

Abstract
The cerebral cortex is subdivided into six layers based on morphological features. The supragranular layers 2/3 (L2/3) contain morphologically and genetically diverse populations of neurons, suggesting the existence of discrete classes of cells. In primates and carnivores L2/3 can be subdivided morphologically, but cytoarchitectonic divisions are less clear in rodents. Nevertheless, discrete classes of cells could exist based on their computational requirement, which might be linked to their associated functional microcircuits. Through in vitro slice recordings coupled with laser-scanning photostimulation we investigated whether L2/3 of male mouse auditory cortex contains discrete subpopulations of cells with specific functional microcircuits. We use hierarchical clustering on the laminar connection patterns to reveal the existence of multiple distinct classes of L2/3 neurons. The classes of L2/3 neurons are distinguished by the pattern of their laminar and columnar inputs from within A1 and their location within L2/3. Cells in superficial L2 show more extensive columnar integration than deeper L3 cells. Moreover, L3 cells receive more translaminar input from L4. In vivo imaging in awake mice revealed that L2 cells had higher bandwidth than L3 cells, consistent with the laminar differences in columnar integration. These results suggest that similar to higher mammals, rodent L2/3 is not a homogenous layer but contains several parallel microcircuits.
SIGNIFICANCE STATEMENT Layer 2/3 of auditory cortex is functionally diverse. We investigated whether L2/3 cells form classes based on their functional connectivity. We used in vitro whole-cell patch-clamp recordings with laser-scanning photostimulation and performed unsupervised clustering on the resulting excitatory and inhibitory connection patterns. Cells within each class were located in different sublaminae. Superficial cells showed wider integration along the tonotopic axis and the amount of L4 input varied with sublaminar location. To identify whether sensory responses varied with sublaminar location, we performed in vivo Ca2+ imaging and found that L2 cells were less frequency-selective than L3 cells. Our results show that the diversity of receptive fields in L2/3 is likely due to diversity in the underlying functional circuits.
Keywords: A1, bandwidth, circuit, cortex, layer 2/3, supragranular
Introduction
The cerebral cortex is subdivided into six layers based on morphological features. In particular, the supragranular layers 2/3 (L2/3) are of interest because they reflect the first hierarchical cortical processing stage after sensory input is received in layer 4 (L4). In the auditory cortex (A1) sound frequency information is represented tonotopically in L4, whereas the frequency organization of supragranular L2/3 has been shown to be more heterogeneous by in vivo two-photon imaging (Bandyopadhyay et al., 2010; Rothschild et al., 2010; Winkowski and Kanold, 2013; Kanold et al., 2014) and patch-clamp recordings (Maor et al., 2016). This functional heterogeneity in L2/3 could be the result of cellular diversity.
Functionally, laminar in vivo recordings in cat L2/3 neurons show differences in their spectrotemporal receptive fields with cells in superficial L2/3, i.e., L2, showing broader frequency tuning than L3 cells (Atencio and Schreiner, 2010) as well as extended temporal responses (Atencio and Schreiner, 2010). Moreover, L2 and L3 cells show differences in interlaminar connection strengths derived from cross-correlation measures (Atencio et al., 2009). These different in vivo responses are likely due to differences in the underlying cells and microcircuits. Layer 2/3 contains multiple distinct cell populations. A1 L2 in cat contains small and medium sized pyramidal neurons, as well as a wide range of nonpyramidal neurons that project locally within L1–L3 and to adjacent auditory areas (Mitani et al., 1985; Winer, 1985). In contrast to L2 pyramidal cells, L3 pyramidal cells have more complex dendritic arbors (Winer, 1984; Ojima et al., 1991), and are the source as well as the target of the majority of local, ipsilateral, and contralateral cortical, as well as thalamic connections (Code and Winer, 1985; Winguth and Winer, 1986). Genetic studies in somatosensory cortex showed that within each layer a considerable genetic diversity exists and that genetic differences are associated with differences in projection patterns with, for example, D8Ertd82e-expressing neurons projecting to S2 and Trpc6-positive neurons projecting to frontal cortex (Sorensen et al., 2015). Because output signals to different areas might contain different information, inputs to these discrete cell classes might also arise from different functional intracortical sources. Although these anatomical and genetic studies reveal morphological differences between cells it is unclear whether these classes of neurons receive similar functional inputs.
