

Abstract
Mitochondrial dysfunction has been implicated in the pathophysiology of neurodegenerative disorders, including multiple sclerosis (MS). To date, the investigation of mitochondrial dysfunction in MS has focused exclusively on neurons, with no studies exploring whether dysregulation of mitochondrial bioenergetics and/or genetics in oligodendrocytes might be associated with the etiopathogenesis of MS and other demyelinating syndromes. To address this question, we established a mouse model where mitochondrial DNA (mtDNA) double-strand breaks (DSBs) were specifically induced in myelinating oligodendrocytes (PLP:mtPstI mice) by expressing a mitochondrial-targeted endonuclease, mtPstI, starting at 3 weeks of age. In both female and male mice, DSBs of oligodendroglial mtDNA caused impairment of locomotor function, chronic demyelination, glial activation, and axonal degeneration, which became more severe with time of induction. In addition, after short transient induction of mtDNA DSBs, PLP:mtPstI mice showed an exacerbated response to experimental autoimmune encephalomyelitis. Together, our data demonstrate that mtDNA damage can cause primary oligodendropathy, which in turn triggers demyelination, proving PLP:mtPstI mice to be a useful tool to study the pathological consequences of mitochondrial dysfunction in oligodendrocytes. In addition, the demyelination and axonal loss displayed by PLP:mtPstI mice recapitulate some of the key features of chronic demyelinating syndromes, including progressive MS forms, which are not accurately reproduced in the models currently available. For this reason, the PLP:mtPstI mouse represents a unique and much needed platform for testing remyelinating therapies.
SIGNIFICANCE STATEMENT In this study, we show that oligodendrocyte-specific mitochondrial DNA double-strand breaks in PLP:mtPstI mice cause oligodendrocyte death and demyelination associated with axonal damage and glial activation. Hence, PLP:mtPstI mice represent a unique tool to study the pathological consequences of mitochondrial dysfunction in oligodendrocytes, as well as an ideal platform to test remyelinating and neuroprotective agents.
Keywords: animal model, demyelination, mitochondria, multiple sclerosis, oxidative phosphorylation, remyelination
Introduction
Multiple sclerosis (MS) is a chronic demyelinating disorder of the CNS affecting ∼2.5 million individuals worldwide. MS manifests in genetically susceptible individuals with environmental factors influencing penetrance and clinical course.
It is still unclear whether MS is caused by a peripheral or a CNS trigger (Dendrou et al., 2015; Axisa and Hafler, 2016). According to the “CNS extrinsic” hypothesis of MS, autoreactive T cells enter the CNS where they activate the innate immune response and cause demyelination. This is well suited in modeling the relapsing–remitting form of MS (Trapp and Nave, 2008; Dendrou et al., 2015) but alone does not accurately fit the pathophysiology of progressive MS where axonal damage, demyelination, and neurodegeneration are often independent of immune cell presence. As such, progressive MS is best described as a syndrome with some pathological aspects that likely are dependent on autoimmune activation from a peripheral trigger, and with others possibly caused by a CNS-specific trigger. This latter pathological mechanism is defined in the so-called “CNS intrinsic” model, where events in the CNS are believed to initiate the disease (Trapp and Nave, 2008; Dendrou et al., 2015; Mahad et al., 2015). In support of this hypothesis, oligodendrocyte death and demyelination have been shown to occur in the absence of T cells, with demyelinating lesions characterized by axonal transection, and histopathologically similar to those observed in neurological disorders like stroke (Lucchinetti et al., 2000; Peterson et al., 2001; Aboul-Enein et al., 2003). This is true in the rodent brain as well, where primary oligodendrocyte death triggers CNS inflammation, followed by a T-cell response and myelin autoantibody production (Traka et al., 2016). Still, a better understanding of the molecular mechanisms underlying neurodegeneration in progressive MS is needed.
Mitochondrial dysfunction has been implicated in the pathophysiology of neurological disorders, including MS (Witte et al., 2014; Cabezas-Opazo et al., 2015; Keogh and Chinnery, 2015; Ryan et al., 2015; Patergnani et al., 2017). Dysfunctional mitochondria contribute to axonal damage and neuronal loss in MS (Dutta et al., 2006; Mahad et al., 2008; Witte et al., 2014) as well as experimental autoimmune encephalomyelitis (EAE; Nikić et al., 2011; Sadeghian et al., 2016). It has also been shown that mitochondrial DNA (mtDNA) mutations could underlie neuronal mitochondrial dysfunction in MS (Kalman et al., 1995; Ban et al., 2008; Andalib et al., 2013). Further evidence pointing at a connection between mitochondrial dysfunction and MS is that demyelination occurs in various mitochondrial diseases including Leber's hereditary optic neuropathy (LHON; Yu-Wai-Man et al., 2011), Friedreich's ataxia (Koeppen and Mazurkiewicz, 2013), and Charcot-Marie-Tooth disease type 2A (Kwong et al., 2006). In LHON, which is caused by mtDNA mutations, patients have increased risk of developing an MS-like syndrome known as Harding's disease (Palace, 2009) and display white matter lesions indistinguishable from those found in MS patients (Matthews et al., 2015), suggesting that mitochondrial dysfunction could be a common pathophysiological pathway in these disorders.
To date, the investigation of mitochondrial dysfunction in MS has focused only on neurons, partially because oligodendrocytes have been shown to be highly glycolytic and relatively resistant to oxidative phosphorylation (OXPHOS) defects (Fünfschilling et al., 2012). Our hypothesis is that, at least in some MS forms, primary oligodendropathy caused by mitochondrial damage and/or dysfunction may result in myelin degradation causing neuroinflammation and secondary activation of autoreactive T cells. Using the PLP:mtPstI mouse model, here we show that mtDNA double-strand breaks (DSBs) cause primary oligodendropathy, which leads to demyelination, axonal injury, and glial activation. On this basis, we believe an investigation of oligodendrocyte-specific mitochondrial dysfunction in MS patients as a possible contributor to MS pathogenesis is warranted. Finally, we show that PLP:mtPstI mice serve as a customizable model of chronic progressive demyelination, and may be used as a platform for testing agents targeting CNS remyelination and repair.
Materials and Methods
Mice
PLP:mtPstI transgenic mice were generated by breeding hemizygous proteolipid protein (PLP)-tetracycline transactivator (tTA) mice (Inamura et al., 2012) with hemizygous mitochondrial-PstI (mtPstI) mice (Fukui and Moraes, 2009), both backcrossed for a minimum of eight to nine generations to a C57BL/6 background. The genotype was confirmed with the following primers: PLP-tTA, forward 5′-CGGAGTTGATCACCTTGGACTTGT-3′, and reverse 5′-TTTCCCATGGTCTCCCTTGAGCTT-3′; and mtPstI, forward 5′-GGAGGGGAAGCTTATGAAG-3′, and reverse 5′-CCGTTGGGGTAGTTATGCAG-3′. The mito-EYFP [enhanced yellow fluorescent protein; C57BL/6-Tg(tetO-COX8A/EYFP)1Ksn/J] reporter line (Chandrasekaran et al., 2006) was obtained from The Jackson Laboratory (catalog #006618) and genotyped with the following primers: transgenic allele, forward 5′-GCTGACCCTGAAGTTCATCTGC-3′, and reverse 5′-CATGATATAGACGTTGTGGCTGT-3′; and internal positive control, forward 5′-CAAATGTTGCTTGTCTGGTG-3′, and reverse 5′-GTCAGTCGAGTGCACAGTTT-3′. In all experiments (including the EAE experiment), control mice were littermates of the PLP:mtPstI transgenic mice, constituting a mix of WT, PLP-tTA, and mtPstI mice. Transgenic and control mice were kept on the same doxycycline (DOX) regimen, and given DOX-containing food (200 mg/kg; Bio-Serv) for variable lengths of time (until weaning age, throughout life, or temporarily) depending on the experiment. Transgenic and control mice were housed together throughout the experimentation. Colonies were housed at the Animal Facility of The Miami Project to Cure Paralysis in a virus-free/antigen-free vivarium with a 12 h light/dark cycle and controlled temperature and humidity, and were provided with water and food ad libitum. Mice were group caged (maximum five per cage) throughout the duration of the experiments. Experiments were performed according to protocols and guidelines approved by the Institutional Animal Care and Use Committee of the University of Miami, and all efforts were made to minimize pain and distress.
Behavioral assays
Open field test.
The open field test was performed in adult male mice in an odor-free, nontransparent square arena, as previously described (Madsen et al., 2016b). The arena was divided into three zones (wall, intermediate, and center), and mouse behavior was recorded over a 5 min period using a high-resolution video camera. The total number of lines crossed, the time spent in each zone, and stereotypical behavior such as rearing were analyzed and expressed as number of events. Mice that did not enter all three zones or cross a minimum of 50 lines during the 5 min trial were excluded.
Rotarod test.
Motor coordination and balance were tested on the accelerating rotarod (10–60 rpm over 10 min period and constant acceleration; Rotamex 4/8, Columbus Instruments) using adult male mice. The test consisted of a 5 d pretraining period (days 1–5) followed by the actual test (day 7). The total time on the accelerating rod was automatically recorded, and nonwalking behaviors (e.g., passive clinging to the rod) were manually corrected for. Mice that could not maintain their balance on the rod for a minimum of 60 s during pretraining were excluded (Lambertsen et al., 2012).
Induction of EAE with myelin oligodendrocyte glycoprotein 35–55 peptide
Active EAE was induced in adult (2–3 months) female PLP:mtPstI and control mice using the myelin oligodendrocyte glycoprotein 35–55 peptide (MOG35–55; BioSynthesis) as previously described (Madsen et al., 2016a). Mice received an intraperitoneal injection of pertussis toxin dissolved in PBS (400 ng/mouse; day 0), followed by subcutaneous injection of MOG35–55 peptide (300 ng/mouse; day 1) emulsified in complete Freund's adjuvant (200 μl total volume), followed by a second intraperitoneal injection of pertussis toxin (400 ng/mouse; day 2). The disease course was assessed daily using a standardized scale from 0 to 6 as follows: 0, no clinical signs; 1, loss of tail tone; 2, flaccid tail; 3, complete hindlimb paralysis; 4, complete forelimb paralysis; 5, moribund; and 6, dead. Increments of 0.5 points were applied for intermediate behaviors.
