

Abstract
The flavin chromophore in blue light using FAD (BLUF) photoreceptors is surrounded by a hydrogen bond network that senses and responds to changes in the electronic structure of the flavin on the ultrafast time scale. The hydrogen bond network includes a strictly conserved Tyr residue, and previously we explored the role of this residue, Y21, in the photoactivation mechanism of the BLUF protein AppABLUF by the introduction of fluorotyrosine (F-Tyr) analogs that modulated the pKa and reduction potential of Y21 by 3.5 pH units and 200 mV, respectively. Although little impact on the forward (dark to light adapted form) photoreaction was observed, the change in Y21 pKa led to a 4,000-fold increase in the rate of dark state recovery. In the present work we have extended these studies to the BLUF protein PixD, where, in contrast to AppABLUF, modulation in the Tyr (Y8) pKa has a profound impact on the forward photoreaction. In particular, a decrease in Y8 pKa by 2 or more pH units prevents formation of a stable light state, consistent with a photoactivation mechanism that involves proton transfer or proton coupled electron transfer from Y8 to the electronically excited FAD. Conversely, the effect of pKa on the rate of dark recovery is markedly reduced in PixD. These observations highlight very significant differences between the photocycles of PixD and AppABLUF, despite their sharing highly conserved FAD binding architectures.
Keywords: PixD, BLUF, Ultrafast infrared, Fluorotyrosine, FAD, Photoactivation, kinetics
Graphical Abstract
Introduction
Flavins are ubiquitous biological cofactors and are found in ~1% of all proteins, usually either as flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD).1 The chemistry associated with FMN and FAD is performed by the 7,8-dimethylisoalloxazine ring, a planar tricyclic aromatic system which can undergo 1 or 2 electron reduction and also participate in the formation of covalent adducts with reactants and amino acid side chains.1–3 The broad distribution of flavoproteins across biological space includes flavin-dependent photoreceptors which utilize the visible electronic transition of the oxidized isoalloxazine ring, centered at around 450 nm, to sense and respond to blue-light.4 Structural changes initiated by light absorption on the picosecond time scale lead to macromolecular reorganization and biological output on the millisecond to second time scale.5 Of central importance in photoreceptor biology is the understanding of the underlying structural dynamics that link events separated by more than ten decades of time. Indeed, the mechanism of photoreceptor signaling is an area of intense experimental and theoretical activity,6–12 stimulated by the recent interest in deploying flavin-dependent photoreceptors as optogenetic devices that enable the in vivo control of biology through non-invasive light signaling.13 However, despite the unique qualities and potential applications of flavoprotein photoreceptors, in many cases the detailed mechanism of photocycling still remains to be elucidated.
Blue-light sensing flavoproteins are found in animals, plants, fungi, and bacteria,14–17 and include the photolyase/cryptochromes, the light-oxygen-voltage (LOV) domain proteins, and the blue light using FAD (BLUF) domain proteins.15, 18 For both the photolyase/cryptochrome and LOV domain photosensors, photoexcitation leads to changes to the flavin that recapitulate common themes in flavin-dependent enzyme reactions: an alteration in oxidation state of the flavin or the formation of a covalent adduct. In contrast, a unifying chemical mechanism linking flavin excitation to photoreceptor activation is yet to emerge for the BLUF protein family. While it is clear that the formation of a flavin adduct is not involved in BLUF domain activation, the role of electron transfer and accompanying changes in flavin redox state remains controversial, with both dark adapted and light adapted forms containing a fully oxidized flavin.
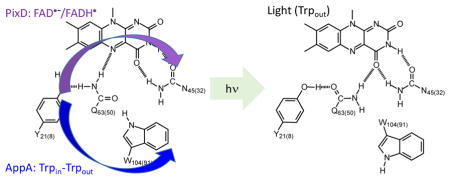
In the BLUF photoreceptors,19 the flavin is surrounded by a hydrogen bonding network that includes conserved Tyr and Gln residues, both of which are essential for photoactivity (Figure 1). In some BLUF protein structures a conserved Trp is within hydrogen bonding distance of the network, and indeed the variable location of the Trp side chain, between this Trpin conformation and a Trpout conformation, supports proposed mechanisms in which a light-induced change in Trp conformation is on the reaction coordinate for photoactivation.7, 20–21
Figure 1. The Hydrogen Bonding Network and Primary Photoactivation Mechanism.

The isoalloxazine ring is surrounded by a conserved hydrogen bonded network that includes a Tyr Y21(8), Gln Q63(50) and Asn N45(32). Also shown is a Trp W104(91) which is shown hydrogen bonded to the Gln residue in the dark state but not in the light state. Movement of the Trp from a Trpin to a Trpout conformation is thought to be a central component of the BLUF photoactivation mechanism although we note that in the X-ray crystal structures of PixD in the dark (PDB: 2HFN)22 and a stable lit states (PDB: 3MZI),7 the W91 sidechain is rotated out of the binding pocket. Photoactivation involves rotation of the Gln side-chain, and in the above mechanism keto-enol tautomerism of the Gln side-chain is shown preceding side-chain rotation.10, 23–25 Residues are numbered based on the sequence of AppA with the residue number in PixD given in parentheses.
All active BLUF domain proteins exhibit a characteristic ~10 nm red-shift in the electronic spectrum of the oxidized flavin upon blue light excitation,26 which has been linked to the formation of an additional hydrogen bond to the flavin C2=O carbonyl group from the side chain of the conserved Gln.5, 23, 27–29 This, in turn, is thought to result from light-induced rotation of the Gln side chain, a proposal that remains a central tenet of BLUF protein photoactivation.5, 30–32 Based on ultrafast infrared spectroscopy, we have proposed that keto-enol tautomerism of the Gln side chain precedes rotation,23 a hypothesis that is supported by theoretical and experimental studies.9–10, 24 Gln rotation may trigger a change in Trp conformation, and we and others have demonstrated that mutagenesis of the Trp uncouples the light-induced changes in the flavin absorption spectrum and hydrogen-bonding from the global changes in BLUF protein structure that accompany photoactivation.8, 33
The light-driven changes in the hydrogen bond network presumably result from alterations in the electronic structure of the flavin caused by photoexcitation. Based on experiments with PixD (Slr1694), which controls phototaxis in the cyanobacterium Synechocystis sp. PCC 6903 through a light-dependent interaction with PixE, it has been shown that electron transfer from the conserved Tyr (Y8 in PixD) to the photoexcited isoalloxazine ring of the flavin results in formation of an anionic semiquinone radical (FAD•−).25, 34–36 This electron transfer is proposed to be followed by the sequential transfer of a proton leading to the neutral flavin semiquinone, FADH•, in which Y8 operates as both the electron as well as a proton donor to the flavin (i.e. FAD* → FAD•− → FADH•).34–37 However, we have previously shown that a metastable radical intermediate state is not formed during photoactivation of the BLUF domain proteins AppABLUF and BlsA, whereas radical formation was clearly observed in PixD.34 We further confirmed that electron transfer did not occur during the photoactivation of AppABLUF using the site-specific incorporation of fluorotyrosine (F-Tyr) analogs into position Y21.21 The observation of radical formation in PixD,34, 38 and in the BLUF protein PapB,39 thus suggests that significant differences exist in the mechanism of photoactivation within the BLUF protein family.
To gain insight into the structural basis for the significant variation in photoactivation mechanism, we superimpose the structures of AppABLUF and PixD in Figure 2. Analysis of the flavin binding pocket reveals that the conserved Tyr and Gln residues, as well as N45 (N32) occupy essentially identical locations in both photoreceptors. Thus, at least on this basis, Y21 in AppABLUF should be able to fulfill the same function in electron and proton transfer as Y8 does in PixD. However, two differences can be observed. In AppABLUF, the FAD C2=O group makes a single hydrogen bond to H44, whereas in PixD two hydrogen bonds are present to N31 (the homolog of H44) and to R65, which is a His (H78) in AppABLUF. We note that the homolog of N31 (H44) in BlsA is a Phe (F32),40 and given that radical formation is not observed in either AppABLUF or BlsA, it is possible that alterations in the environment of C2=O may modulate the BLUF photocycle. The other major difference is the position of the conserved Trp, which is in the Trpin conformation in AppABLUF (W104) and in the Trpout conformation in PixD (W91). Since the X-ray structure of BlsA is not available, a comparison with this protein cannot be drawn. However, it is possible that the position of the Trp influences the ability of the Tyr to participate in proton coupled electron transfer (PCET), as proposed recently based on computational studies.11
Figure 2. The Flavin Binding Pocket of.

