signaling Technology, Inc.). Immunoreactive bands were visualized using SupersignalTM West Pico chemiluminescent substrate (PIERCE) according to the manufacturer’s directions.
Effect of a-MBM-DOXs on drug sensitivity ofOVCAR8/ADR cells
OVCAR8 and OVCAR8/ADR cells were seeded into 96-well plates at a final concentration of 1 × 104 cells/well in a culture medium (100 μL), and incubated overnight before treatment. The culture medium was then removed and replaced with new medium containing free DOX, ctrl-MBM-DOX, or a-MBM-DOXs. After 72-h incubation, cell viability was measured using a MTT assay.
Supplementary Material
Acknowledgments
This study was supported by National Natural Science Foundation of China (Nos. 81671753 and 81271606), the Fundamental Research Funds for the Central universities (Nos. JB151201, JBG161202, and QR15003), the national 1000 Young Talents Program of China, and the Intramural Research Program (IRP) of NIBIB/NIH.
Footnotes
Supporting information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Ruili Zhang, China-Japan Union Hospital, Jilin University, Changchun, Jilin, 130033 China. Engineering Research Center of Molecular-imaging and Neuro-imaging of ministry of education, School of Life Science and Technology, Xidian University, Xi’an, Shaanxi 710126, China. Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland, 20892 USA.
Shi Gao, China-Japan Union Hospital, Jilin University, Changchun, Jilin, 130033 China.
Prof. Zhongliang Wang, Engineering Research Center of Molecular-imaging and Neuro-imaging of ministry of education, School of Life Science and Technology, Xidian University, Xi’an, Shaanxi 710126, China
Dr. Da Han, Center for Research at Bio/Nano Interface, University of Florida, Gainesville, FL 32611 USA
Lin Liu, China-Japan Union Hospital, Jilin University, Changchun, Jilin, 130033 China.
Prof. Qingjie Ma, China-Japan Union Hospital, Jilin University, Changchun, Jilin, 130033 China
Prof. Weihong Tan, Center for Research at Bio/Nano Interface, University of Florida, Gainesville, FL 32611 USA
Prof. Jie Tian, Engineering Research Center of Molecular-imaging and Neuro-imaging of ministry of education, School of Life Science and Technology, Xidian University, Xi’an, Shaanxi 710126, China
Prof. Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland, 20892 USA
References
- 1.Pauwels EK, Erba P, Mariani G, Gomes CM. Drug News Perspect. 2007;20:371. doi: 10.1358/dnp.2007.20.6.1141496. [DOI] [PubMed] [Google Scholar]
- 2.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Nat Rev Drug Discov. 2006;5:219. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman MM, Fojo T, Bates SE. Nat Rev Cancer. 2002;2:48. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 4.Duhem C, Ries F, Dicato M. Oncologist. 1996;1:151. [PubMed] [Google Scholar]
- 5.Dassow H, Lassner D, Remke H, Preiss R. Int J Clin Pharmacol Ther. 2000;38:209. doi: 10.5414/cpp38209. [DOI] [PubMed] [Google Scholar]
- 6.Nadali F, Pourfathollah AA, Alimoghaddam K, Nikougoftar M, Rostami S, Dizaji A, Azizi E, Zomorodipour A, Ghavamzadeh A. Hematology. 2007;12:393. doi: 10.1080/10245330701283991. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Wang Z, Liu D, Yan X, Wang F, Niu G, Yang M, Chen X. Angew Chem Int Ed. 2014;53:1997. doi: 10.1002/anie.201309985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi KY, Silvestre OF, Huang X, Min KH, Howard GP, Hida N, Jin AJ, Carvajal N, Lee SW, Hong JI, Chen X. ACS Nano. 2014;8:4559. doi: 10.1021/nn500085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu H, Hait WN, Yang JM. Cancer Res. 2003;63:1515. [PubMed] [Google Scholar]
- 10.Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Adv Drug Deliv Rev. 2013;65:1866. doi: 10.1016/j.addr.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu J, Zhao W, Liu H, Marquez R, Huang Y, Zhang Y, Li J, Xie W, Venkataramanan R, Xu L, Li S. J Control Release. 2014;196:272. doi: 10.1016/j.jconrel.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee Y, Graeser R, Kratz F, Geckeler KE. Adv Funct Mater. 2011;21:4211. [Google Scholar]