Recent in vitro studies using laser-scanning photostimulation (LSPS) in mouse primary auditory cortex slices started to analyze the spatial pattern of functional intracortical inputs to L2/3 neurons in auditory cortex. Comparing the intralaminar and interlaminar excitatory circuits between superficial L2 and deep L3 neurons showed that L2 neurons received more L6 input from within the home column than L3 neurons, whose L6 input was displaced out of the tonotopic column (Oviedo et al., 2010). Moreover, comparing intralaminar functional connection patterns using slices of different orientations revealed that A1 L2/3 neurons showed differential integration with respect to the tonotopic axis (Oviedo et al., 2010; Watkins et al., 2014). Fine sublaminar differences in interlaminar cortical circuitry have also been reported in the primary somatosensory barrel cortex (Shepherd and Svoboda, 2005; Bureau et al., 2006; Staiger et al., 2015). Although clear cytoarchitectonic borders are absent in mice, these prior studies a priori divided L2/3 into a more superficial L2 and deeper L3 and did not characterize inhibitory inputs, which can crucially contribute to the functional response identity of L2/3 neurons (Tao et al., 2017).
Here, by combining in vitro whole-cell patch-clamp recordings with LSPS and using an unbiased clustering approach, we systematically investigated whether cells within L2/3 of A1 receive excitatory and inhibitory input from different intracortical sources to functionally identify laminar circuit motifs within a population of supragranular A1 cells. The hierarchical clustering of the individual laminar connectivity maps revealed that cells could be classified into multiple groups based on their inputs from either L5/6 or L4. Although some cells received input from a broad area along the tonotopic axes other cells received inputs from a focal area that could be displaced from the tonotopic (columnar) location of the cells. We also observed that areas providing excitatory inputs to a cell could be mutually exclusive with areas giving inhibitory inputs; this likely reflects functionally specific circuits. The populations that were separated by clustering varied by their depth relative to the pia, and thus represent different sublayers. Moreover, in vivo two-photon imaging of tone-evoked activity in awake mice revealed that L2 cells had higher bandwidth than L3 cells, consistent with the laminar differences in columnar integration.
Together our results show that the supragranular layer of mouse auditory cortex contains a functionally heterogeneous population of neurons and suggests a functional subdivision of L2/3 containing multiple circuits but without any obvious cytoarchitectonic laminar borders.
Materials and Methods
All procedures followed the University of Maryland College Park animal use regulations. Male C57BL/6J mice (Jackson Laboratories) were raised in 12 h light/dark conditions. A fraction of cells included here were previously used in population averages in a prior study (Meng et al., 2015).
Slice preparation.
Mice (∼P28) are deeply anesthetized with isofluorane (Halocarbon). A block of brain containing A1 and the medial geniculate nucleus (MGN) is removed and thalamocortical slices (500–600 μm thick) are cut on a vibrating microtome (Leica) in ice-cold ACSF containing the following (in mm): 130 NaCl, 3 KCl, 1.25 KH2PO4, 20 NaHCO3, 10 glucose, 1.3 MgSO4, and 2.5 CaCl2, pH 7.35–7.4, in 95%O2–5%CO2. For A1 slices the cutting angle is ∼15° from the horizontal plane (lateral raised; Cruikshank et al., 2002; Zhao et al., 2009). Slices are incubated for 1 h in ACSF at 30°C and then kept at room temperature. For recording, slices are held in a chamber on a fixed stage microscope (Olympus BX51) and superfused (2–4 ml/min) with high-Mg ACSF recording solution at room temperature to reduce spontaneous activity in the slice. The recording solution contained the following (in mm): 124 NaCl, 5 KCl, 1.23 NaH2PO4, 26 NaHCO3, 10 glucose, 4 MgCl2, and 4 CaCl2. The location of the recording site in A1 was identified by landmarks (Cruikshank et al., 2002; Zhao et al., 2009) and care was taken to record at a similar position in A1 based on the location relative to the hippocampus.
Electrophysiology.