AppABLUF and PixD
The structures of AppABLUF (PDB: 1YRX; yellow) and PixD (PDB: 2HFO; slate) have been overlaid to compare and contrast the hydrogen bonding network that surrounds the isoalloxazine ring of the FAD chromophore. The numbering for AppABLUF is used for residues that are conserved between the two proteins with the residue number in PixD given in parentheses. Superposition was performed using pymol,41 using the main chain Cα atoms of 1YRX (117 residues) and Chain A from 2HFO (150 residues) and gave an overall rmsd of 0.7 over 76 atoms. Hydrogen bond distances are in Å.
In the current work, we have probed the specific role of the Y8 hydroxyl group in PixD photoactivation by replacing this residue with F-Tyr analogs that modulate the pKa and reduction potential by 3.5 pH units and 200 mV, respectively. Although little impact was observed on the forward light driven reaction in AppABLUF,21 the F-Tyr analogues are shown here to have a profound impact on the photoactivation mechanism of PixD. In wild-type PixD there is evidence from ultrafast infrared spectroscopy for the sequential formation of FAD•− followed by FADH• on the reaction coordinate for light state formation. In 3-F-Tyr (3-FY8) PixD, sequential electron and proton transfer is no longer observed, with FAD•− and FADH• appearing simultaneously, while the proteins substituted with 2,3-diF-Tyr (2,3-F2Y8), 3,5-diF-Tyr (3,5-F2Y8), or 2,3,5,-triF-Tyr (2,3,5-F3Y8) do not progress to a stable light state. In contrast, for the light to dark recovery, the F-Tyr analogs resulted in a dramatically enhanced recovery rate in AppABLUF, an effect which is here shown to be much less marked in PixD. These observations highlight fundamental differences between the photocycles of PixD and AppABLUF.
Experimental Methods
Materials
3-Fluorophenol and 2,6-difluorophenol were purchased from Sigma-Aldrich. 2,3-Difluorophenol was purchased from Acros Organics. 2,3,6-Trifluorophenol was purchased from Oakwood Chemical. Pyridoxal-5′-phosphate, and flavin adenine dinucleotide were purchased from Sigma-Aldrich.
F-Tyr synthesis using Tyrosine Phenol Lyase (TPL)
Tyrosine phenol lyase (TPL) was prepared and used for the synthesis of the F-Tyr analogs as previously described.21
Expression and purification of PixD
Expression of PixD was performed using a plasmid containing the gene for PixD cloned into the pOPINE vector so that the expressed protein included a C-terminal His-tag. BL21(DE3) Escherichia coli cells were transformed by the PixD C-6xHis construct (pOPINE vector) and plated on LB-Agar containing 50 mg/L ampicillin (Amp). Overnight cultures were prepared by inoculating 10 mL Luria Broth (LB) media containing 50 mg/L Amp with a single colony and incubating the culture at 37 °C at 250 rpm. Subsequently 5 mL of the overnight culture was used to inoculate 1L of LB media containing 50 mg/L Amp in a 4L flask. The culture was shaken at 37 °C and 250 rpm until the cells reached an OD600 of about 0.8, after which the temperature was lowered to ~ 18 °C. After ~30 min, 0.8 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to induce protein expression, and the culture was then incubated for ~ 16 hr in the dark. Cells were harvested by centrifugation at 4,225 x g and 4 °C and stored in the dark at −20 °C until needed. Purification of PixD was performed under minimal illumination. The cell pellets were thawed and resuspended in 40 mL of 50 mM NaH2PO4, pH 8.0 buffer containing 10 mM NaCl (buffer A). Then, 200 μL of 50 mM phenylmethanesulphonylfluoride (PMSF) was added together with 14 μL of β-mercaptoethanol, and the cells were lysed using a high-pressure cell disruptor (Constant Systems, Model E1061) at 30 kpsi whilst maintaining the cell disruptor vessel at 4 °C. Cell debris was then removed by ultracentrifugation at 126,264 x g and 4 °C for 90 min, after which the supernatant was incubated with 10 mg of FAD for 45 min on ice. PixD was then purified using Ni-NTA affinity chromatography. After applying the supernatant to a 25 mL column containing 5 mL of resin, the column was washed with 100 mL of buffer A. A second wash was performed with 100 mL of buffer A containing 10 mM imidazole, followed by 100 mL of buffer A containing 20 mM imidazole. Protein was eluted using buffer A containing 250 mM imidazole. The eluted protein was dialyzed against buffer A overnight. The purity of the protein was determined by SDS-PAGE electrophoresis. To exchange the protein into D2O, the samples were first frozen in liquid N2 and lyophilized overnight, followed by 2–3 cycles of resuspension in D2O and lyophilization.
Incorporation of F-Tyr residues using orthogonal aminoacyl-tRNA synthetases
F-Tyr analogs 3-FY, 2,3-F2Y, 3,5-F2Y and 2,3,5-F3Y were incorporated into position 8 of PixD using two orthogonal polyspecific aminoacyl-tRNA synthetases, E3 and E11, generously provided by Professor Joanne Stubbe.42 Site-directed mutagenesis was first used to convert the codon for Y8 to the amber codon (TAG) in PixD using the primers in Table S1. A C-terminal 6xHis-tag PixD construct was used (pOPINE C-6His), so that only protein with a F-Tyr incorporated at position 8 was able to bind to the Ni-NTA affinity purification resin. The PixD plasmid with the Y8-TAG mutation was co-transformed into BL21(DE3) E. coli cells together with either the E3 or E11 pEVOL plasmid, and plated on LB agar containing both Amp (50 mg/L) and Cam (50 mg/L) to select cells harboring both plasmids. A colony from the LB/Amp/Cam plate was used to start a 10 mL overnight culture, which was subsequently used to inoculate 500 mL 2x YT media containing Amp and Cam. The culture was grown until an OD600 of ~ 0.4, and a F-Tyr analog dissolved in NaOH was added to the media to give a final concentration of ~1 mM. After 30 min, 0.05% w/v arabinose was added to induce expression of the E3 or E11 synthetase. The culture was incubated at 37 °C (250 rpm) until the OD600 reached ~1.0, and then 0.8 mM IPTG was added to the media. After incubating overnight at 30 °C (250 rpm), the cells were harvested and protein was purified using the same protocol as that described for the purification of PixD above. Incorporation of F-Tyr amino acids was analyzed by ESI MS/MS mass spectrometry of tryptic digests using a Thermo Scientific Orbitrap Fusion Lumos instrument.
UV-Vis Spectroscopy
Absorption spectra of all protein samples were obtained at 20 °C using a Cary 100 Bio UV-Vis spectrometer. The concentration of the protein samples was kept at around 80 μM in either H2O or D2O buffer (50 mM NaH2PO4, 10 mM NaCl, pH/D 8.0). Dark-adapted spectra were obtained first and then the samples were illuminated with ~500 mW of 455 (± 10) nm light for ~ 1 min in order to generate the light state.
Time-resolved UV-Vis spectroscopy
Absorption spectra of protein samples were obtained using an Ocean Optics USB2000+ spectrometer described previously.21 Spectra of dark adapted PixD and the F-Tyr-substituted variants were first obtained, and then the sample window (5 cm2 area) was irradiated with ~500 mW of 455 (± 10) nm light until the photostationary state was generated. Conversion to the light state was monitored by taking absorption spectra of the samples in the IR cell. Recovery of the dark state was then monitored after illumination was discontinued.
Ultrafast Time-Resolved Infrared Spectroscopy
Ultrafast time-resolved IR (TRIR) spectra were measured at the STFC Central Laser Facility with ~ 100 fs time resolution.43 TRIR spectra were acquired at 20 °C from 1300 – 1800 cm−1 at a resolution of 3 cm−1 per pixel. Data were obtained using a 50 μm path length flow cell of volume < 1 ml, which was also rastered in the excitation beam in order to minimize unwanted secondary photochemistry processes such as photobleaching, photodegradation and photoconversion. The excitation beam of the 450 nm 100 fs 5 kHz pulses was focused to a spot size of ~ 100 μm and the pulse energy was kept below 400 nJ to avoid buildup of the light state. Transient difference spectra (pump on – pump off) were recorded using the IR probe at time delays between 1 ps and 2 ns. After the measurements were recorded, the extent of photoconversion was shown to be negligible using absorbance spectroscopy. Spectra were calibrated relative to the IR transmission of a pure cis stilbene standard sample placed at the sample position. TRIR spectra of light-adapted PixD were recorded while continuously illuminating the sample cell with ~200 mW of 450 nm light from an LED. The cell was rastered during data collection.