- 13.Lu Y, Aimittee A, Langer R, Gu Z. Nat Rev Mater. 2016;1:16075. [Google Scholar]
- 14.Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Nat Rev Drug Discov. 2011;10:521. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]
- 15.Whitehead KA, Langer R, Anderson DG. Nat Rev Drug Discov. 2009;8:129. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tseng YC, Mozumdar S, Huang L. Adv Drug Delivery Rev. 2009;61:721. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YS, Hung YC, Liau I, Huang G. Nanoscale Res Lett. 2009;4:858. doi: 10.1007/s11671-009-9334-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharifi S, Behzadi S, Laurent S, Laird Forrest M, Stroeve P, Mahmoudi M. Chem Soc Rev. 2012;41:2323. doi: 10.1039/c1cs15188f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun TM, Du JZ, Yao YD, Mao CQ, Dou S, Huang SY, Zhang PZ, Leong KW, Song EW, Wang J. ACS Nano. 2011;5:1483. doi: 10.1021/nn103349h. [DOI] [PubMed] [Google Scholar]
- 20.MacDiarmid JA, Amaro-Mugridge NB, Madrid-Weiss J, Sedliarou I, Wetzel S, Kochar K, Brahmbhatt VN, Phillips L, Pattison ST, Petti C, Stillman B, Graham RM, Brahmbhatt H. Nat Biotechnol. 2009;27:643. doi: 10.1038/nbt.1547. [DOI] [PubMed] [Google Scholar]
- 21.Ren Y, Wang Y, Zhang Y, Wei D. Mol Pharm. 2008;5:579. doi: 10.1021/mp800001j. [DOI] [PubMed] [Google Scholar]
- 22.Lo YL, Liu Y, Tsai JC. Biomed Pharmacother. 2013;67:261. doi: 10.1016/j.biopha.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Wu C, Chen T, Han D, You M, Peng L, Cansiz S, Zhu G, Li C, Xiong X, Jimenez E, Yang CJ, Tan W. ACS Nano. 2013;7:5724. doi: 10.1021/nn402517v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu L, Wu C, You M, Han D, Chen T, Zhu G, Jiang J, Yu R, Tan W. J Am Chem Soc. 2013;135:12952. doi: 10.1021/ja406252w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiao G, Zhuo L, Gao Y, Yu L, Li N, Tang B. Chem Commun. 2011;47:7458. doi: 10.1039/c1cc11490e. [DOI] [PubMed] [Google Scholar]
- 26.Chinen AB, Guan CM, Ferrer JR, Barnaby SN, Merkel TJ, Mirkin CA. Chem Rev. 2015;115:10530. doi: 10.1021/acs.chemrev.5b00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14:303. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 28.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Science. 2006;312:1027. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 29.Chen T, Wu CS, Jimenez E, Zhu Z, Dajac JG, You M, Han D, Zhang X, Tan W. Angew Chem Int Ed. 2013;52:2012. doi: 10.1002/anie.201209440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seferos DS, Giljohann DA, Hill HD, Prigodich AE, Mirkin CA. J Am Chem Soc. 2007;129:15477. doi: 10.1021/ja0776529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, Zhang R, Wang Z, Wang HF, Wang Y, Zhao J, Wang F, Li W, Niu G, Kiesewetter DO, Chen X. ACS Nano. 2014;8:12386. doi: 10.1021/nn505047n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song S, Liang Z, Zhang J, Wang L, Li G, Fan C. Angew Chem Int Ed. 2009;48:8670. doi: 10.1002/anie.200901887. [DOI] [PubMed] [Google Scholar]
- 33.Li N, Chang C, Pan W, Tang B. Angew Chem Int Ed. 2012;51:7426. doi: 10.1002/anie.201203767. [DOI] [PubMed] [Google Scholar]
- 34.Wei T, Liu J, Ma H, Cheng Q, Huang Y, Zhao J, Huo S, Xue X, Liang Z, Liang XJ. Nano Lett. 2013;13:2528. doi: 10.1021/nl400586t. [DOI] [PubMed] [Google Scholar]
- 35.Wang C, Cheng L, Liu Z. Biomaterials. 2011;32:1110. doi: 10.1016/j.biomaterials.2010.09.069. [DOI] [PubMed] [Google Scholar]
- 36.Ma Y, Wang Z, Zhang M, Han Z, Chen D, Zhu Q, Gao W, Qian Z, Gu Y. Angew Chem Int Ed. 2016;55:3304. doi: 10.1002/anie.201509182. [DOI] [PubMed] [Google Scholar]
- 37.Wu Y, Sefah K, Liu H, Wang R, Tan W. Proc Natl Acad Sci USA. 2010;107:5. doi: 10.1073/pnas.0909611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elsabahy M, Nazarali A, Foldvari M. Curr Drug Deliv. 2011;8:235. doi: 10.2174/156720111795256174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.