Whole-cell recordings are performed with a patch-clamp amplifier (Multiclamp 700B, Molecular Devices) using pipettes with input resistance of 4–9 MΩ. Data acquisition is performed by National Instruments AD boards and custom software (Ephus; Suter et al., 2010) adapted to our setup. Voltages were corrected for an estimated junction potential of 10 mV. Electrodes are filled with the following (in mm): 115 cesium methanesulfonate (CsCH3SO3), 5 NaF, 10 EGTA, 10 HEPES, 15 CsCl, 3.5 MgATP, and 3 QX-314, pH 7.25, 300 mOsm. Biocytin or neurobiotin (0.5%) is added to the electrode solution as needed. Series resistances were typically 20–25 MΩ.
Photostimulation.
Caged glutamate [N-(6-nitro-7-coumarylmethyl)-l-glutamate (Ncm-Glu); 0.5–1 mm; Kao, 2006; Muralidharan et al., 2016] is added to the ACSF. Without UV light, this compound has no effect on neuronal activity (Kao, 2006; Muralidharan et al., 2016). UV laser light (500 mW, 355 nm, 100 kHz repetition rate; DPSS; 1 ms pulses) is split by a 33% beam splitter (CVI Melles Griot), attenuated and shuttered by a Pockels Cells (Conoptics) and laser shutter (NM Laser), and coupled into a microscope via scan mirrors (Cambridge Technology) and a dichroic mirror. The laser beam in LSPS enters the slice axially through the objective (Olympus 10×, 0.3NA/water) and has a diameter of <20 μm. Laser power at the sample is <25 mW. We typically stimulated up to 40 × 35 sites spaced 30 μm apart, enabling us to probe areas of 1 mm2. Stimuli are applied at 0.5–1 Hz. Acquisition was controlled by the Ephus software suite (Suter et al., 2010). Analysis is performed essentially as described previously (Meng et al., 2014, 2015) with custom software written in MATLAB. Activation profiles of neurons were produced by recording in cell-attached mode while mapping the same region and recording action potentials. To detect monosynaptically-evoked PSCs we detected peak PSC amplitudes in an ∼50 ms time window after the stimulation. We measured both peak amplitude and transferred charge. Transferred charge was measure by integrating the PSC. Traces containing a short-latency (<8 ms “direct”) response were discarded from the synaptic analysis (black patches in color-coded maps) as were traces that contained longer latency inward currents of long duration (>100 ms). These currents could sometimes be seen in locations surrounding (<50 μm) areas that gave a direct response. Occasionally, some of the 'direct' responses contained synaptically-evoked responses that we did not separate out, leading to an underestimation of local short-range connections. Cells that did not show any large (>100 pA) direct responses were excluded from the analysis as these could be astrocytes. It is likely that the observed PSCs at each stimulus location represent the activity of multiple presynaptic cells. Layer boundaries were determined from the infrared pictures. To avoid bias, for each cell we manually determined the boundaries twice and used the average values. We confirmed our boundaries by comparing with the expression of laminar markers (Fig. 1C).
Figure 1.

LSPS to map intracortical connections to L2/3 cells. A, Left, Infrared image of brain slice with patch pipette on layer 2/3 neuron. Blue dots indicate stimulation grid. Scale bar, 200 μm. Cortical layers are identified based on the DIC image. Right, Position of recorded neurons within layer 2/3. Plotted is the relative position within layer 2/3 with 0 referring to the border with layer 4 and 100 referring to the border with layer 2. B, Whole-cell voltage-clamp recordings at holding potentials of −70 mV (top left) or 0 mV (top right) distinguish between photostimulation-evoked excitatory and inhibitory currents, respectively. Shown are traces obtained with photostimulation at different locations. Solid blue line indicates time of photostimulation; dashed blue line marks 8 ms poststimulus, which is the minimal latency for synaptic responses; and dashed green line marks 50 ms, the end of the analysis window. C, Left, The distributions of upper and lower borders of L4 (upper: cyan; lower: purple) and L5 (upper: purple; lower: gray) for each mapped L2/3 neuron determined from DIC images. Right, The relative location of layer borders (L2/3–L4: cyan; L4–L5: purple; L5–L6: gray). The traces on the right indicate expression profiles of L2/3 marker MATN2 (cyan), L4 marker RORb (purple), and L5 marker ETV1 (gray) in coronal slices from mice at P56. Images for L2/3, L4, and L5 markers are from Allen Brain Atlas image (Image for Matn2 marker: series ID 73817421, image ID 73773513; Image for RORb marker: series ID 79556597, image ID 79561062; Image for ETV1 marker: series ID 72119595, image ID 72017783-31). D, Excitatory and inhibitory LSPS maps from 3 cells in L2/3. Pseudocolor encodes PSC charge at each stimulus location. Direct responses indicated were set to 0 (black). White filled circle marks the soma location. Horizontal bars indicate layer borders. Note that maps can be diverse, but that all cells received input from within L2/3 and 4. Moreover, some cells show no overlap of regions from which excitation and inhibition emerge.