Time Resolved Multiple Probe Spectroscopy (TRMPS)
TRMPS spectra were also obtained at the STFC Central Laser Facility.44 The TRMPS method has been described,44 and previously used by us to analyze the photoactivation of BLUF and LOV domain proteins.8 Spectra were measured at 20 °C between 100 fs to 200 μs after excitation of FAD at 450 nm. A rastered flow cell was used, and data were acquired using a 450 nm pump operated at 0.6–0.8 μJ per pulse and a repetition rate of 1 kHz. After the measurements were recorded, the extent of photoconversion was shown to be negligible using absorbance spectroscopy. The spectral resolution was 3 cm−1 and the temporal resolution was 200 fs. A typical measurement was acquired during 45 min of data collection. All samples were prepared at 1–2 mM concentration in D2O buffer (50 mM NaH2PO4, 10 mM NaCl, pD 8.0). Spectra were calibrated relative to the IR transmission of a pure cis stilbene standard sample placed at the sample position. Data were analyzed globally using the sequential model with Glotaran software package.45
Results and Discussion
TRIR of the Wild-type PixD Dark State
Complex multiexponential kinetics have been previously reported during the decay of the transient electronic spectrum of PixD, consistent with the sequential formation of radical intermediates during photoactivation.34–35, 37, 46–47 Kennis and co-workers used transient electronic spectroscopy to observe the formation of radical states during photoactivation of PixD,38, 47 while our recent publication confirmed the observation of a flavin radical as well as complex kinetics leading to light state formation in PixD, and contrasted this with the absence of observable radical intermediates in AppABLUF, which had overall slower photoactivation kinetics.34 The initial step in the PixD photocycle was characterized using ultrafast time-resolved infrared (TRIR) spectroscopy which probes changes in the infrared spectrum of both the chromophore and protein following photoexcitation. In the present work, we first reexamine the TRIR spectra of wild-type PixD, acquired at higher S/N than in our previous study, as a foundation for exploring the impact of F-Tyr substitution on the PixD photocycle.
The newly measured TRIR spectra (Figure 3A) were subjected to global analysis using Glotaran,45 in which the kinetic traces at all measured wavenumbers were simultaneously fit to a sum of exponential decays. The resulting decay associated spectra, generated by assuming parallel decay of all the components, are presented in the supplementary information (Figure S1). The corresponding evolution associated spectra (EAS) of wild-type PixD are shown in Figure 3B, where the EAS assume a sequential reaction scheme: for the wild-type photoreceptor, four time constants are needed to adequately describe the data (Table 1).
Figure 3. TRIR Spectra of the Wild-type PixD Dark State.

(A) TRIR spectra of wild-type PixD (B) EAS of wild-type PixD from a global fit of the TRIR data in (A). Transients assigned to FADH• (1535 cm−1) and FAD•− (1515 cm−1) are shown in the inset. (C) Kinetic traces of the excited state decay (1380 cm−1) and the ground state recovery (1547 cm−1, shown inverted for comparison). The overlay demonstrates slower recovery of the ground state compared to the decay of the excited state. (D) Kinetic traces show the rise and decay of bands assigned to FAD radicals in PixD. Raw data are shown as solid lines while global fitting results are shown as dashed lines.
Table 1.
Time constants from TRIR Data for PixD and n-FY8 substituted PixDa
| τ1/ps | τ2/ps | τ3/ps | τ4/ps | <τ>/psd | |
|---|---|---|---|---|---|
| PixDb | 2.5±0.1 | 20±0.5 | 110±2 | 525±15 | 96 |
| 3-FY8 PixDb | 4.5±0.3 | 33±1 | 217±5 | 1300±100 | 347 |
| 2,3-F2Y8 PixDc | 3±1 | 60±1 | 1395±45 | infinite | 917 |
| 3,5-F2Y8 PixDc | 2.5±0.5 | 54±1 | 965±20 | infinite | 640 |
| 2,3,5-F3Y8 PixDc | 4.5±1 | 48±1 | 1030±30 | infinite | 730 |
|
| |||||
| PixD Light Stateb | 3±0.2 | 24±0.5 | 563±7 | infinite | 23 |
Unless noted, the TRIR data are from the dark state of the wild-type and F-Tyr PixD variants. Data were globally analyzed using Glotaran.45
Four time constants were needed to adequately describe the data for the dark and light states of PixD and 3-FY8 PixD.
Very long kinetics were observed for the 2,3-F2Y8, 3,5-F2Y8 and 2,3,5-F3Y8 substituted proteins, and only three time constants were needed to describe the data for these proteins, followed by an ‘infinite’ lifetime extending beyond the range of the TRIR measurement.
Mean lifetime of the excited state decay calculated from the global analysis at 1380 cm−1.
Major negative bands (bleaches) are observed at 1700, 1638, 1580, and 1547 cm−1 in Figure 3A, B which are assigned to the C4=O and C2=O carbonyl groups of the flavin (1700 and 1638 cm−1) and C-N vibrations of the isoalloxazine ring.23, 48–49 In addition to these bleaches, which appear instantaneously after photoexcitation (<100 fs), positive bands (transients) are also observed at 1420 and 1380 cm−1 which correspond to the excited state (ES) of the flavin (FAD*). The change in intensity of the 1380 and 1547 cm−1 bands as a function of time can thus be used to quantify excited state decay and ground state recovery, respectively. In Figure 3C it can be seen that the excited state decays more rapidly than recovery of the ground state, consistent with a mechanism that involves excited state relaxation to an intermediate species before ground state recovery.
Previously we employed model studies to assign a transient at 1528 cm−1 in the TRIR spectrum of PixD to the formation of an FAD radical.34, 50 The improvement in signal quality of the current data enables us to clearly observe two features in this region at around 1515 and 1535 cm−1 (Figure 3A, B), which have different kinetics (Figure 3D) and are assigned to FAD•− and FADH•, respectively, based on TRIR spectra of the anionic and neutral semiquinones formed by model flavins and glucose oxidase.50 The 1515 cm−1 transient appears on a timescale of 2.5 ps and is assigned to formation of the FAD•−. As the transient at ~1515 cm−1 is decaying on the 20 ps timescale, a new transient is formed around 1535 cm−1 which we associate with the formation of FADH• by proton transfer (Figure 3A, B). The transient at 1535 cm−1 decays in a bi-exponential manner with time constants of 110 ps and 525 ps, leading ultimately to the formation of the PixD light state. The final TRIR spectrum (2 ns) is characteristic of the light state and shows a transient at 1690 cm−1 which can be assigned to the C4=O flavin carbonyl group, red shifted from the position observed in the dark state (1700 cm−1) as a consequence on an increase in hydrogen bonding concomitant with photoactivation.48, 51–53 In Figure 3 it can be seen that FADH• forms before complete decay of the excited state, FAD*. This, together with the observation of multiexponential kinetics, suggests the presence of an inhomogeneous population of states imperfectly resolved in the sequential analysis, possibly resulting from a distribution of ground state conformations around the flavin chromophore. Both electron and proton transfer reactions are very distance dependent and sensitive to their environment, so photokinetics will reveal inhomogeneity which is not apparent in crystallography. However, there is clear evidence for FAD radical formation in wild-type PixD as reported previously,34–35 and significantly, we are now able to resolve the radical formation into distinct electron and proton transfer phases.
To support the assignments discussed above, we also obtained TRIR spectra of the Y8F PixD mutant (Figure S2). Although Y8F PixD is photoinactive, excited state quenching also occurs, and is similar in magnitude to that observed for wild-type PixD, and again at a faster rate than the rate of ground state recovery (1380 cm−1 v 1547 cm−1). However, in the region where we observe features associated with FAD radicals in wild-type PixD, only one transient is observed, at 1520 cm−1. Based on the possibility that the 1520 cm−1 band might be comprised of unresolved contributions from FAD•− and FADH•, we measured the kinetics at 1515 and 1530 cm−1. However, no difference was observed in the evolution of the signal at these two frequencies, suggesting that only one transfer process occurs in the Y8F mutant. Since the source of the electron is most likely W91, and since this residue cannot donate a proton, we conclude that in Y8F the 1520 cm−1 band is associated with the flavin semiquinone anion, supporting the assignment of the band at 1515 cm−1 in wild-type PixD to FAD•−.