Immunohistochemistry
Mice were transcardially perfused with 0.1 m PBS followed by 4% paraformaldehyde (PFA) in 0.1 m PBS. Spinal cord, brain, and optic nerve were dissected out and postfixed in 4% PFA in 0.1 m PBS for 2 h followed by cryoprotection in 0.1 m PBS plus 20% sucrose. Spinal cord tissue was cryostat sectioned into 14- or 30-μm-thick serial sections. Sections were blocked with 5% normal goat serum in 0.1 m PBS plus 0.4% Triton-X for 1 h and incubated overnight at 4°C with primary antibodies against GFP (chicken, 1:1000; Abcam), APC (CC1 clone, mouse, 1:500; Millipore), Olig2 (rabbit, 1:500; Millipore), Iba1 (rabbit, 1:500; Wako), GFAP (rabbit,1:800; Dako), and microtubule associated protein 2 (MAP2; rabbit, 1:500; Millipore). Positive staining was visualized with secondary species-specific fluorescent antibodies (goat anti-mouse Alexa Fluor 488 and 594, goat anti-rabbit Alexa Fluor 488 and 594, goat anti-chicken Alexa Fluor488, all diluted 1:750; Invitrogen). Cytochrome c oxidase I (COX1) was identified with anti-MTCO1-Alexa Fluor 488 antibody (mouse, 1:500; Abcam). Lastly, sections were stained with DAPI (1:1000; stock, 1 mg/ml; Invitrogen) for 5 min, coverslipped, and imaged with an Olympus FluoView 1000 Confocal Microscope or with a Zeiss Axiovert A1 Fluorescence Microscope.
Quantification of glial cells in the spinal cord
Total Iba1+ microglia and Olig2+CC1+ oligodendrocytes were estimated in the thoracic spinal cord by stereology. Five 30-μm-thick serial sections taken 300 μm apart were analyzed. Positive cells were counted at 63× magnification with a Zeiss Axiovert A1 fluorescence microscope by a blinded investigator. The total number of positive cells per cubic millimeter was estimated using Stereo Investigator Software (MBF Bioscience) to ensure unbiased counting and systematic random sampling. GFAP intensity was quantified in the thoracic spinal cord from images acquired at 10× magnification with identical exposure time, analyzed using ImageJ after background subtraction, and expressed as intensity per square micrometer.
Isolation of O4+ oligodendrocytes
Mice were transcardially perfused with PBS, and brains and spinal cords were harvested and dissociated into single-cell suspensions using the Adult Brain Dissociation Kit (Miltenyi) according to manufacturer protocols. After red blood cell lysis, cells were resuspended in PBS plus 0.5% BSA, and oligodendrocytes were isolated by positive selection with O4+ magnetic microbeads (Miltenyi) in combination with LS columns (Miltenyi).
Real time RT-PCR
Total RNA was isolated using the RNeasy Micro Kit (Qiagen) and cleaned of genomic DNA by on-column DNase digestion. For isolated oligodendrocytes, 30 ng of RNA was reversed transcribed with Sensiscript Reverse Transcriptase Kit (Qiagen), whereas for sciatic nerves 200 ng of RNA was reversed transcribed with Omniscript Reverse Transcriptase Kit (Qiagen). In parallel, reactions without reverse transcriptase (no-RT) were set up to estimate possible genomic DNA contamination. Real-time PCR amplification was performed with PowerUp SYBR Green PCR Master Mix (Invitrogen) in an ABI 7300 Real-Time PCR System (Thermo Fisher Scientific). COX1 was amplified with 5′-AGGCTTCACCCTAGATGACACA-3′ (forward) and 5′-GTAGCGTCGTGGTATTCCTGAA-3′ (reverse) primers; PstI with 5′-GATTACAAGCCCAATTCCC-3′ (forward) and 5′-ATTGTCCTTGGAGTTGGTG-3′ (reverse) primers; and β-actin with 5′-TAGACTTCGAGCAGGAGATGG-3′ (forward) and 5′-CAAGAAGGAAGGCTGGAAAAGAG-3′ (reverse) primers. Relative gene expression was calculated with the ΔΔCt method after normalization to β-actin and subtraction of no-RT expression.
Assessment of mtDNA content and mtDNA deletions
Mice were perfused with cold PBS, and total DNA was isolated from whole spinal cord homogenates by phenol/chloroform extraction. Genes were amplified using Maxima SYBR Green/ROX qPCR Master Mix (Fermentas) in a CFX96 Realtime PCR system (BIO-RAD). The following primers were used for amplification: COX1, 5′-AGGCTTCACCCTAGATGACACA-3′ (forward) and 5′-GTAGCGTCGTGGTATTCCTGAA-3′ (reverse); ND1, 5′-CAGCCTGACCCATAGCCATA-3′ (forward) and 5′-ATTCTCCTTCTGTCAGGTCGAA-3 (reverse); and genomic β-actin, 5′-GCGCAAGTACTCTGTGTGGA-3′ (forward) and 5′-CATCGTACTCCTGCTTGCTG-3′ (reverse). COX1 mtDNA content was calculated with the ΔΔCt method after normalization to genomic DNA content (β-actin). Data were corrected for relative PCR amplification efficiency using BIO-RAD CFX Manager Software (version 3.0). To detect mtDNA deletions, which we measured by the occurrence of mtDNA recombination, primers flanking the following PstI restriction sites were designed: PstI-8300, 5′-TTGCCCACTTCCTTCCACAAG-3′; and PstI-12812, 5′-TCGGATGTCTTGTTCGTCTG-3′.
Estimation of demyelinated white matter volume
Thoracic spinal cord sections were permeabilized with 0.1 m PBS plus 0.4% Triton-X and stained with FluoroMyelin Red Fluorescent Myelin Stain (1:400; Invitrogen) for 20 min at room temperature. Sections were blocked with 5% normal goat serum in 0.1 m PBS plus 0.4% Triton-X for 1 h and colabeled with anti-GFAP as described above. Five 30-μm-thick serial sections taken 300 μm apart were used for the assessment. The entire white matter and the demyelinated areas were traced at 5× magnification with a Zeiss Axiovert A1 Fluorescence Microscope. Total and demyelinated white matter volume were estimated using Neurolucida Software (MBF Bioscience).
Immunohistochemistry in flat-mounted retinas and quantification of retinal ganglion cell numbers
Tissues were processed as previously described (Brambilla et al., 2012). Briefly, eyes were enucleated and immersion fixed in 4% PFA, pH 7.4, for 1 h. Retinas were then removed and cryoprotected overnight in 30% sucrose. Before staining, retinas were subjected to three freeze–thaw cycles, rinsed in PBS, and blocked for 1 h in 0.1 m Tris buffer containing 5% donkey serum and 0.1% Triton X-100. After overnight incubation with Cy3-conjugated anti-NeuN antibody (1:500; Millipore), retinas were flat mounted, coverslipped, and imaged with a Leica TSL AOBS SP5 Confocal Microscope equipped with Leica LAS AF software. Quantification of NeuN+ RGCs was performed with ImageJ by a blinded investigator on 20× images randomly collected from the four retinal quadrants at the same eccentricity (1–1.5 mm from the optic disk). Retinal GFAP intensity was analyzed using ImageJ after background subtraction and was expressed as GFAP intensity per square micrometer.
Western blotting
Mice were transcardially perfused with chilled 0.1 m PBS, and the brain and spinal cord were immediately dissected out and snap frozen in liquid nitrogen. The cord was split vertically into the two columns. One sample was homogenized in 300 μl of RIPA buffer (with phosphatase inhibitor cocktail 1, Sigma-Aldrich; and Complete Protease Inhibitor Cocktail, Roche Diagnostics), and 20 μg of proteins were loaded per sample and run on an 11% SDS-PAGE gel. Proteins were transferred to a 0.2 μm nitrocellulose membrane, and unspecific binding was blocked with 5% nonfat milk or 5% BSA in TBS-T for 1 h at room temperature. The membrane was then incubated with primary antibodies against PstI (rabbit, 1:1000; Moraes laboratory), myelin basic protein (MBP; rat, 1:5000; Millipore), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; rabbit, 1:2500; Abcam) at 4°C overnight. Proteins were stained with HRP-conjugated species-specific secondary antibodies (all 1:2000; GE Healthcare) and visualized with Super Signal West Pico Chemiluminescent substrate (Thermo Fisher Scientific). Bands were quantified with Quantity One software (BIO-RAD). Data were normalized to GAPDH and expressed as relative protein expression (in arbitrary units) or as a percentage of 2-month-old control mice.
Toluidine blue analysis and electron microscopy
PFA-fixed 1 mm segments of the thoracic spinal cord were postfixed overnight in 2% glutaraldehyde plus 100 mm sucrose in 0.15 m phosphate buffer before incubation with 2% OsO4 for 1 h. Following dehydration in graded ethanol solutions, tissues were embedded in epoxy resin (Embed, Electron Microscopy Sciences). Semithin (1-μm-thick) sections were cut using a Leica Ultracut E Microtome and were stained with 1% toluidine blue solution. Samples were then examined by light microscopy. The number of toluidine blue-stained myelinated or degenerated axons as well as infiltrating cells were estimated using the Stereo Investigator Software (MBF Bioscience). From the same specimens, ultrathin sections (60–90 nm thick) were cut for electron microscopy (EM) and imaged with a Philips CM10 Transmission Electron Microscope.
Quantification of axon diameter/fiber diameter ratios and assessment of remyelinating axons
For evaluating the g-ratio (axon diameter/fiber diameter ratio) of axons in the thoracic spinal cord, EM micrographs were acquired at 5.2K magnification using the Philips CM10 Transmission Electron Microscope (Brambilla et al., 2011). A grid was placed over the entire spinal cord section, and one picture was acquired randomly from each quadrant of both white and gray matter. For each animal, a minimum of 25 images was analyzed. On each micrograph, the fiber diameter and axon diameter of each single axon were measured using ImageJ software. Due to the irregular shapes of the elements, the approximate diameter was calculated as the average of two diameters measured for each element.
Cell isolation for flow cytometry
Following transcardial perfusion with PBS, spinal cords and spleens were harvested and placed in cold HBSS (with Mg2+ and Ca2+). Samples were mechanically dissociated into single-cell suspensions through a 70 μm cell strainer and washed in HBSS. The spleen samples were spun at 400 × g for 5 min, supernatant was removed, and red blood cells were lysed in 2 ml of lysis buffer (eBioscience) according to manufacturer instructions. Cells were then resuspended in flow cytometry buffer (FCB, eBioscience) and stained as described below. Leukocytes infiltrating into the spinal cord were isolated by negative selection of single-cell spinal cord suspensions with Myelin Removal Beads II using the LS magnetic columns as described in the manufacturer protocol (Miltenyi). Similar to the splenocytes, spinal cord cells were resuspended in FCB and stained as described below. The number of viable cells was determined by Trypan Blue exclusion assay using the TC20 Automated Cell Counter (BIO-RAD).