TRIR of the Wild-type PixD Light State
We also obtained TRIR spectra of PixD in the light state (Figure 4). A bleach is formed instantaneously at 1685 cm−1, which is assigned to the C4=O of the flavin. The position of this band is consistent with the transient observed at 1690 cm−1 in the final EAS of the dark-adapted protein. The assignment of the 1685 cm−1 band is also consistent with the proposed mechanism of BLUF protein photoactivation, in which an additional hydrogen bond is formed to the C4=O in the light state, and the red shift in the C4=O frequency from 1700 cm−1 has been observed previously in PixD,48 as well as in other BLUF proteins, such as AppABLUF.23, 51–53
Figure 4. TRIR of the Wild-type PixD Light State.
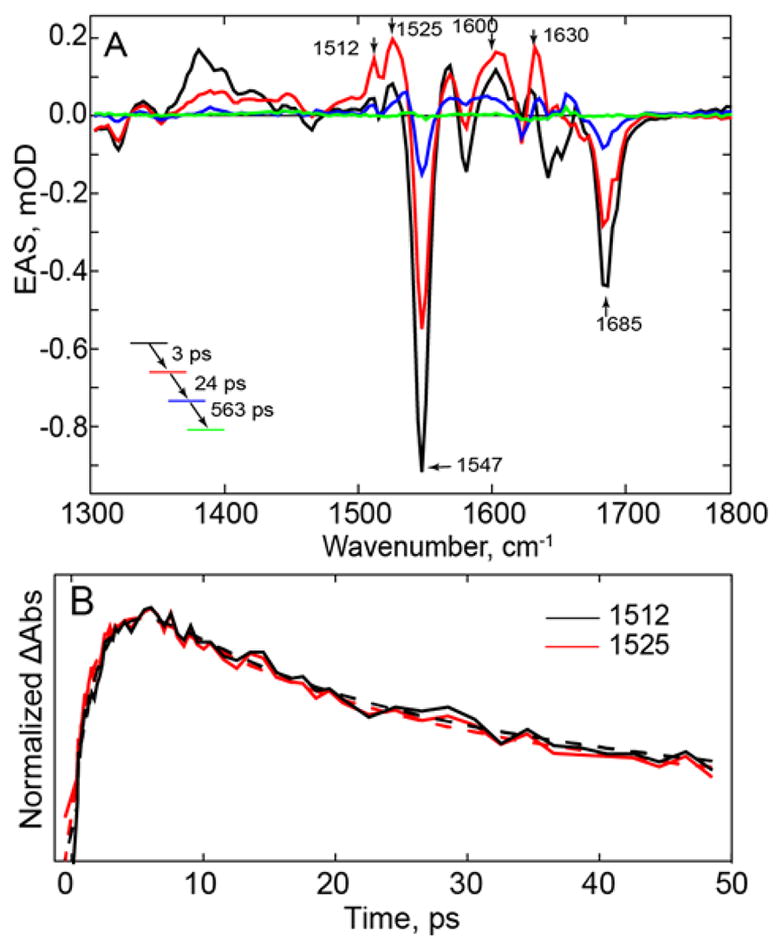
(A) EAS of light adapted PixD generated by constant illumination with 450 nm light focused on the sample in the TRIR experiment. (B) Comparison of the kinetics of the bands at 1512, 1525, cm−1. Raw data are shown as solid lines while global fitting results are shown as dashed lines.
The excited state of the light state decays significantly faster than the corresponding excited state of the dark state and indeed has completely decayed during the time scale of the TRIR experiment, where three time constants are now needed to adequately describe the data (Table 1). It has been proposed that in the light state of PixD a concerted electron proton transfer (CEPT) takes place directly leading to the formation of FADH•.35, 37 In Figure 4, bands at 1512, and 1525 cm−1 are observed within 3 ps, which are assigned to the FAD radical. However, unlike the dark state, the bands associated with the radical do not evolve at different rates but have the same kinetics, consistent with a concerted process such as CEPT. Thus, contrary to the dark state, where we observe FAD* → FAD•− → FADH•, in the light state FAD* decays directly to FADH•. These data agree well with the mechanism proposed by Mathes et al.37
Effect of Y8 fluorination on PixD photoactivation
Y8 in PixD is essential for photoactivity and has been proposed to function as both the electron and proton donor in the concerted formation of the FAD radical state (PCET). To probe the role of Y8 in PCET, we site-specifically replaced this residue with 3-FY8, 2,3-F2Y8, 3,5-F2Y8, and 2,3,5-F3Y8 in order to modulate both the redox potential and pKa. The F-Tyr analogs were synthesized and incorporated into the protein,21 and ESI MS/MS analysis did not detect any native Tyr in the F-Tyr substituted proteins indicating that the F-Tyr content of each variant was ≥99% (Figure S3).
Electronic Spectrum
We first examined the electronic absorption spectrum of each PixD variant. In Figure 5A the dark state spectrum of each F-Tyr substituted PixD is compared with wild-type PixD and FAD in solution. The electronic spectra of the proteins are essentially identical and are characterized by a long wavelength shoulder that is not resolved in the solution spectrum of FAD. Thus, we can conclude that F-Tyr substitution has not perturbed the FAD binding pocket in a way that alters the electronic spectrum.
Figure 5. Electronic Spectra of Wild-type PixD and the F-Tyr PixD Variants.

(A) Comparison of the dark state absorbance spectra of wild-type PixD and the F-Tyr PixD variants. (B) Comparison of the light state absorbance spectra of wild-type PixD and the F-Tyr PixD variants acquired immediately after ~10 s of ~500 mW illumination of the entire 5 cm2 of the sample window with 450 nm light.
We next analyzed the change in the spectrum caused by photoexcitation. In BLUF proteins, irradiation with blue light results in a ~10 nm red shift in the 450 nm FAD peak, as is observed for wild-type PixD in Figure 5B. Light induced changes are also observed for all the F-Tyr PixD variants with the exception of 2,3,5-F3Y8-PixD, although the magnitude of the red shift resolved in these steady-state measurements is apparently lower for 3-FY8, 2,3-F2Y8 and 3,5-F2Y8 PixD.
TRIR and TRMPS of the Forward Photoreaction
The TRIR spectra of the F-Tyr-substituted dark adapted PixD proteins are shown in Figure 6 where it can be seen that the major bleaches and transients observed in the initial (2 ps) spectrum of the wild-type PixD are conserved amongst all the substituted proteins. The similarity of the 2 ps TRIR spectra (Figure 6) across all proteins indicates that the ground state environment of the flavin has not been perturbed by modification of Y8, consistent with the similarity in the dark state absorption spectra (Figure 5). However, a small (4 cm−1) increase in the C4=O flavin carbonyl model, from 1700 to 1704 cm−1, is observed in the spectra of the F-Tyr PixD proteins, which may indicate that the hydrogen bond to the C4=O carbonyl is slightly weakened due to the incorporation of F-Tyr analogs, although the effect is not a function of pKa.
Figure 6. TRIR Spectra Wild-type PixD and the F-Tyr PixD Variants.

TRIR Spectra of wild-type PixD and the F-Tyr PixD variants at 2, 40, 200, 1000, and 3400 ps for (A) wild-type, (B) 3-FY8 PixD, (C) 2,3-F2Y8 PixD, (D) 3,5-F2Y8 PixD and (E) 2,3,5-F3Y8 PixD.
The TRIR spectra at later times demonstrate that alteration in the reduction potential and/or pKa of Y8 has a dramatic impact on the forward photoreaction (Figure 6), in contrast to the BLUF protein AppABLUF where replacement of the conserved Y21 with F-Tyr analogs results in only a small ~3-fold change in the kinetics for photoactivation over the entire pKa range.21 3-FY8 PixD exhibits the highest similarity to the wild-type protein although the rate of spectral evolution is described by significantly longer time constants compared to wild-type PixD (Table 1). The light state, characterized by the transient at 1690 cm−1 forms within 230 ps, although a considerable amount of the excited state is still present at this stage, again consistent with the presence of multiple conformations and an associated distribution of rates. The transients at 1515 and 1535 cm−1 are also still observed for 3-FY8 PixD suggesting that both the electron and proton transfer steps occur, albeit more slowly than for wild-type PixD. This observation is consistent with the studies of Kennis and coworkers, who also observed an increase in the average excited state lifetime in 3-FY8 PixD,35 a result which broadly recapitulates similar studies on ribonucleotide reductase, where an increase in reduction potential leads to a decrease in the rate of ribonucleotide reduction.54 In our studies on AppABLUF, we considered the impact of F-Tyr substitution on the TRIR spectra using Marcus theory to analyze a sequential scheme involving charge separation (kcs) followed by charge recombination (kcr) and concluded that either kcs ≪ kcr since no radical intermediate could be detected, or that the reaction proceeded without population of a distinct charge transfer intermediate. Consistent with the absence of electron transfer in the AppABLUF mechanism, F-Tyr substitution only had a small (3-fold) effect on the overall rate of light state formation.21 Although the situation is more complex in PixD, since both electron and proton transfer are clearly involved, in contrast to AppABLUF, the data do indicate that the rate of charge separation must be faster than recombination, since a flavin radical clearly forms on photoactivation (Figure 3). In addition, the observation that the rate of radical formation and decay are both decreased by fluorination suggests that both kcs and kcr are in the Marcus inverted region.
The TRIR spectra of 2,3-F2Y8, 3,5-F2Y8, and 2,3,5-F3Y8 PixD differ more dramatically from the wild-type data than those of 3-FY8 PixD (Figure 6). The most significant differences include the lack of a distinct 1535 cm−1 transient, assigned to FADH•, and the lack of the 1690 cm−1 transient assigned to formation of the light state. TRMPS spectra (Figure S4) show that no further evolution occurs following the final TRIR spectrum, and the absence of the 1690 cm−1 transient therefore shows that F-Tyr substitution prevents the formation of a detectable population of light state, which contrasts sharply with AppABLUF, where light state formation persists for all F-Tyr analogs. In addition, the kinetics for the spectral evolution that can be observed for 2,3-F2Y8, 3,5-F2Y8, and 2,3,5-F3Y8 PixD are significantly slower compared to 3-FY8 and wild-type PixD, and only three distinct lifetimes are now required to fit the nanosecond domain TRIR data (Table 1).
The transient assigned to FAD•− is still observed at 1515 cm−1 for 2,3-F2Y8, 3,5-F2Y8 and 2,3,5-F3Y8 PixD (Figure 6). Analysis of the kinetics for this band show both rise and decay components (Figure S5), consistent with the assignment to an intermediate, although differences between the rates of excited state decay (1380 cm−1) and ground state recovery (1550 cm−1) (Figure S6 and S7) are less obvious for the di- and tri-substituted F-Tyr PixD variants than for the wild-type or 3-FY8 PixD. Finally, in the TRMPS data for 3,5-F2Y8, and 2,3,5-F3Y8 PixD (Figure S4), a feature around 1490 cm−1 can be observed, indicative of triplet state formation that is absent in wild-type PixD; this result is consistent with an increase in lifetime of the singlet excited state, allowing intersystem crossing to compete. Due to the slower excited state decay in 3-FY8, 2,3-F2Y8, 3,5-F2Y8, and 2,3,5-F3Y8 PixD (Figure S6) we also observe a significant increase in flavin fluorescence with an ~3-fold increase in quantum yield between the wild-type protein and the F-Tyr substituted PixD variants (Table S2).
Taken together, the data suggest that the change in reduction potential and/or pKa of Y8 upon di- and tri-fluoro substitution apparently halts the photocycle after FAD•− is formed. A simple explanation for the lack of further progression of the photocycle is that the pKa of 2,3-F2Y8, 3,5-F2Y8, and 2,3,5-F3Y8 has dropped sufficiently that this residue is no longer protonated and thus cannot transfer a proton to FAD•−, supporting the importance of PCET in the photoactivation of PixD. The proton transfer is evidently critical for trapping the light state in which Q50 has rotated to H-bond to the C4=O carbonyl group.
Recovery of the Dark State
The electronic spectra of wild-type PixD and the F-Tyr PixD variants indicate that photoexcitation leads to the formation of a red-shifted absorption spectrum for all proteins except 2,3,5-F3Y8 PixD (Figure 5), which is evidence for the formation of the PixD light state in those cases. However, under the same illumination conditions the red shift is clearly less marked for 2,3-F2Y8 and 3,5-F2Y8 PixD than for wild-type and 3-FY8 PixD. This is consistent with the slower excited state decay, indicating a reduced quenching rate constant for light state formation, thus allowing other processes (such as intersystem crossing) to compete. One apparent paradox is that for all PixD variants, except 2,3,5-F3Y8 PixD, the red shift in the absorption spectra indicates light-state formation, whilst, based on the TRIR spectra, we concluded that the photocycle for 2,3-F2Y8 and 3,5-F2Y8, as well as 2,3,5-F3Y8 PixD stalled after the initial electron transfer event. This suggests that the light state populated during the continuous illumination, generating the red-shifted electronic spectra, is not sufficiently stable to result in an observable signal in the single-shot TRIR spectra of 2,3-F2Y8 and 3,5-F2Y8 PixD. To test this hypothesis, we obtained TRIR spectra under continuous illumination (Figure 7). In the TRIR spectra of both wild-type PixD and 3-FY8 PixD, continuous illumination at 450 nm leads to the observation of a bleach at 1686–1689 cm−1 which is assigned to the C4=O carbonyl group of the light state, consistent with the transient observed at ~1690 cm−1 in the pump-probe TRIR spectra (Figures 3, 4 and 6). However, in 2,3-F2Y8 PixD two bands are observed, at 1686 and 1700 cm−1 while in the 3,5-F2Y8 and 2,3,5-F3Y8 PixD proteins the major band in this region is observed at 1704 cm−1, with a barely resolvable shoulder at around 1686 cm−1 in the spectrum of 3,5-F2Y8 PixD. These data indicate that the light state can be partially populated in 2,3-F2Y8 PixD, and possibly in 3,5-F2Y8 PixD, under continuous illumination, consistent with the steady state spectra; however, such a light state may be unstable with respect to reversion to the dark state. One plausible explanation is the generation of an increasing fraction of a population of PixD which is incapable of light state formation with decreasing Y8 pKa.
Figure 7. TRIR Spectra of Wild-type PixD and the F-Tyr PixD Variants Under Continuous Illumination.
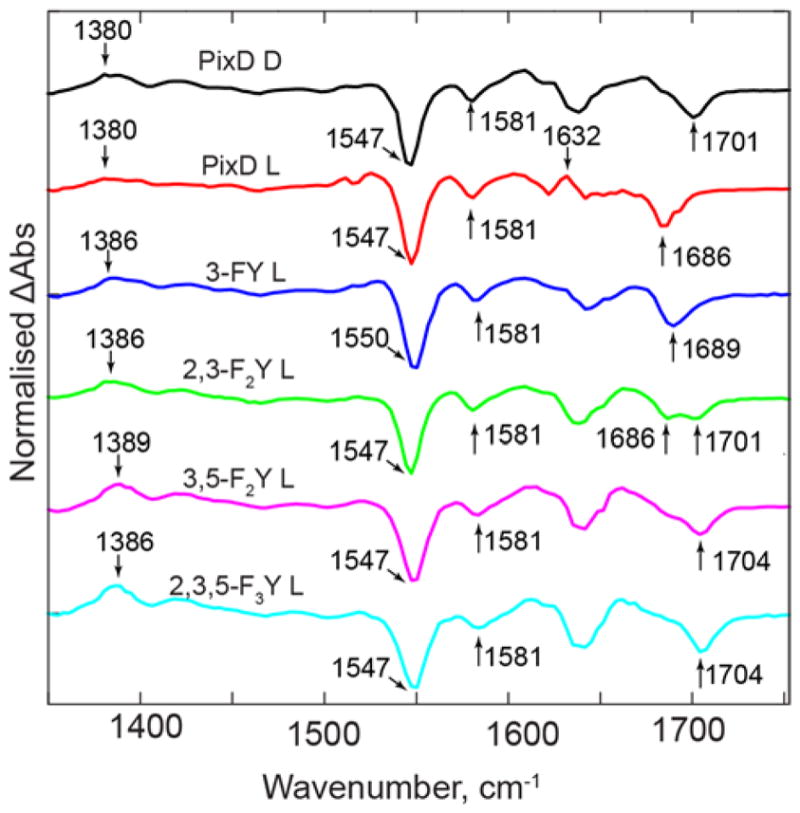
TRIR spectra were obtained whilst continuously illuminating the protein samples with ~200mW of 450 nm light (L). The top spectrum is the TRIR spectrum of wild-type PixD obtained using the standard pump-probe approach (dark state, D). A key observation is the change in position of the C4=O bleach from 1701 cm−1 in the PixD dark state to 1686 cm−1 upon formation of the light state. Continuous illumination of the F-Tyr substituted PixD variants results in full conversion of 3-FY8 PixD, but only partial population of the light state in 2,3-F2Y8 PixD, and possibly in 3,5-F2Y8 PixD. In addition, there is no light state signature in 2,3,5-F3Y8 PixD.