Immunolabeling and flow cytometric analysis
Cells were resuspended in 100 μl of FCB, blocked with 2 μl of TruStainFcX (anti-mouse CD16/32 FcR block, BioLegend) for 10 min at 4°C, and stained for 30 min at 4°C with the following antibodies: APC-Cy7-anti-CD45 (1:200; BioLegend), FITC-anti-CD4 (1:200; eBioscience), PerCP-Cy5.5-anti-CD8a (1:200; eBioscience), PE-Cy7-anti-CD25 (1:200; eBioscience), PE-anti-CD45R/B220 (1:200; BioLegend), and eFluor450-anti-CD11b (1:200; eBioscience). Cell suspensions were then fixed in 1% PFA for 1 h and, finally, resuspended in 500 μl of FCB. Samples were analyzed with an LSRII Flow Cytometer (BD Biosciences) equipped with FACS-Diva 6.0 Software (BD Biosciences).
Anti-MOG and anti-PLP antibody detection
Blood samples were collected directly from the heart before transcardial perfusion with PBS or PFA. The blood was allowed to coagulate for 30 min at room temperature, and serum was isolated by centrifugation for 10 min at 10,000 rpm. Serum samples were diluted 1:2 and analyzed for the presence of antibodies (IgGs) against MOG and PLP. Preblocked ELISA plates coated with recombinant mouse MOG1–125 (AnaSpec) or PLP178–191 (AnaSpec) peptides were used for detection. The assay was performed in duplicate and according to the manufacturer protocol. Briefly, 100 μl of serum were loaded into each well and incubated for 30 min at room temperature. Wells were washed and incubated with peroxidase-conjugated antibodies against mouse IgG for 1 h at room temperature. The reaction was developed by adding 50 μl of tetramethylbenzidine to each well, and the optical density was measured at 450 λ using a spectrophotometer. The relative anti-MOG and anti-PLP IgG concentrations were estimated based on the manufacturer-provided standard and expressed as nanograms per milliliter.
Experimental design and statistical analysis
All statistical analyses were conducted with GraphPad Prism 5. Statistical tests and specific parameters are detailed in each individual figure legend. Female mice were used for all experiments unless otherwise specified, and no sex differences were observed in the PLP:mtPstI mouse with respect to PstI expression, behavior, or histopathological outcome.
In Figure 1, the data are reported as follows: Figure 1E, n = 3–7/group, Student's t test between 6-month-old control and PLP:mtPstI mice; Figure 1F, n = 3–5/group, Student's t test between 2- and 4-month-old PLP:mtPstI mice; Figure 1G, n = 3–4/group, Student's t test between control and PLP:mtPstI mice; and Figure 1K, the weight was recorded in male mice, n = 3–7/group, Student's t test between genotypes at individual time points.
Figure 1.
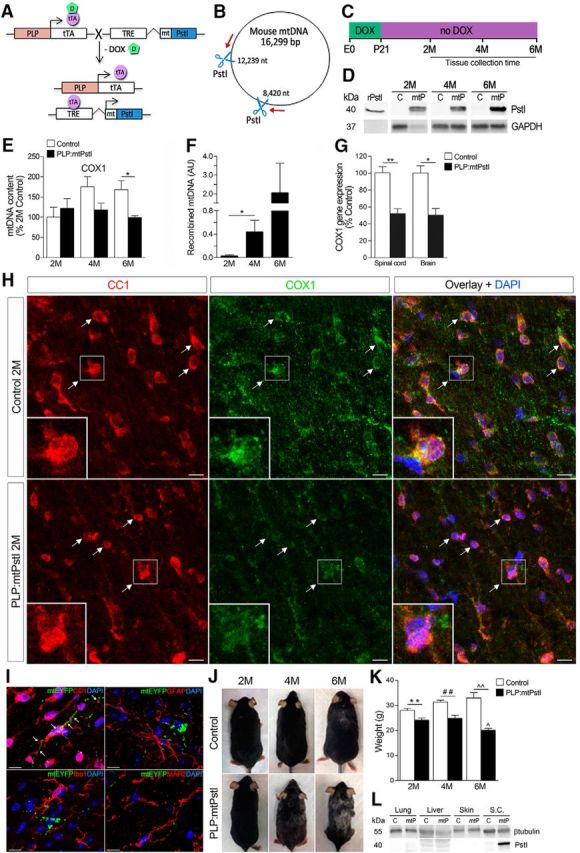
Generation and characterization of PLP:mtPstI mice. A, Schematic of the generation of PLP:mtPstI mice. PLP-tTA mice expressing the tTA under the control of the PLP promoter were crossed with mice expressing a mtPstI downstream of a TRE. In the absence of DOX (−DOX) tTA binds to TRE, initiating PstI transcription (tet-off system). B, Diagram of mouse circular mtDNA with PstI restriction sites (8420 and 12,239 nt, blue scissors) and primers (red arrows) for the amplification of recombined mtDNA fragments generated following PstI cleavage. C, Experimental timeline: mice received DOX until P21 and were sacrificed at 2, 4, or 6 months of age. D, PstI protein expression in the spinal cords of 2-, 4-, and 6-month-old PLP:mtPstI (mtP) and control (c) mice; recombinant PstI was included as positive control. E, Real-time PCR quantification of COX1 in the spinal cord of 2-, 4-, and 6-month-old mice as a measure of mtDNA depletion. Results are normalized to genomic β-actin and are expressed as percentage of 2-month-old controls; n = 3–7/group; *t(4) = 3.031, p = 0.0387, t test. F, Real-time PCR quantification of recombined mtDNA in PLP:mtPstI mice as a measure of mtDNA deletions; n = 4–5/group; *t(7) = 2.407, p = 0.047, t test. AU, Arbitrary units. G, Real-time PCR quantification of COX1 gene expression in oligodendrocytes from spinal cords and brains of 4-month-old control and PLP:mtPstI mice; n = 3–4/group; *t(4) = 4.259, p = 0.0131, t test; **t(6) = 5.131, p = 0.0022, t test. H, Confocal images of the spinal cord white matter of 2M control and PLP:mtPstI mice colabeled with CC1 and mitochondrial COX1; white arrows show localization of COX1 in CC1+ mature oligodendrocytes. Scale bars, 10 μm. I, Representative confocal images from the thoracic spinal cords of 2-month-old PLP:mitoEYFP mice after 5 weeks of reporter induction (EYFP). Sections were colabeled for CC1, GFAP, Iba1 and MAP2. Scale bar, 10 μm. J, Progressive phenotypic changes in PLP:mtPstI mice induced at P21 compared with control mice. K, Assessment of body weight in PLP:mtPstI mice induced at P21 and in control mice; **t(10) = 3.319, n = 5–7, p = 0.0078, t test; ##t(5) = 4.145, n = 3–4, p = 0.009, t test; ∧∧t(10) = 4.134, n = 4–8, p = 0.002, t test; ∧F(2,10) = 7.586, p = 0.0099, ANOVA, Bonferroni's multiple-comparison test: 2- vs 6-month-old PLP:mtPstI mice, t = 3.188, p < 0.05; and 4- vs 6-month-old PLP:mtPstI mice, t = 3.582, p < 0.05. L, Representative Western blot of PstI expression in non-neural tissues. M, Month.
In Figure 2A–F, all behavioral assessments were conducted using male mice. The data for Figure 2 are reported as follows: Figure 2A, n = 4–9/group, statistical comparison between groups or genotypic differences over time were assessed with a one-way ANOVA followed by Bonferroni's multiple-comparison test; Figure 2B,C, n = 4–12/group, one-way ANOVA followed by Bonferroni's multiple-comparison test to compare over time, and Student's t test to compare matching time points; Figure 2G, n = 3–4/group, Student's t test between control and PLP:mtPstI mice at each time point; Figure 2H, n = 3–6/group, Student's t test between control and PLP:mtPstI mice at each time point; Figure 2K,M, data were fitted to a linear regression, and slopes and intersections were compared; Figure 2L,N, Student's t test between control and PLP:mtPstI mice; and Figure 2P, Student's t test between control and PLP:mtPstI mice at each time point.
Figure 2.