Based on the conclusion that the alteration in the electronic spectra upon photoexcitation represented light state formation, we monitored the time-dependent recovery of the absorption spectra to quantify the rate of dark state recovery. It has already been noted that the rate of recovery in wild-type PixD is substantially greater than for AppABLUF (Table 2). For 3-FY8 and 2,3-F2Y8 PixD this rate is further accelerated 6 and 7 fold, consistent with formation of a light state which is increasingly unstable with respect to dark state recovery in F-Tyr PixD. Further, as was observed for AppABLUF, reduction in the Y8 pKa monotonically accelerates the rate of dark state recovery (Table 2). However, the relative increase in the rate of recovery is much less marked in PixD: in H2O, the rate increases 970-fold between wild-type AppABLUF and 3,5-F2Y21 AppABLUF,21 compared to only 15-fold for PixD, while in D2O the change in rate is 850-fold for AppABLUF and 6-fold for PixD. The corresponding Brønsted plots for PixD have shallower slopes of 0.43 in H2O and 0.28 in D2O (Figure 8), compared to slopes of ~1 for AppABLUF.21 Thus, alteration of the Y8 pKa has a smaller effect on the rate of dark state recovery in PixD compared to AppABLUF. However, the observation of large normal solvent isotope effects on the rate of recovery for both PixD (2–11) and AppABLUF (2–9), together with Brønsted α values of 0.43 and 1, respectively, indicates that two proteins have similar recovery mechanisms which differ primarily in the degree of proton transfer from the conserved Tyr (Y21 or Y8) in the rate limiting transition state.
Table 2.
Dark state recovery rate constantsa
| PixD | AppABLUFb | ||||||
|---|---|---|---|---|---|---|---|
| pKac | kH2O (s−1) | kD2O (s−1) | sied | kH2O (s−1) | kD2O (s−1) | sied | |
| Wild-type | 9.9 | 0.038 ± 0.004 | 0.010 ± 0.0003 | 3.7 ± 0.1 | 0.00065 ± 0.00006 | 0.00013 ± 0.00001 | 5.0 ± 1 |
| 3-FY | 8.4 | 0.22 ± 0.012 | 0.023 ± 0.0008 | 9.5 ± 0.1 | 0.034 ± 0.002 | 0.0039 ± 0.0001 | 8.8 ± 0.7 |
| 2,3-F2Y | 7.8 | 0.27 ± 0.013 | 0.047 ± 0.0065 | 5.8 ± 0.1 | 0.22 ± 0.01 | 0.040 ± 0.001 | 5.5 ± 0.2 |
| 3,5-F2Y | 7.2 | 0.58 ± 0.06 | 0.058 ± 0.0036 | 9.9 ± 0.2 | 0.63 ± 0.02 | 0.11 ± 0.01 | 5.8 ± 0.3 |
| 2,3,5-F3Ye | 6.4 | ND | ND | ND | 2.6 ± 0.1 | 1.15 ± 0.03 | 2.2 ± 0.1 |
Recovery rate constants were obtained from the change in absorption spectra once irradiation at 455 nm had been terminated by fitting the data to a single exponential function. Errors are based on measurements made in triplicate or quadruplicate.
AppABLUF. Recovery data are for the BLUF domain of AppA and are taken from Gil et al.21
The pKa is that for the amino acid in solution.54
sie, solvent isotope effect.
ND, not detected. For 2,3,5-F3Y8 PixD no light state was detected.
Figure 8. Rate of Dark State Recovery for Wild-type PixD and the F-Tyr PixD Variants.

(A) Brønsted plots in H2O and D2O. The log of recovery rate constant –log(k) plotted against corresponding -pKa. The rate increases 15.4-fold from wild-type (pKa 9.9) to 3,5-F2Y8 PixD (pKa 7.2) with a slope of 0.43 ± 0.04 (R2 0.99), in D2O the rate increases 5.6-fold from wild-type to 3,5-F2Y8 PixD with a slope of 0.28 ± 0.03 (R2 0.972). (B) Comparisons of the dark state recovery rate for PixD and AppABLUF, data measured in H2O, the values for AppABLUF taken from Gil et al.21
Comparison of the PixD and AppABLUF Photocycles
In the BLUF proteins only the forward photoreaction is light driven, while the recovery of the dark state occurs in the absence of illumination. Thus, it may be assumed that photoactivation and dark state recovery follow different reaction pathways. In both PixD and AppABLUF, the conserved Tyr (Y8 or Y21) is essential for reactivity, and in both cases replacement of this Tyr with, for example Phe, leads to a loss of photoactivity. However, despite these overall similarities, profound differences are found in the photocycles of PixD and AppABLUF. In PixD there is clear evidence for coupled electron and proton transfer resulting in the intermediate formation of a flavin radical during photoactivation. In contrast, radical formation is not apparent in the photoactivation of AppABLUF. Modulation of the pKa and reduction potential of the conserved Tyr adds fine detail that allows a deeper comparison of AppABLUF and PixD. Thus, in addition to the lack of radical formation in AppABLUF, replacement of Y21 with F-Tyr analogs has little effect on AppABLUF activation. In contrast, and in agreement with a radical mechanism, changes to the pKa and reduction potential of Y8 have a major impact on PixD activation, where 2,3-F2Y8, 3,5-F2Y8 and 2,3,5-F3Y8 PixD are seemingly unable to proceed past the initial electron transfer event to create a significant long-lived population of light state, and 2,3,5-F3Y8 PixD is unable to populate the light state at all, even under continuous illumination.
In contrast to major differences in the mechanism of photoactivation, the recovery of the dark state in AppABLUF and PixD appear to follow essentially the same mechanism, although there are large absolute differences in the recovery rates. Large normal isotope effects are observed on the rate of recovery for both proteins, and in each case the increase in acidity of the conserved Tyr results in an increase in the observed rate constant, albeit that the change in rate between wild-type and 3,5-F2Tyr proteins (pKa 9.9 to 7.2) is 15-fold for PixD compared to 970-fold for AppABLUF. In AppABLUF, a Brønsted analysis indicated that complete transfer of a proton occurred in the rate limiting transition state on the reaction coordinate for dark state recovery. Based on this result, we proposed a mechanism of recovery in which proton transfer from Y21 to Q63 played a key role in keto-enol tautomerism and subsequent rotation of Q63.21 In PixD, the Brønsted coefficient (α) is 0.43 compared to 1 in AppABLUF indicating an earlier transition state in the recovery of dark adapted PixD and thus that proton transfer involving Y8 has a less significant role in the mechanism of recovery. In addition, the differences in α indicate that the transition state in AppABLUF is similar in structure to that of the dark state whereas the transition state in PixD more closely resembles the photoactivated state of the protein.55
It is difficult to rationalize the different roles for the conserved Tyr in AppABLUF and PixD, given the similarity in the position of Y21 and Y8 from the X-ray structures. However, the flavin binding pockets in the two proteins are not strictly conserved. For instance, in PixD the flavin C2=O group is hydrogen-bonded to two amino acids, N31 and R65, whereas in AppA the C2=O is only hydrogen bonded to H44, the homolog of N31. Analysis of existing data for other BLUF proteins suggests that the presence of two hydrogen bonds to the C2=O may correlate with radical formation. The N31, R65 hydrogen bonding motif is repeated in TePixD (Tll0078),56 which also forms a radical during the photocycle.57 In contrast, while N31 is replaced by an Arg in BlrB, a second hydrogen bond donor to C2=O is not present,6 and there is no evidence for radical formation during photoactivation of this BLUF protein.58 X-ray structures are not available for BlsA and PapB, however sequence alignment suggests that two hydrogen bonds may be present in PapB, where N31 is conserved and R65 is replaced by a Lys, while only one hydrogen bond is possible in BlsA where the residue homologous to N31 is a Phe. Again, while radical formation is detected during the photocycle of PapB,39 there is no evidence for a radical intermediate in BlsA.34 Secondly, as noted in the introduction, W91 is predominantly in the Trpout conformation in the PixD X-ray structures, in contrast to AppA where both Trpin and Trpout conformations are populated.5, 31 Although the X-ray structures do not rule out the possibility that the Trp side chain can adopt multiple conformations, including Trpin and Trpout, Mehlhorn et al,59 have suggested that light-induced changes in the BLUF β-sheet may be communicated from the flavin via another residue (N32) rather than W91, based on the supposition that W91 remains in the Trpout conformation throughout the PixD photocyle. Thus, if W91 is not hydrogen bonded to Q50 in the dark state, then keto-enol tautomerism and rotation of Q50 may play a less critical role in stabilizing the PixD light state, hence reducing the impact of the Y8 pKa on dark state recovery. Finally, it should be noted that the studies on AppABLUF, PixD, PapB and BlsA have been conducted in the absence of their output partners, PpsR, PixE and PapA,60 respectively (the output partner for BlsA has not yet been identified), and indeed in AppABLUF, without the C-terminal domain of the full length AppA protein. Thus it remains to be seen how protein-protein interactions in physiologically relevant complexes of the BLUF proteins might affect the photocycles that have been so thoroughly characterized in the isolated BLUF domains. Our observation that relatively small changes in structure modify the mechanism of photoactivation suggest that this might be a fruitful line of research.