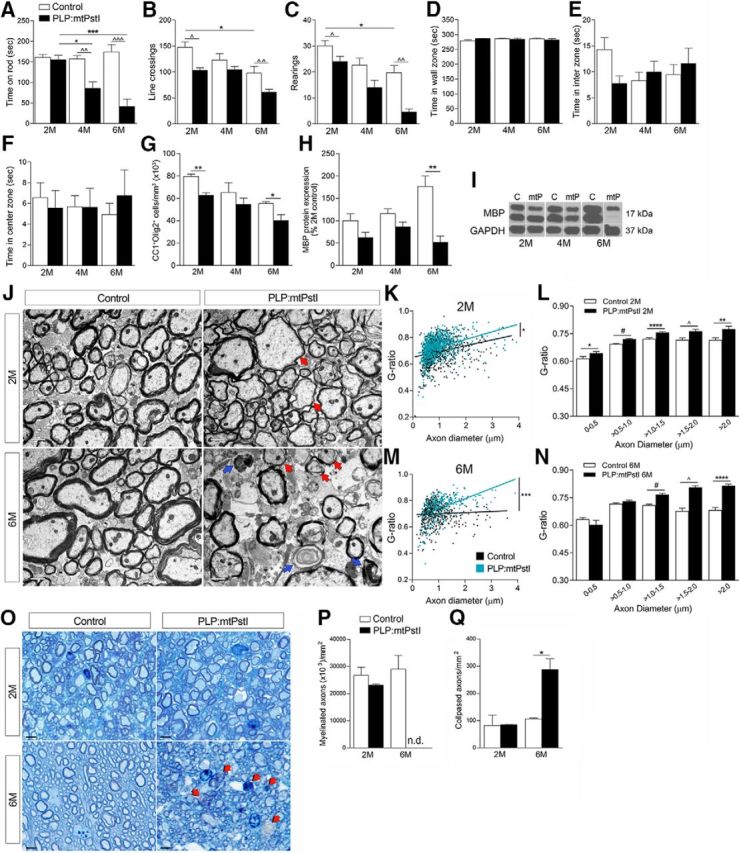
Sustained mtDNA DBSs in oligodendrocytes correlates with worsening of the locomotor phenotype, oligodendrocyte loss, demyelination, and axonal damage in the spinal cord. A, Balance and motor coordination evaluated with the rotarod test and expressed as time spent on the rod; n = 4–9/group; F(5,36) = 13.52, p < 0.0001, ANOVA, Bonferroni's multiple-comparison test: *t = 3.610, p < 0.05; ***t = 5.874, p < 0.001; ∧∧t = 3.625, p < 0.01; ∧∧∧t = 6.681, p < 0.001. B–F, Spontaneous activity and anxiety-like behaviors assessed with the open field test by measuring the number of lines crossed (B; *F(2,20) = 4.692, p = 0.0213, ANOVA, Bonferroni's multiple-comparison test: *t = 3.063, p < 0.05; ∧t(10) = 2.358, p = 0.0401, t test; ∧∧t(6) = 5.131, p = 0.0022, t test); rearings (C; F(3,20) = 16.47, p < 0.0001, ANOVA, Bonferroni's multiple-comparison test: *t = 3.470, p < 0.05; ∧t = 2.492, p < 0.05; ∧∧t = 4.052, p < 0.01); time spent in wall (D); interzone (E); and center zone (F); n = 4–12/group. G, Stereological quantification of CC1+Olig2+ oligodendrocytes in the thoracic spinal cord of control and PLP:mtPstI mice; n = 3–4/group, *t(5) = 3.120, n = 3–4, p = 0.0262, t test; **t(4) = 5.284, n = 3, p = 0.0062, t test. H, Quantification of MBP protein expression in the spinal cord; results are normalized to GAPDH and expressed as the percentage of 2-month-old controls; n = 3–10/group, **t(14) = 3.334, n = 6–10, p = 0.0049, t test. I, Representative MPB Western blots. C, Control; mtP, PLP:mtPstI. J, Representative electron micrographs of the thoracic spinal cord lateral columns of control and PLP:mtPstI mice. Red arrows, Thinly myelinated or demyelinated axons; blue arrows, degenerated axons. Scale bars, 1 μm. K, M, Assessment of the g-ratio at 2 (K) and 6 (M) months; values were plotted versus the corresponding axon diameter. Data were fitted to a linear regression, and the slopes were significantly different between genotypes. Slopes: 2-month-old control (0.0408 ± 0.0053, r2 = 0.060) vs 2-month-old PLP:mtPstI mice (0.0567 ± 0.0047, r2 = 0.1417). *p = 0.027; 6-month-old control (0.0086 ± 0.0071, r2 = 0.0029) vs 6-month-old PLP:mtPstI mice (0.0077 ± 0.0081, r2 = 0.2474). L, N, Quantification of the g-ratio at 2 (L) and 6 (N) months grouped by increasing axon diameter; L: *t(218) = 2.175, p = 0.031; #t(974) = 6.011, p = 0.0004, t test; ****t(388) = 4.814, p < 0.0001; ∧t(118) = 3.122, p = 0.002; **t(218) = 2.175, p = 0.031, t test; N: #t(183) = 6.347, p < 0.0001; ∧t(61) = 7.486, p < 0.0001; ****t(41) = 7.159, p < 0.0001, t test. O, Toluidine blue staining of semithin thoracic spinal cord sections of control and PLP:mtPstI mice. Red arrows, Degenerated collapsed axons. Scale bars, 5 μm. P, Q, Stereological quantification of myelinated (P) and collapsed (Q) axons in the spinal cord white matter of control and PLP:mtPstI mice; n = 3/group, *t(2) = 4.540, n = 3, p = 0.0452, t test. M, Month, n.d., not determined.
In Figure 3, the data are reported as follows: Figure 3B, n = 3/group, Student's t test was applied between genotypes, and one-way ANOVA followed by Bonferroni's multiple-comparison test was used for comparisons over time; Figure 3C, n = 3–4/group, Student's t test between genotypes at each time point; Figure 3E, n = 5/group, Student's t test; and Figure 3G, n = 3/group, Student's t test between genotypes at each time point.
Figure 3.
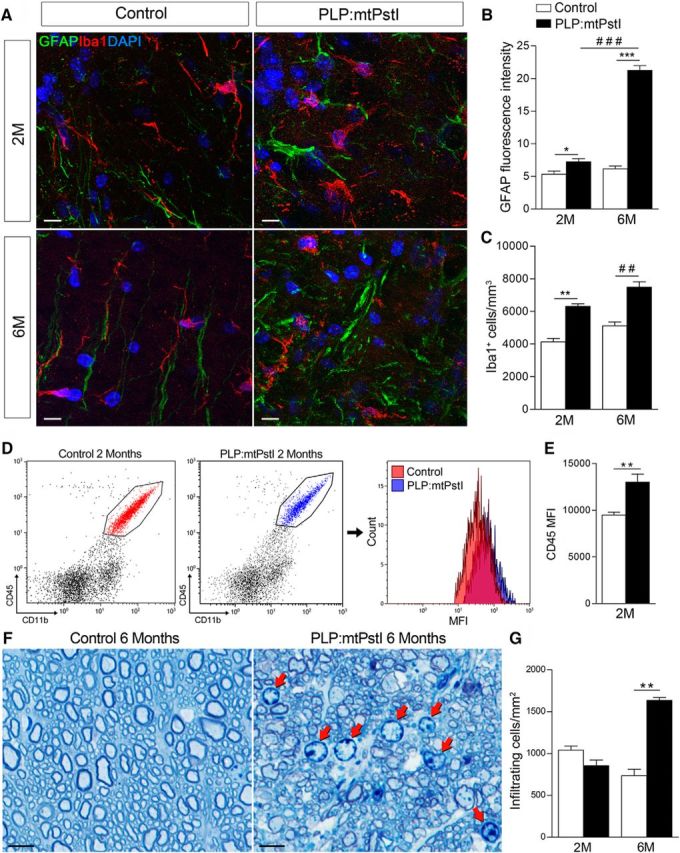
Sustained mtDNA DBSs in oligodendrocytes lead to astrocyte and microglia activation. A, Representative confocal images of GFAP+ astrocytes and Iba1+ microglia in the thoracic spinal cord white matter of control and PLP:mtPstI mice. Scale bar, 10 μm. B, Quantification of GFAP fluorescence intensity in the spinal cord thoracic segment as a measure of astrogliosis; n = 3/group. *t(4) = 2.893, n = 3, p = 0.0444, t test; ***t(4) = 17.62, n = 3, p < 0.0001, t test; ###F(3,8) = 195.5, p < 0.0001, ANOVA, Bonferroni's multiple-comparison test: t = 18.37, p < 0.001. C, Stereological quantification of Iba1+ cells in the spinal cord; n = 3–4/group. **t(3) = 7.207, n = 3, p = 0.0055, t test; ##t(5) = 5.897, n = 3–4, p = 0.0020, t test. D, Representative flow cytometry plots of CD45+CD11b+ microglia in the spinal cord of control and PLP:mtPstI mice at 2 months showing population frequency and CD45 mean fluorescence intensity (MFI). E, Quantification of the CD45 MFI; n = 5/group. **t(8) = 3.805, n = 5, p = 0.0052, t test. F, Toluidine blue staining of semithin thoracic spinal cord sections from control and PLP:mtPstI mice at 6 months. Red arrows show infiltrating immune cells. Scale bars, 5 μm. G, Quantification of infiltrating immune cells in the spinal cord white matter of control and PLP:mtPstI mice; n = 3/group. **t(2) = 10.68, p = 0.0086, t test. M, Month.
The data in Figure 4 are reported as follows: Figure 4B, n = 4–5/group, Student's t test between genotypes at each time point; Figure 4C, n = 7/group, Student's t test between genotypes at each time point; and Figure 4E, n = 3–5, Student's t test between genotypes at each time point.
Figure 4.
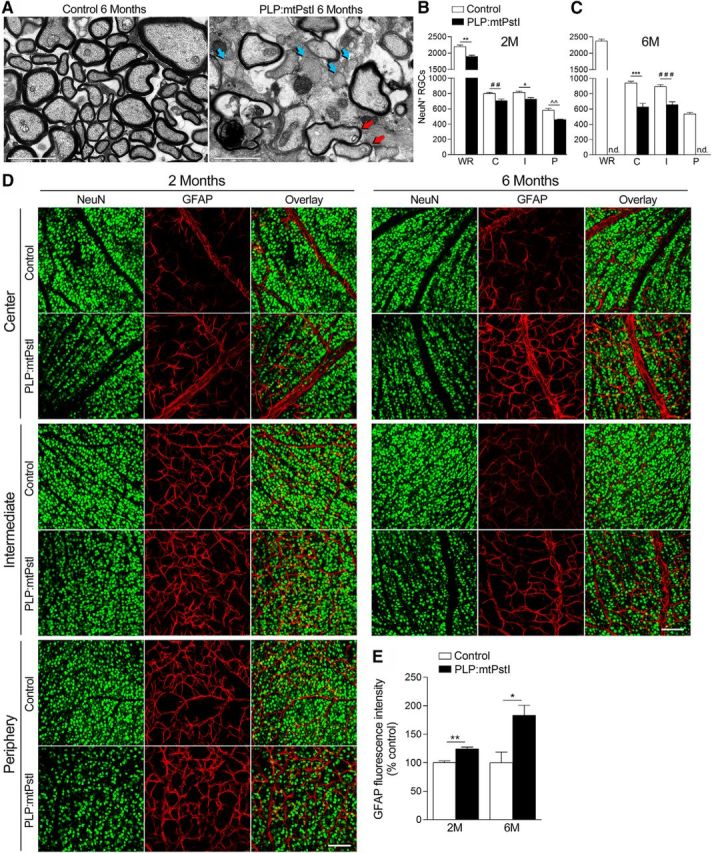
Sustained mtDNA DBSs in oligodendrocytes induce tissue damage in optic nerve and retina. A, Representative electron micrographs of the optic nerve of 6-month-old control and PLP:mtPstI mice. Red arrows indicate remyelinated axons and blue arrows bare demyelinated axons. Scale bars, 2 μm. B, C, Quantification of NeuN+ RGCs in the retina of 2-month-old control and PLP:mtPstI mice (B; WR, whole retina; C, center retina; I, intermediate retina; P, peripheral retina. *t(7) = 3.484, n = 4–5, p = 0.0102, t test; **t(7) = 4.794, n = 4–5, p = 0.0020, t test; ##t(7) = 3.544, n = 4–5, p = 0.0094, t test; ∧∧t(7) = 4.295, n = 4–5, p = 0.0036, t test) and of 6-month-old control and PLP:mtPstI mice (C; ***t(12) = 6.063, n = 7, p < 0.0001, t test; ###t(12) = 5.087, n = 7, p = 0.0003, t test). D, Representative confocal images of flat mounted retinas of control and PLP:mtPstI mice showing NeuN+ and GFAP+ cells in the center, middle, and peripheral zones of the retina. Scale bars, 100 μm. E, Quantification of GFAP fluorescence intensity in the retina expressed as a percentage of the corresponding control (*t(5) = 3.176, n = 3–4, p = 0.0247, t test; **t(7) = 5.056, n = 4–5, p = 0.0015, t test). M, Month.
In Figure 5, data are reported as follows: Figure 5B,D, data were fitted to a linear regression, and slopes and intersection were compared; Figure 5C,E, Student's t test between control and PLP:mtPstI mice; and Figure 5F–I, n = 3/group, Student's t test between control and PLP:mtPstI mice at each time point.
Figure 5.

mtPstI expression in the peripheral nervous system does not affect peripheral myelination. A, Representative electron micrographs of the sciatic nerve of 2- and 6-month-old control and PLP:mtPstI mice. Scale bars, 2 μm. B, D, Assessment of the g-ratio at 2 (B) and 6 (D) months; values were plotted vs the corresponding axon diameter. Data were fitted to a linear regression, and the slopes were not significantly different between genotypes. Slopes: 2-month-old controls (0.0070 ± 0.0029, r2 = 0.026) vs 2-month-old PLP:mtPstI mice (0.0055 ± 0.0029, r2 = 0.0160); 6-month-old controls (0.0087 ± 0.0021, r2 = 0.074) vs 6-month-old PLP:mtPstI mice (0.0070 ± 0.0018, r2 = 0.0684). C, E, G-ratio of myelinated fibers at 2 (C) and 6 (E) months grouped by increasing axon diameter. *t(90) = 2.317, p = 0.023, t test. F, Real-time PCR quantification of ND1 in the sciatic nerve of 2- and 6-month-old mice as a measure of mtDNA depletion. Results are normalized to genomic β-actin and expressed as the percentage of 2-month-old controls. G, Real-time PCR quantification of recombined mtDNA in the sciatic nerve as measure of mtDNA deletions. H, Real-time RT-PCR quantification of mtPstI gene expression in the sciatic nerve. I, Real time RT-PCR quantification of COX1 gene expression in the sciatic nerve. AU, Arbitrary units; M, month; n.d., nondetectable.
In Figure 6, the data are reported as follows: Figure 6E, n = 3/group, Student's t test; and Figure 6I,K,L, n = 4–5/group, Student's t test.
Figure 6.

Brief transient induction of mtDNA DSBs in oligodendrocytes leads to the production of myelin autoantibodies and a biphasic EAE response. A, Experimental setup: DOX was withdrawn for 3 weeks, from week 5 to week 8, and resumed 2 d before EAE induction. All mice, both transgenics and controls, were subjected to the same regimen. Controls were littermates of PLP:mtPstI mice, constituting a mix of WT, PLP-tTA, and mtPstI mice. B, Western blot assessment of PstI protein expression in the spinal cord at 8 weeks, immediately after 3 weeks of DOX withdrawal. C, Control mice; mtP, PLP:mtPstI mice. Recombinant PstI was included as a positive control. C, Real-time PCR quantification of recombined mtDNA in PLP:mtPstI mice as a measure of mtDNA deletion. n = 3/group. D, Real-time PCR quantification of the mitochondrial-specific genes COX1 and ND1 in the spinal cords of PLP:mtPstI and control mice at 8 weeks as a measure of mtDNA depletion. Results are normalized to genomic β-actin and expressed as a percentage of the corresponding control; n = 3/group. E, F, ELISA detection of anti-MOG (E; *t(4) = 4.183, n = 3, p = 0.0139, t test) and anti-PLP (F) antibodies in the serum of PLP:mtPstI and control mice at 8 weeks. G, Clinical course of MOG35–55-induced EAE. Results are expressed as daily mean scores ± SEM of 8–12 mice from 3 independent experiments. The subacute to chronic phase of the disease (20–40 dpi, starting from peak disease) in PLP:mtPstI mice is significantly different from that in controls; ***p < 0.001, Mann–Whitney test. H, Representative confocal images of the thoracic spinal cord labeled with anti-GFAP (green) and FluroMyelin (red). Scale bars, 400 μm. I, Stereological estimation of the demyelinated white matter volume based on FluroMyelin staining. Results are expressed as a percentage of total white matter; *t(7) = 3.168, n = 4–5, p = 0.0157, t test. J, Toluidine blue staining of semithin thoracic spinal cord sections. Green arrows, collapsed axons; red arrows, infiltrating immune cells. Scale bars, 5 μm. K, L, Stereological estimation of collapsed axons (K; *t(7) = 2.362, n = 4–5, p = 0.0502, t test) and infiltrating cells (L; *t(7) = 3.105, n = 4–5, p = 0.0172, t test) in the spinal cord white matter. AU, Arbitrary units; n.d., nondetectable; W, week; dpi, days post induction; WM, white matter.
Results
Generation and characterization of PLP:mtPstI mice
To investigate the effect of mtDNA damage on oligodendrocyte function we bred PLP-tTA mice expressing the tTA under the control of the oligodendrocyte-specific PLP promoter (Inamura et al., 2012) to tetracycline responsive element (TRE)-mtPstI mice expressing the endonuclease PstI in mitochondria (mtPstI; Fukui and Moraes, 2009). In the resulting PLP:mtPstI mice, the PstI gene, cloned downstream of a mitochondrial targeting sequence, is inducibly transcribed upon DOX removal and the protein product is localized to mitochondria (Fig. 1A). Since mouse mtDNA contains two PstI restriction sites (Fig. 1B), PstI expression results in mtDNA DSBs. Although some of the cleaved mtDNA is recombined into shorter mtDNAs, deleting the region between the PstI sites, the majority of the cleaved molecules is degraded, leading to mtDNA depletion (Fukui and Moraes, 2009). We assessed mtDNA damage in mice deprived of DOX at postnatal day 21 (P21) and sacrificed at 2, 4, and 6 months of age (Fig. 1C). Western blot analysis of the spinal cord showed a time-dependent increase of mtPstI in PLP:mtPstI mice and no expression in control littermates (Fig. 1D). This resulted in reduced mtDNA levels at 6 months (Fig. 1E) and in the presence of mtDNA deletions (Fig. 1F). PstI-dependent mtDNA DSBs caused reduced expression of mtDNA-coded COX1 in oligodendrocytes isolated from spinal cord and brain (Fig. 1G). This resulted in a reduced COX1 protein level in oligodendrocytes, with diminished COX1 immunoreactivity in PLP:mtPstI mice already evident at 2 months (Fig. 1H, white arrows). COX1 is an essential component of complex IV of the mitochondrial electron transport chain, and low steady-state levels have been associated with OXPHOS dysfunction (Diaz et al., 2005).
To confirm the specificity of mtPstI expression in oligodendrocytes, we crossed our PLP-tTA mice with TRE-mtEYFP reporter mice (Chandrasekaran et al., 2006). Five weeks after induction (age 2 months), EYFP was localized only to CC1+ oligodendrocytes, not Iba1+ microglia, GFAP+ astrocytes, or MAP2+ neurons (Fig. 1I), confirming previous observations (Inamura et al., 2012) that this PLP-tTA mouse line drives gene expression specifically to mature oligodendrocytes. mtPstI expression led to a reduction in body size and mass (Fig. 1J,K), accompanied by hair graying. As this could indicate mtPstI expression outside the nervous system, we measured mtPstI by Western blot in various non-neural tissues but found no signal in any of the organs tested (Fig. 1L).
Sustained mtDNA DSBs in oligodendrocytes correlate with worsening of the locomotor phenotype
Assessment of motor coordination with the rotarod test in mice induced at P21 showed that PLP:mtPstI mice performed worse than controls at 4 months and deteriorated further at 6 months (Fig. 2A). PLP:mtPstI mice also displayed progressive reduction of spontaneous activity measured with the open field test (Fig. 2B, Movie 1, mouse with red tail mark, 4 months of age). This was accompanied by gait alterations, trunk instability, and loss of tail tone, leading to a stiff and wobbly walking behavior (Movie 1). Stereotypical behavior was altered as well, with a reduction in the number of rearings (Fig. 2C), likely due to a lack of weight support by the hindlimbs (Movie 1). Anxiety-related behaviors, measured as time spent in each region of the open field, were not altered (Fig. 2D–F).
Video showing the abnormal motor phenotype of PLP:mtPstI mice (red tag) at 4 months (DSBs induced at 3 weeks), compared to aged-matched controls.
Sustained mtDNA DSBs in oligodendrocytes correlate with oligodendrocyte loss, demyelination, and axonal damage in the spinal cord
Since mtDNA DSBs can lead to cell death (Pickrell et al., 2011), we investigated whether mtPstI-dependent DSBs of mtDNA resulted in oligodendrocyte loss. In the spinal cord, the number of CC1+Olig2+ oligodendrocytes was reduced in PLP:mtPstI mice beginning at 2 months (Fig. 2G). This was paralleled by a reduction in MBP content (Fig. 2H,I). EM analysis showed myelin abnormalities, with thinly myelinated or fully demyelinated axons (Fig. 2J, red arrows). Myelin thickness, quantified by measuring the g-ratio, was mildly but significantly reduced already at 2 months, and most severely at 6 months (Fig. 2K,M), as shown by the increase in g-ratio. This was also evident as g-ratios were plotted versus axons grouped by diameter size (Fig. 2L,N). Such thinning could be due to oligodendrocyte death, and, in some cases, it might be the sign of axons undergoing spontaneous remyelination. At 6 months, delayed axonal degeneration was prominent, likely caused by the loss of myelin support (Fig. 2J, blue arrows). Indeed, intact and degenerated axons were not different from control mice at 2 months, but were higher at 6 months (Fig. 2M–P). At this time, axonal damage was so severe that a reliable identification and quantification of axons with intact myelin was not possible (Fig. 2N). It should be noted that at 2 months we did not find axonal damage or observe any shift in the distribution of axonal diameters. At 6 months, however, we observed a drop in the frequency of mid-size axons in PLP:mtPstI mice. This may simply depend on the fact that these axons are by far the most numerous, hence the probability increases that they are the target of degeneration. Nevertheless, we cannot exclude the possibility that some undergo swelling as a result of cellular stress and consequently shift to a higher diameter size, increasing the g-ratio measurement.