Conclusion
The role of Y8 in the photocycle of the BLUF protein PixD has been probed via a combination of unnatural amino acid mutagenesis and ultrafast infrared spectroscopy. In the wild-type protein, the TRIR spectra are consistent with a photoactivation mechanism in which electron-proton transfer from Y8 occurs sequentially, with electron transfer (2.5 ps) to the excited FAD* first generating FAD•− followed by proton transfer (20 ps) to generate the neutral semiquinone FADH•. This mechanism is quite different to that operating in AppABLUF, where no radical intermediates are observed and the excited state decay is longer. Replacement of Y8 with F-Tyr analogs that increase the reduction potential of the phenolic group reduce the rate of electron transfer. In addition, for the F-Tyr analogs with pKa values in solution below 8 (2,3-F2Y8, 3,5-F2Y8 and 2,3,5-F3Y8 PixD), no progression of the photocycle is observed after formation of the FAD anionic semiquinone FAD•−. Assuming that the F-Tyr analogs have similar pKa values in the protein, this observation confirms that Y8 is the source of both electron and proton in the PCET mechanism, since at pH 8 the Tyr analogs will be deprotonated. Although no stable light state can be detected in the TRIR spectra of 2,3-F2Y8, 3,5-F2Y8 and 2,3,5-F3Y8 PixD, a photostationary population of light state can be generated in 2,3-F2Y8 and 3,5-F2Y8 PixD indicating that the reaction coordinate leading to the light state is still accessible, but that the light state reversion to the ground state is accelerated. In contrast to the profound impact on the forward photoreaction, F-Tyr substitution has a more modest effect on the rate of dark state recovery. These observations differ fundamentally from the impact of F-Tyr substitution on the photocyle of AppABLUF where the change in pKa and reduction potential have only a minor effect on excited state decay, but increase the rate of dark state recovery 4,000-fold. Thus, despite high levels of structural conservation in the FAD binding pocket, profound differences can exist between the photophysics of BLUF proteins. For AppABLUF and PixD, the identity of the residues that bind to the C2=O carbonyl group and/or the position of the conserved Trp (W91 and W104) in the two proteins may play a fundamental role in determining the function of the conserved Tyr in the photocycle.
Supplementary Material
Acknowledgments
Funded by the EPSRC (EP/G002916 to SRM) and NSF (CHE-1223819 to PJT). We are grateful to STFC for access to the ULTRA laser facility. We are grateful to Professor Ray Owens for assistance in protein preparation and access to the Oxford Protein Production Facility – UK, and to Arthur Makarenko at CSHL for assistance with the mass spectrometry analysis. JI was supported by an NIH Chemistry-Biology Interface training grant (T32GM092714). AL is a Bolyai Janos Research Fellow and was supported by OTKA NN113090.
Footnotes
Supporting Information Available: ESI MS/MS analysis of fluorotyrosine incorporation into PixD. Additional TRMPS and TRIR datasets. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Piano V, Palfey BA, Mattevi A. Trends Biochem Sci. 2017;42:457. doi: 10.1016/j.tibs.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Massey V. FASEB J. 1995;9:473. doi: 10.1096/fasebj.9.7.7737454. [DOI] [PubMed] [Google Scholar]
- 3.Ghisla S, Massey V. Eur J Biochem. 1989;181:1. doi: 10.1111/j.1432-1033.1989.tb14688.x. [DOI] [PubMed] [Google Scholar]
- 4.Heelis PF. Chem Soc Rev. 1982;11:15. [Google Scholar]
- 5.Anderson S, Dragnea V, Masuda S, Ybe J, Moffat K, Bauer C. Biochemistry. 2005;44:7998. doi: 10.1021/bi0502691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung A, Domratcheva T, Tarutina M, Wu Q, Ko WH, Shoeman RL, Gomelsky M, Gardner KH, Schlichting I. Proc Natl Acad Sci U S A. 2005;102:12350. doi: 10.1073/pnas.0500722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan H, Dragnea V, Wu Q, Gardner KH, Bauer CE. Biochemistry. 2011;50:6365. doi: 10.1021/bi200701d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brust R, Lukacs A, Haigney A, Addison K, Gil A, Towrie M, Clark IP, Greetham GM, Tonge PJ, Meech SR. J Am Chem Soc. 2013;135:16168. doi: 10.1021/ja407265p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Udvarhelyi A, Domratcheva T. J Phys Chem B. 2013;117:2888. doi: 10.1021/jp400437x. [DOI] [PubMed] [Google Scholar]
- 10.Domratcheva T, Hartmann E, Schlichting I, Kottke T. Sci Rep. 2016;6:22669. doi: 10.1038/srep22669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goyal P, Hammes-Schiffer S. Proc Natl Acad Sci U S A. 2017;114:1480. doi: 10.1073/pnas.1621393114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hall CR, Heisler IA, Jones GA, Frost JE, Gil AA, Tonge PJ, Meech SR. Chem Phys Lett. 2017;683:365. [Google Scholar]
- 13.Stierl M, Stumpf P, Udwari D, Gueta R, Hagedorn R, Losi A, Gartner W, Petereit L, Efetova M, Schwarzel M, Oertner TG, Nagel G, Hegemann P. J Biol Chem. 2011;286:1181. doi: 10.1074/jbc.M110.185496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomelsky M, Klug G. Trends Biochem Sci. 2002;27:497. doi: 10.1016/s0968-0004(02)02181-3. [DOI] [PubMed] [Google Scholar]
- 15.Masuda S. Plant Cell Physiol. 2013;54:171. doi: 10.1093/pcp/pcs173. [DOI] [PubMed] [Google Scholar]
- 16.Cao Z, Livoti E, Losi A, Gartner W. Photochem Photobiol. 2010;86:606. doi: 10.1111/j.1751-1097.2010.00724.x. [DOI] [PubMed] [Google Scholar]
- 17.Tang Y, Cao Z, Livoti E, Krauss U, Jaeger KE, Gartner W, Losi A. Photochem Photobiol Sci. 2010;9:47. doi: 10.1039/b9pp00075e. [DOI] [PubMed] [Google Scholar]
- 18.Kennis JT, Groot ML. Curr Opin Struct Biol. 2007;17:623. doi: 10.1016/j.sbi.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 19.van der Horst MA, Hellingwerf KJ. Acc Chem Res. 2004;37:13. doi: 10.1021/ar020219d. [DOI] [PubMed] [Google Scholar]
- 20.Okajima K, Yoshihara S, Fukushima Y, Geng X, Katayama M, Higashi S, Watanabe M, Sato S, Tabata S, Shibata Y, Itoh S, Ikeuchi M. J Biochem. 2005;137:741. doi: 10.1093/jb/mvi089. [DOI] [PubMed] [Google Scholar]
- 21.Gil AA, Haigney A, Laptenok SP, Brust R, Lukacs A, Iuliano JN, Jeng J, Melief EH, Zhao RK, Yoon E, Clark IP, Towrie M, Greetham GM, Ng A, Truglio JJ, French JB, Meech SR, Tonge PJ. J Am Chem Soc. 2016;138:926. doi: 10.1021/jacs.5b11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan H, Anderson S, Masuda S, Dragnea V, Moffat K, Bauer C. Biochemistry. 2006;45:12687. doi: 10.1021/bi061435n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stelling AL, Ronayne KL, Nappa J, Tonge PJ, Meech SR. J Am Chem Soc. 2007;129:15556. doi: 10.1021/ja074074n. [DOI] [PubMed] [Google Scholar]