Collectively, these data demonstrate that mtDNA DSBs are sufficient to cause primary oligodendropathy, leading to cell death, demyelination, and axonal damage.
Oligodendropathy in PLP:mtPstI mice induces glial activation
Injury to the CNS initiates glia-driven inflammation, which contributes to propagating and exacerbating damage (Amor et al., 2014). Since mtDNA DSBs caused oligodendrocyte loss and accumulation of tissue damage, we investigated whether this triggered CNS inflammation by assessing astroglial and microglial responses (Fig. 3A). Astrogliosis, measured by GFAP expression, was increased in PLP:mtPstI mice, especially at 6 months (Fig. 3B). Similarly, at 2 and 6 months PLP:mtPstI mice showed microgliosis with higher numbers of Iba1+ microglia in the spinal cord (Fig. 3C) and elevated microglial expression of CD45, which is suggestive of increased activation (Fig. 3D,E). At 6 months, we also observed the presence of peripheral immune cells in the spinal cords of PLP:mtPstI mice, which was not found in WT mice (Fig. 3F, red arrows, G).
These data show that primary oligodendropathy caused by mtDNA DSBs induces CNS inflammation driven by resident glial cells, which is the likely cause of secondary recruitment of peripheral immune cells to the CNS.
Sustained mtDNA DSBs in oligodendrocytes induce tissue damage in the optic nerve and retina
To determine whether tissue damage due to mtDNA DSBs in oligodendrocytes occurred in other CNS regions, we evaluated myelin and axon integrity in the optic nerve, a highly affected area in MS. EM analysis showed demyelination and axonal loss in PLP:mtPstI mice induced from P21, most evident at 6 months (Fig. 4A). Axons possibly undergoing remyelination with thin and highly compact myelin could also be found (Fig. 4A, red arrows). Optic nerve degeneration was paralleled by pathological changes in the retina. Indeed, the number of NeuN+ RGCs was already reduced at 2 months in PLP:mtPstI mice in all regions of the retina (Fig. 4B–D). At 6 months, RGC loss increased further in the central and intermediate regions and was virtually complete in the peripheral region, where tissue damage was so extensive that cells could not be counted (Fig. 4C,D). RGC death was accompanied by astrogliosis, which was measured by increased GFAP fluorescence (Fig. 4D,E).
mtPstI expression in the peripheral nervous system does not affect peripheral myelination
The PLP-driven tTA transgene has been found to be expressed in Schwann cells (SCs; Inamura et al., 2012). This was expected based on previous reports showing the expression of PLP in this cell population (Puckett et al., 1987). Hence, to determine whether in PLP:mtPstI mice Schwann cell expression of mtPstI could alter peripheral myelin integrity, we assessed myelin thickness/compaction by measuring the g-ratio in the sciatic nerve (Fig. 5A–E). Despite mtPstI being expressed at appreciable levels (even though lower than in the CNS; Fig. 5H), no differences were found between PLP:mtPstI mice induced from P21 and corresponding controls both at 2 and 6 months of age (Fig. 5B–E). Morphologically, no abnormalities were observed (Fig. 5A). To determine whether mtPstI effectively caused mtDNA DSBs, we quantified by real-time PCR the mitochondrial-specific gene ND1 as an index of mtDNA depletion (Fig. 5F) and the presence of recombined mtDNA as an index of mtDNA deletions (Fig. 5G). We measured a minimal presence of mtDNA deletions (Fig. 5G) and no mtDNA depletion (Fig. 5F) even at 6 months, indicating the mtPstI produced only causes minimal mtDNA DSBs. The lack of mtDNA depletion translated into normal expression of mitochondrial-specific genes, as shown by COX1 quantification, which we found unchanged compared with controls (Fig. 5I). This suggests that OXPHOS is unaffected and can help explain why we did not measure any alterations in myelin integrity (Fig. 5A–E), indicating that SC survival is not compromised in our model.
Brief transient induction of mtDNA DSBs in oligodendrocytes leads to production of myelin-specific antibodies
To determine whether even limited damage to mtDNA was sufficient to cause cellular distress in oligodendrocytes and increase CNS vulnerability to demyelinating injury, we induced brief transient expression of mtPstI in oligodendrocytes by DOX deprivation for only 3 weeks, starting at 5 weeks of age (Fig. 6A). At 8 weeks, as mice were restored to a DOX diet, we assessed mtPstI protein expression in the spinal cord and detected it in PLP:mtPstI mice but not in controls (Fig. 6B). Low levels of mtDNA deletions could be observed by the presence of PCR-detectable recombined mtDNA in PLP:mtPstI mice (Fig. 6C), but not by changes in mtDNA content (Fig. 6D). This brief mtPstI induction led to the production of myelin-specifc antibodies, as anti-MOG and anti-PLP IgGs were detectable in the serum of PLP:mtPstI mice (Fig. 6E,F). These data indicate that brief transient mtDNA DSBs can cause oligodendrocyte distress sufficient to induce an immune response.
Brief transient induction of mtDNA DSBs in oligodendrocytes leads to exacerbation of chronic EAE
To test whether a relatively brief induction of mtDNA DSBs in oligodendrocytes altered the CNS response to injury, we induced EAE in mice deprived of DOX for 3 weeks and 2 d after being restored to a DOX diet (Fig. 6A). PLP:mtPstI mice showed a delay in reaching the peak EAE score (Fig. 6G; Table 1). Despite such a delay, the peak disease phenotype in PLP:mtPstI mice was more pronounced than in controls (Fig. 6G, Table 1), and peak EAE scores were sustained chronically with significant disease exacerbation compared with control mice. The increase in clinical score of ∼0.5 displayed by PLP:mtPstI mice in the chronic phase of EAE has important functional implications. Controls, with an average chronic score of <3, retained full forelimb movement and only had partial hindlimb paralysis, whereas PLP:mtPstI mice, with an average chronic score of ∼3.5, had significant impairment of forelimb function in addition to complete hindlimb paralysis. This was associated with a higher degree of demyelination (Fig. 6H,I), more severe axonal loss (Fig. 6J,K), and a significant increase in immune cell infiltration (Fig. 6J,L) into the spinal cord.
Table 1.
EAE parameters in controls versus PLP:mtPstI mice after brief and transient induction of mtDNA DSBs
| Group | Incidence | Onset (d)a | Peak disease (score) | Peak disease (d) | CDI |
|---|---|---|---|---|---|
| Controls | 86.7% (13 of 15) | 14.31 ± 0.89* | 3.48 ± 0.11 | 15.58 ± 1.03** | 78.64 ± 6.70 |
| PLP:mPstI | 72.7% (8 of 11) | 20.63 ± 1.74 | 3.75 ± 0.37 | 20.63 ± 1.84 | 84.31 ± 14.35 |
aOnset is considered as the day after the first disease manifestation; CDI = Cumulative Disease Index; n = 8–13/group.
*t(19) = 3.582, p = 0.0020, t test;
**t(18) = 2.584, p = 0.0187, t test.
Discussion
Mitochondrial dysfunction has been implicated in MS pathophysiology. To date, studies in postmortem MS tissues and preclinical models have focused exclusively on neurons, showing that defective mitochondria contribute to axonal degeneration and neuronal loss, participating in disease progression and irreversible functional impairment (Dutta et al., 2006; Mahad et al., 2008; Nikić et al., 2011; Witte et al., 2014). Despite demyelination being one of the primary pathological features of MS, whether mitochondrial dysfunction in oligodendrocytes might play a role in disease etiology and progression has never been investigated. This is surprising considering that pattern III MS lesions have been associated with defects in complex IV of the mitochondrial respiratory chain not only in axons, but also in oligodendrocytes (Mahad et al., 2008).
To address this void, we generated the PLP:mtPstI mouse model where mtDNA DSBs can be timely and reversibly induced by controlling the expression of the mtPstI endonuclease. This is an excellent tool to help us understand how much oligodendrocyte rely on mtDNA integrity and OXPHOS for survival and to carry out their physiological functions. By continuous mtPstI induction, we showed that mtDNA DSBs caused oligodendrocyte loss, demyelination, and axonal injury. These are features characteristically found in chronic demyelinating disorders, including progressive MS forms, and are recapitulated by the PLP:mtPstI mouse more closely than by any in vivo model currently available (e.g., EAE and cuprizone).
Since mtDNA encodes 13 key proteins of the OXPHOS system, alterations to mtDNA cause impairment of respiratory function, compromising the main energy production mechanism and causing cell death (Pinto and Moraes, 2015). Even though we could not directly measure OXPHOS in our model because of the low percentage of oligodendrocytes in a mixed cell population, we did observe reduced oligodendroglial COX1. A recent report by Wang et al. (2016) in which the mitochondrial m-AAA protease coded by the Afg3l2 gene was deleted in oligodendrocytes supports the idea that OXPHOS is important for oligodendrocyte survival, even if they are mainly glycolytic cells that are relatively resistant to OXPHOS defects (Fünfschilling et al., 2012). Although no leakage of mtPstI in the nucleus was observed in our model (Pinto et al., 2017), the study by Wang et al. (2016) further minimizes concerns that small amounts of mtPstI could leak into the nucleus, as the phenotype they observed by altering a mitochondrial protease was remarkably similar to the one in our model where mtDNA was cleaved.
A defect in OXPHOS is also the mechanism proposed to cause cuprizone-induced oligodendrocyte death, as decreased activity of respiratory chain complexes was found in mitochondria of cuprizone-treated oligodendrocytes (Pasquini et al., 2007). Furthermore, in vitro exposure of differentiated oligodendrocytes to complex IV inhibitors caused apoptotic cell death (Ziabreva et al., 2010). Notably, the small population of surviving oligodendrocytes consisted mostly of undifferentiated cells that did not exhibit overt alterations, suggesting a higher vulnerability of mature oligodendrocytes to OXPHOS impairment (Ziabreva et al., 2010).