- 24.Domratcheva T, Grigorenko BL, Schlichting I, Nemukhin AV. Biophys J. 2008;94:3872. doi: 10.1529/biophysj.107.124172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lukacs A, Haigney A, Brust R, Zhao RK, Stelling AL, Clark IP, Towrie M, Greetham GM, Meech SR, Tonge PJ. J Am Chem Soc. 2011;133:16893. doi: 10.1021/ja2060098. [DOI] [PubMed] [Google Scholar]
- 26.Masuda S, Bauer CE. Cell. 2002;110:613. doi: 10.1016/s0092-8674(02)00876-0. [DOI] [PubMed] [Google Scholar]
- 27.Kraft BJ, Masuda S, Kikuchi J, Dragnea V, Tollin G, Zaleski JM, Bauer CE. Biochemistry. 2003;42:6726. doi: 10.1021/bi030055o. [DOI] [PubMed] [Google Scholar]
- 28.Masuda S, Hasegawa K, Ishii A, Ono TA. Biochemistry. 2004;43:5304. doi: 10.1021/bi049836v. [DOI] [PubMed] [Google Scholar]
- 29.Hasegawa K, Masuda S, Ono TA. Biochemistry. 2004;43:14979. doi: 10.1021/bi048671n. [DOI] [PubMed] [Google Scholar]
- 30.Unno M, Masuda S, Ono TA, Yamauchi S. J Am Chem Soc. 2006;128:5638. doi: 10.1021/ja060633z. [DOI] [PubMed] [Google Scholar]
- 31.Jung A, Reinstein J, Domratcheva T, Shoeman RL, Schlichting I. J Mol Biol. 2006;362:717. doi: 10.1016/j.jmb.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 32.Grinstead JS, Hsu ST, Laan W, Bonvin AM, Hellingwerf KJ, Boelens R, Kaptein R. Chembiochem. 2006;7:187. doi: 10.1002/cbic.200500270. [DOI] [PubMed] [Google Scholar]
- 33.Masuda S, Hasegawa K, Ono TA. Plant Cell Physiol. 2005;46:1894. doi: 10.1093/pcp/pci208. [DOI] [PubMed] [Google Scholar]
- 34.Lukacs A, Brust R, Haigney A, Laptenok SP, Addison K, Gil A, Towrie M, Greetham GM, Tonge PJ, Meech SR. J Am Chem Soc. 2014;136:4605. doi: 10.1021/ja4121082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mathes T, van Stokkum IH, Stierl M, Kennis JT. J Biol Chem. 2012;287:31725. doi: 10.1074/jbc.M112.391896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laptenok SP, Lukacs A, Brust R, Haigney A, Gil A, Towrie M, Greetham GM, Tonge PJ, Meech SR. Faraday Discuss. 2015;177:293. doi: 10.1039/c4fd00189c. [DOI] [PubMed] [Google Scholar]
- 37.Mathes T, Zhu JY, van Stokkum IHM, Groot ML, Hegemann P, Kennis JTM. J Phys Chem Lett. 2012;3:203. [Google Scholar]
- 38.Gauden M, van Stokkum IHM, Key JM, Luhrs DC, Van Grondelle R, Hegemann P, Kennis JTM. Proc Natl Acad Sci U S A. 2006;103:10895. doi: 10.1073/pnas.0600720103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujisawa T, Takeuchi S, Masuda S, Tahara T. J Phys Chem B. 2014;118:14761. doi: 10.1021/jp5076252. [DOI] [PubMed] [Google Scholar]
- 40.Brust R, Haigney A, Lukacs A, Gil A, Hossain S, Addison K, Lai CT, Towrie M, Greetham GM, Clark IP, Illarionov B, Bacher A, Kim RR, Fischer M, Simmerling C, Meech SR, Tonge PJ. J Phys Chem Lett. 2014;5:220. doi: 10.1021/jz4023738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The PyMOL Molecular Graphics System, Version 1.8. Schrodinger, LLC; [Google Scholar]
- 42.Minnihan EC, Young DD, Schultz PG, Stubbe J. J Am Chem Soc. 2011;133:15942. doi: 10.1021/ja207719f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greetham GM, Burgos P, Cao Q, Clark IP, Codd PS, Farrow RC, George MW, Kogimtzis M, Matousek P, Parker AW, Pollard MR, Robinson DA, Xin ZJ, Towrie M. Appl Spectrosc. 2010;64:1311. doi: 10.1366/000370210793561673. [DOI] [PubMed] [Google Scholar]
- 44.Greetham GM, Sole D, Clark IP, Parker AW, Pollard MR, Towrie M. Rev Sci Instrum. 2012;83:103107. doi: 10.1063/1.4758999. [DOI] [PubMed] [Google Scholar]
- 45.Snellenburg JJ, Laptenok SP, Seger R, Mullen KM, van Stokkum IHM. J Stat Softw. 2012;49:1. [Google Scholar]
- 46.Bonetti C, Mathes T, van Stokkum IHM, Mullen KM, Groot ML, van Grondelle R, Hegemann P, Kennis JTM. Biophys J. 2008;95:4790. doi: 10.1529/biophysj.108.139246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonetti C, Stierl M, Mathes T, van Stokkum IH, Mullen KM, Cohen-Stuart TA, van Grondelle R, Hegemann P, Kennis JT. Biochemistry. 2009;48:11458. doi: 10.1021/bi901196x. [DOI] [PubMed] [Google Scholar]
- 48.Hasegawa K, Masuda S, Ono TA. Plant Cell Physiol. 2005;46:136. doi: 10.1093/pcp/pci003. [DOI] [PubMed] [Google Scholar]
- 49.Haigney A, Lukacs A, Zhao RK, Stelling AL, Brust R, Kim RR, Kondo M, Clark I, Towrie M, Greetham GM, Illarionov B, Bacher A, Romisch-Margl W, Fischer M, Meech SR, Tonge PJ. Biochemistry. 2011;50:1321. doi: 10.1021/bi101589a. [DOI] [PubMed] [Google Scholar]
- 50.Lukacs A, Zhao RK, Haigney A, Brust R, Greetham GM, Towrie M, Tonge PJ, Meech SR. J Phys Chem B. 2012;116:5810. doi: 10.1021/jp2116559. [DOI] [PubMed] [Google Scholar]
- 51.Laan W, van der Horst MA, van Stokkum IH, Hellingwerf KJ. Photochem Photobiol. 2003;78:290. doi: 10.1562/0031-8655(2003)078<0290:icotpp>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 52.Unno M, Sano R, Masuda S, Ono TA, Yamauchi S. J Phys Chem B. 2005;109:12620. doi: 10.1021/jp0522664. [DOI] [PubMed] [Google Scholar]
- 53.Masuda S, Hasegawa K, Ono TA. Biochemistry. 2005;44:1215. doi: 10.1021/bi047876t. [DOI] [PubMed] [Google Scholar]
- 54.Seyedsayamdost MR, Yee CS, Reece SY, Nocera DG, Stubbe J. J Am Chem Soc. 2006;128:1562. doi: 10.1021/ja055927j. [DOI] [PubMed] [Google Scholar]
- 55.Szabo A. Proc Natl Acad Sci U S A. 1978;75:2108. doi: 10.1073/pnas.75.5.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kita A, Okajima K, Morimoto Y, Ikeuchi M, Miki K. J Mol Biol. 2005;349:1. doi: 10.1016/j.jmb.2005.03.067. [DOI] [PubMed] [Google Scholar]
- 57.Nagai H, Fukushima Y, Okajima K, Ikeuchi M, Mino H. Biochemistry. 2008;47:12574. doi: 10.1021/bi8010187. [DOI] [PubMed] [Google Scholar]
- 58.Mathes T, van Stokkum IH, Bonetti C, Hegemann P, Kennis JT. J Phys Chem B. 2011;115:7963. doi: 10.1021/jp201296m. [DOI] [PubMed] [Google Scholar]
- 59.Mehlhorn J, Lindtner T, Richter F, Glass K, Steinocher H, Beck S, Hegemann P, Kennis JT, Mathes T. J Phys Chem Lett. 2015;6:4749. doi: 10.1021/acs.jpclett.5b02245. [DOI] [PubMed] [Google Scholar]
- 60.Kanazawa T, Ren S, Maekawa M, Hasegawa K, Arisaka F, Hyodo M, Hayakawa Y, Ohta H, Masuda S. Biochemistry. 2010;49:10647. doi: 10.1021/bi101448t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.