A report by Fünfschilling et al. (2012) showed that postmyelination oligodendrocytes are not as dependent on OXPHOS for survival as other cells, including nonmyelinating oligodendrocytes in vitro, since cytochrome oxidase defects did not cause oligodendrocyte death or demyelination in mice. They convincingly showed that myelinating oligodendrocytes survived by relying on glycolysis, which provided the energy supply in the absence of functional OXPHOS. In contrast, we did observe a progressive death of myelinating oligodendrocytes that paralleled the increased accumulation of mtDNA damage and depletion. This suggests that in our model either glycolysis is not sufficient to compensate for the impaired OXPHOS or other mechanisms independent of a compromised electron transport chain, but initiated by mtDNA DSBs, are responsible for oligodendrocyte death. In support of the latter hypothesis, it has been shown that mtDNA, its fragments, and other mitochondrial components (e.g., formyl peptides) act as nonmicrobial damage-associated molecular patterns capable of activating innate immunity and inflammation (Krysko et al., 2011) by binding to Toll-like receptor 9 (TLR9; Zhang et al., 2010; Wei et al., 2015). Along this line, it was recently shown that mtDNA released into the cytosol as a consequence of mitochondrial stress can activate the stimulator of interferon genes (STING) pathway and the production of cytokines (Barber, 2015; West et al., 2015). Hence, it is plausible that mtDNA fragments accumulated in the oligodendrocytes of induced PLP:mtPstI mice could activate the TLR9 and/or STING pathways and compromise oligodendrocyte survival.
Another possible mechanism of oligodendrocyte death is the accumulation of reactive oxygen species (ROS). Increased ROS production by mitochondrial oxidases and other cellular sources is implicated in demyelination and neurodegeneration in MS and EAE (Smith et al., 1999; van Horssen et al., 2008; Haider et al., 2011; Fischer et al., 2012; Ohl et al., 2016). Since oligodendrocytes are highly vulnerable to ROS-mediated death due to low levels of cellular glutathione and high levels of iron, which catalyzes hydrogen peroxide conversion into hydroxyl radicals (McTigue and Tripathi, 2008), it is believed that ROS are key contributors to oligodendrocyte death and demyelination (McTigue and Tripathi, 2008). ROS increase in dysfunctional mitochondria with impaired electron transport, and susceptible cells (e.g., stem cells) are especially sensitive to ROS generated by mtDNA mutations (Hämäläinen et al., 2015) or DSBs (Pinto et al., 2017). On this basis, it is reasonable to hypothesize that excessive ROS production in PLP:mtPstI mice could lead to oligodendrocyte death and demyelination.
We cannot exclude that toxic metabolites accumulating as a result of mitochondrial dysfunction may cause axonal damage independently of oligodendrocyte death. This idea is corroborated by the work of Viader et al. (2011, 2013), who demonstrated that mitochondrial metabolism disruption by Tfam loss of function in myelinating SCs causes the accumulation of toxic acylcarnitine lipid intermediates. These do not affect SC survival but induce axonal damage that only at later stages leads to SC death and demyelination as secondary consequence of the primary axonopathy. It is plausible that in our model mtDNA DSBs may cause axonal damage not only secondary to oligodendrocyte death, but as a primary event as well.
The consequence of oligodendrocyte death in PLP:mtPstI mice is glial reactivity. Since this is typically associated with the release of proinflammatory molecules such as cytokines and chemokines (Pekny et al., 2016; Gualtierotti et al., 2017), we presume that it is the production of such mediators by reactive glia that promotes the recruitment of immune cells into the CNS, which we showed by histological analysis. To assess the type and degree of activation of the adaptive immune response in our model, we are now conducting a detailed cell profiling by flow cytometry both in the CNS and in peripheral immune organs. This is the focus of a separate comprehensive study.
Following just a brief period of mtDNA DSBs, we detected the presence of myelin-specific antibodies. It should be noted that in MS the pathogenic function of myelin-specific antibodies is controversial. A number of early reports identified anti-MOG antibodies in MS patients; however, the accuracy of the detection methods has since been questioned. Recent studies found anti-MOG antibodies to be transient and restricted to acute disseminated encephalomyelitis and pediatric MS (Ramanathan et al., 2016). In our model, such antibodies are likely produced against myelin antigens released from distressed oligodendrocytes, supporting the idea of secondary adaptive immune activation in response to a CNS-intrinsic trigger.
It has been suggested that mitochondrial dysfunction, including mtDNA mutations, may alter the risk of the development of various diseases, including MS (Hudson et al., 2014; Keogh and Chinnery, 2015). Therefore, we devised a short protocol of mtDNA damage to test whether relatively mild mitochondrial distress, not sufficient to cause cell death or obvious phenotypical alterations, could affect the response to neuroimmune disease. PLP:mtPstI mice with brief transient induction of mtDNA DSBs showed expression of mtPstI, which caused low levels of mtDNA deletions but was not sufficient to cause mtDNA depletion, indicating that OXPHOS function was mostly intact. Induced with EAE, PLP:mtPstI mice developed a more severe chronic phenotype than control mice, suggesting that mild mitochondrial alterations, even if they are per se are nonpathogenic, may exacerbate concomitant neurologic conditions or constitute a predisposing factor.
In summary, our findings demonstrate that mtDNA DSBs cause primary oligodendropathy, which triggers demyelination and irreversible neurological deficit. This raises the possibility that mtDNA damage or OXPHOS dysfunction in oligodendrocytes might play a pathogenic role in chronic demyelinating diseases, possibly including certain forms of MS. We believe this warrants an in-depth investigation of mitochondrial bioenergetics in the oligodendrocytes of MS patients. Importantly, with the PLP:mtPstI mouse model we now have a sophisticated in vivo tool to address the dependence of oligodendrocytes on mitochondrial function. Indeed, to our knowledge, this is the only model where the severity and timing of oligodendrocyte-specific mtDNA damage can be controlled, allowing for modulation of the degree of oligodendrocyte death, demyelination, and functional impairment. Additionally, induced PLP:mtPstI mice recapitulate the hallmarks of demyelinating syndromes associated with neuronal degeneration more accurately than the models currently available. This is especially relevant as the lack of appropriate preclinical models is widely regarded as one of the main reasons for the failure of producing effective remyelinating therapies for chronic demyelinating syndromes. We believe the PLP:mtPstI mouse can fill this void and provide an ideal platform for testing remyelinating and neuroprotective agents.
Footnotes
We thank Margaret Bates and Vania Almeida at the Electron Microscopy Core Facility of The Miami Project To Cure Paralysis for technical assistance with the toluidine blue staining and EM imaging. We also thank Melissa Carballosa-Gautam at the Imaging Core Facility of The Miami Project To Cure Paralysis for assistance with stereological counting. This work was supported by National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) Grants NS-084303-01A1 and 1R01-NS-094522-01 (R.B.); The Miami Project To Cure Paralysis and the Buoniconti Fund (R.B.); The Danish Multiple Sclerosis Society (P.M.M.); Fonden til Lægevidenskabens Fremme (P.M.M.); Foundation for Research in Neurology (P.M.M.); NIH Grant NEI R01-EY027311 (D.I.); NIH Grants NEI 5R01-EY-010804 and NIA 1R01-AG-036871; and NINDS Grant 1R01-NS-079965 (C.T.M.).
The authors declare no competing financial interests.
References
- Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Brück W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H (2003) Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol 62:25–33. 10.1093/jnen/62.1.25 [DOI] [PubMed] [Google Scholar]
- Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D, van Noort JM (2014) Inflammation in neurodegenerative diseases—an update. Immunology 142:151–166. 10.1111/imm.12233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andalib S, Talebi M, Sakhinia E, Farhoudi M, Sadeghi-Bazargani H, Motavallian A, Pilehvar-Soltanahmadi Y (2013) Multiple sclerosis and mitochondrial gene variations: a review. J Neurol Sci 330:10–15. 10.1016/j.jns.2013.04.018 [DOI] [PubMed] [Google Scholar]
- Axisa PP, Hafler DA (2016) Multiple sclerosis: genetics, biomarkers, treatments. Curr Opin Neurol 29:345–353. 10.1097/WCO.0000000000000319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban M, Elson J, Walton A, Turnbull D, Compston A, Chinnery P, Sawcer S (2008) Investigation of the role of mitochondrial DNA in multiple sclerosis susceptibility. PLoS One 3:e2891. 10.1371/journal.pone.0002891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. (2015) STING: infection, inflammation and cancer. Nat Rev Immunol 15:760–770. 10.1038/nri3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Ashbaugh JJ, Magliozzi R, Dellarole A, Karmally S, Szymkowski DE, Bethea JR (2011) Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 134:2736–2754. 10.1093/brain/awr199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Dvoriantchikova G, Barakat D, Ivanov D, Bethea JR, Shestopalov VI (2012) Transgenic inhibition of astroglial NF-κB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J Neuroinflammation 9:213. 10.1186/1742-2094-9-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas-Opazo FA, Vergara-Pulgar K, Pérez MJ, Jara C, Osorio-Fuentealba C, Quintanilla RA (2015) Mitochondrial dysfunction contributes to the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev 2015:509654. 10.1155/2015/509654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran K, Hazelton JL, Wang Y, Fiskum G, Kristian T (2006) Neuron-specific conditional expression of a mitochondrially targeted fluorescent protein in mice. J Neurosci 26:13123–13127. 10.1523/JNEUROSCI.4191-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dendrou CA, Fugger L, Friese MA (2015) Immunopathology of multiple sclerosis. Nat Rev Immunol 15:545–558. 10.1038/nri3871 [DOI] [PubMed] [Google Scholar]
- Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT (2005) Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet 14:2737–2748. 10.1093/hmg/ddi307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, Gudz T, Macklin WB, Lewis DA, Fox RJ, Rudick R, Mirnics K, Trapp BD (2006) Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol 59:478–489. 10.1002/ana.20736 [DOI] [PubMed] [Google Scholar]
- Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, Lassmann H (2012) NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135:886–899. 10.1093/brain/aws012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Moraes CT (2009) Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet 18:1028–1036. 10.1093/hmg/ddn437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, Nave KA (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485:517–521. 10.1038/nature11007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualtierotti R, Guarnaccia L, Beretta M, Navone SE, Campanella R, Riboni L, Rampini P, Marfia G (2017) Modulation of neuroinflammation in the central nervous system: role of chemokines and sphingolipids. Adv Ther 34:396–420. 10.1007/s12325-016-0474-7 [DOI] [PubMed] [Google Scholar]
- Haider L, Fischer MT, Frischer JM, Bauer J, Höftberger R, Botond G, Esterbauer H, Binder CJ, Witztum JL, Lassmann H (2011) Oxidative damage in multiple sclerosis lesions. Brain 134:1914–1924. 10.1093/brain/awr128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hämäläinen RH, Ahlqvist KJ, Ellonen P, Lepistö M, Logan A, Otonkoski T, Murphy MP, Suomalainen A (2015) mtDNA mutagenesis disrupts pluripotent stem cell function by altering redox signaling. Cell Rep 11:1614–1624. 10.1016/j.celrep.2015.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson G, Gomez-Duran A, Wilson IJ, Chinnery PF (2014) Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet 10:e1004369. 10.1371/journal.pgen.1004369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamura N, Sugio S, Macklin WB, Tomita K, Tanaka KF, Ikenaka K (2012) Gene induction in mature oligodendrocytes with a PLP-tTA mouse line. Genesis 50:424–428. 10.1002/dvg.20808 [DOI] [PubMed] [Google Scholar]
- Kalman B, Lublin FD, Alder H (1995) Mitochondrial DNA mutations in multiple sclerosis. Mult Scler 1:32–36. 10.1177/135245859500100106 [DOI] [PubMed] [Google Scholar]
- Keogh MJ, Chinnery PF (2015) Mitochondrial DNA mutations in neurodegeneration. Biochim Biophys Acta 1847:1401–1411. 10.1016/j.bbabio.2015.05.015 [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Mazurkiewicz JE (2013) Friedreich ataxia: neuropathology revised. J Neuropathol Exp Neurol 72:78–90. 10.1097/NEN.0b013e31827e5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P (2011) Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 32:157–164. 10.1016/j.it.2011.01.005 [DOI] [PubMed] [Google Scholar]
- Kwong JQ, Beal MF, Manfredi G (2006) The role of mitochondria in inherited neurodegenerative diseases. J Neurochem 97:1659–1675. 10.1111/j.1471-4159.2006.03990.x [DOI] [PubMed] [Google Scholar]
- Lambertsen KL, Gramsbergen JB, Sivasaravanaparan M, Ditzel N, Sevelsted-Møller LM, Oliván-Viguera A, Rabjerg M, Wulff H, Köhler R (2012) Genetic KCa3.1-deficiency produces locomotor hyperactivity and alterations in cerebral monoamine levels. PLoS One 7:e47744. 10.1371/journal.pone.0047744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000) Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47:707–717. 10.1002/1531-8249(200006)47:6%3C707::AID-ANA3%3E3.0.CO%3B2-Q [DOI] [PubMed] [Google Scholar]
- Madsen PM, Motti D, Karmally S, Szymkowski DE, Lambertsen KL, Bethea JR, Brambilla R (2016a) Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J Neurosci 36:5128–5143. 10.1523/JNEUROSCI.0211-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen PM, Clausen BH, Degn M, Thyssen S, Kristensen LK, Svensson M, Ditzel N, Finsen B, Deierborg T, Brambilla R, Lambertsen KL (2016b) Genetic ablation of soluble tumor necrosis factor with preservation of membrane tumor necrosis factor is associated with neuroprotection after focal cerebral ischemia. J Cereb Blood Flow Metab 36:1553–1569. 10.1177/0271678X15610339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad DH, Trapp BD, Lassmann H (2015) Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 14:183–193. 10.1016/S1474-4422(14)70256-X [DOI] [PubMed] [Google Scholar]
- Mahad D, Ziabreva I, Lassmann H, Turnbull D (2008) Mitochondrial defects in acute multiple sclerosis lesions. Brain 131:1722–1735. 10.1093/brain/awn105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews L, Enzinger C, Fazekas F, Rovira A, Ciccarelli O, Dotti MT, Filippi M, Frederiksen JL, Giorgio A, Küker W, Lukas C, Rocca MA, De Stefano N, Toosy A, Yousry T, Palace J, Network M (2015) MRI in Leber's hereditary optic neuropathy: the relationship to multiple sclerosis. J Neurol Neurosurg Psychiatry 86:537–542. 10.1136/jnnp-2014-308186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Tripathi RB (2008) The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem 107:1–19. 10.1111/j.1471-4159.2008.05570.x [DOI] [PubMed] [Google Scholar]
- Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, Brück W, Bishop D, Misgeld T, Kerschensteiner M (2011) A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 17:495–499. 10.1038/nm.2324 [DOI] [PubMed] [Google Scholar]
- Ohl K, Tenbrock K, Kipp M (2016) Oxidative stress in multiple sclerosis: central and peripheral mode of action. Exp Neurol 277:58–67. 10.1016/j.expneurol.2015.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palace J. (2009) Multiple sclerosis associated with Leber's hereditary optic neuropathy. J Neurol Sci 286:24–27. 10.1016/j.jns.2009.09.009 [DOI] [PubMed] [Google Scholar]
- Pasquini LA, Calatayud CA, Bertone Uña AL, Millet V, Pasquini JM, Soto EF (2007) The neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochem Res 32:279–292. 10.1007/s11064-006-9165-0 [DOI] [PubMed] [Google Scholar]
- Patergnani S, Fossati V, Bonora M, Giorgi C, Marchi S, Missiroli S, Rusielewicz T, Wieckowski MR, Pinton P (2017) Mitochondria in multiple sclerosis: molecular mechanisms of pathogenesis. Int Rev Cell Mol Biol 328:49–103. 10.1016/bs.ircmb.2016.08.003 [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M, Messing A, Steinhäuser C, Lee JM, Parpura V, Hol EM, Sofroniew MV, Verkhratsky A (2016) Astrocytes: a central element in neurological diseases. Acta Neuropathol 131:323–345. 10.1007/s00401-015-1513-1 [DOI] [PubMed] [Google Scholar]
- Peterson JW, Bö L, Mörk S, Chang A, Trapp BD (2001) Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 50:389–400. 10.1002/ana.1123 [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Pinto M, Hida A, Moraes CT (2011) Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson's disease. J Neurosci 31:17649–17658. 10.1523/JNEUROSCI.4871-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto M, Moraes CT (2015) Mechanisms linking mtDNA damage and aging. Free Radic Biol Med 85:250–258. 10.1016/j.freeradbiomed.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto M, Pickrell AM, Wang X, Bacman SR, Yu A, Hida A, Dillon LM, Morton PD, Malek TR, Williams SL, Moraes CT (2017) Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS-dependent but p53/p21-independent manner. Cell Death Differ 24:288–299. 10.1038/cdd.2016.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckett C, Hudson L, Ono K, Friedrich V, Benecke J, Dubois-Dalcq M, Lazzarini RA (1987) Myelin-specific proteolipid protein is expressed in myelinating Schwann cells but is not incorporated into myelin sheaths. J Neurosci Res 18:511–518. 10.1002/jnr.490180402 [DOI] [PubMed] [Google Scholar]
- Ramanathan S, Dale RC, Brilot F (2016) Anti-MOG antibody: the history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun Rev 15:307–324. 10.1016/j.autrev.2015.12.004 [DOI] [PubMed] [Google Scholar]
- Ryan BJ, Hoek S, Fon EA, Wade-Martins R (2015) Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci 40:200–210. 10.1016/j.tibs.2015.02.003 [DOI] [PubMed] [Google Scholar]
- Sadeghian M, Mastrolia V, Rezaei Haddad A, Mosley A, Mullali G, Schiza D, Sajic M, Hargreaves I, Heales S, Duchen MR, Smith KJ (2016) Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci Rep 6:33249. 10.1038/srep33249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Kapoor R, Felts PA (1999) Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol 9:69–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Podojil JR, McCarthy DP, Miller SD, Popko B (2016) Oligodendrocyte death results in immune-mediated CNS demyelination. Nat Neurosci 19:65–74. 10.1038/nn.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nave KA (2008) Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 31:247–269. 10.1146/annurev.neuro.30.051606.094313 [DOI] [PubMed] [Google Scholar]
- van Horssen J, Schreibelt G, Drexhage J, Hazes T, Dijkstra CD, van der Valk P, de Vries HE (2008) Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic Biol Med 45:1729–1737. 10.1016/j.freeradbiomed.2008.09.023 [DOI] [PubMed] [Google Scholar]
- Viader A, Golden JP, Baloh RH, Schmidt RE, Hunter DA, Milbrandt J (2011) Schwann cell mitochondrial metabolism supports long-term axonal survival and peripheral nerve function. J Neurosci 31:10128–10140. 10.1523/JNEUROSCI.0884-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viader A, Sasaki Y, Kim S, Strickland A, Workman CS, Yang K, Gross RW, Milbrandt J (2013) Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron 77:886–898. 10.1016/j.neuron.2013.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Jacquemyn J, Murru S, Martinelli P, Barth E, Langer T, Niessen CM, Rugarli EI (2016) The mitochondrial m-AAA protease prevents demyelination and hair greying. PLoS Genet 12:e1006463. 10.1371/journal.pgen.1006463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Shao B, He Z, Ye T, Luo M, Sang Y, Liang X, Wang W, Luo S, Yang S, Zhang S, Gong C, Gou M, Deng H, Zhao Y, Yang H, Deng S, Zhao C, Yang L, Qian Z, et al. (2015) Cationic nanocarriers induce cell necrosis through impairment of Na(+)/K(+)-ATPase and cause subsequent inflammatory response. Cell Res 25:237–253. 10.1038/cr.2015.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520:553–557. 10.1038/nature14156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte ME, Mahad DJ, Lassmann H, van Horssen J (2014) Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med 20:179–187. 10.1016/j.molmed.2013.11.007 [DOI] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Griffiths PG, Chinnery PF (2011) Mitochondrial optic neuropathies-disease mechanisms and therapeutic strategies. Prog Retin Eye Res 30:81–114. 10.1016/j.preteyeres.2010.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:104–107. 10.1038/nature08780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziabreva I, Campbell G, Rist J, Zambonin J, Rorbach J, Wydro MM, Lassmann H, Franklin RJ, Mahad D (2010) Injury and differentiation following inhibition of mitochondrial respiratory chain complex IV in rat oligodendrocytes. Glia 58:1827–1837. 10.1002/glia.21052 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video showing the abnormal motor phenotype of PLP:mtPstI mice (red tag) at 4 months (DSBs induced at 3 weeks), compared to aged-matched controls.