Hierarchical clustering.
To separate the different spatial patterns of input to L2/3 cells we performed an unsupervised hierarchical cluster analysis using Wards's linkage (minimum variance) method (MATLAB 2015b, Statistics toolbox, MathWorks) on the dataset that contains all individual fractional integrated excitatory and inhibitory charges evoked from inside L2/3, L4, L5, and L6 to each L2/3 neuron. Figure 3A shows a schematic diagram of an example fractional charge. We only included cells that had both excitatory and inhibitory connection maps.
Figure 3.
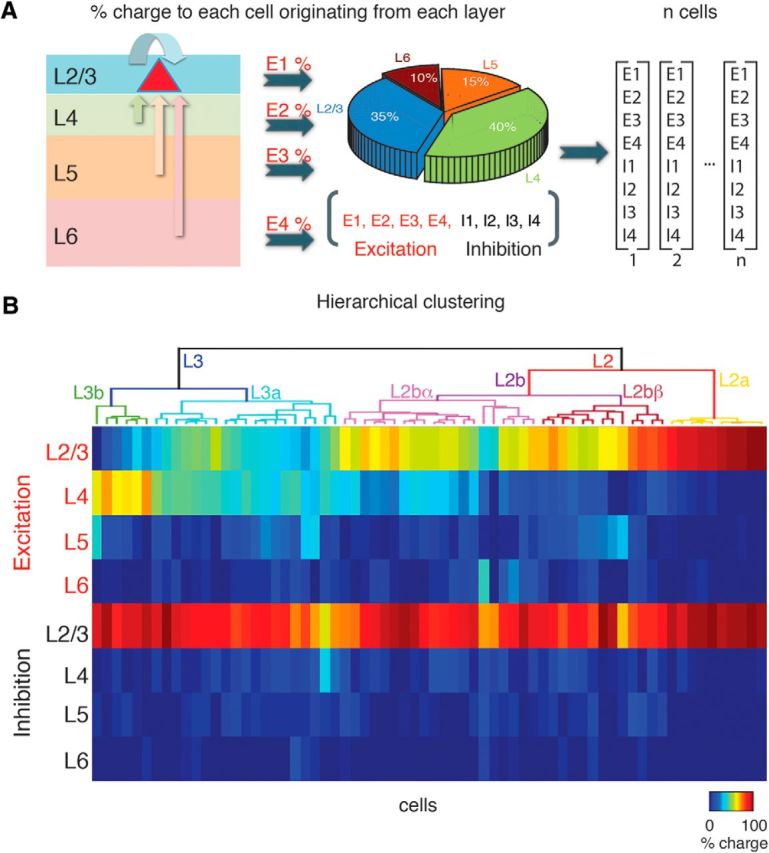
Hierarchical clustering of laminar connections to L2.3 neurons. We identified the variety of intracortical inputs by the fraction of connection charge a particular cell received from each layer. A, Schematic diagram shows the fraction of total input charge (excitatory or inhibitory) a neuron in L2/3 received. Pie chart shows the fractional amount of input and 8-element vector below illustrates clustering variables. B, Dendrograms following unsupervised hierarchical cluster analysis of L2/3 neurons based on the two 4-element vectors, one for the fractional excitatory charge and one for the fractional inhibitory charge from L2/3, L4, L5, and L6. The height of each U-shape tree branches represents the distance between the two clusters being connected. The color code indicates the fraction of charge received from each layer for excitatory and inhibitory inputs respectively.
Display of average maps.
Based upon the segregation from the linkage method, individual excitatory or inhibitory functional connection maps inside each group were aligned to the cell body position and averaged across all the cells in the group. These average maps allow us to visualize the spatial pattern in each group. The connection probability for each relative spatial position is calculated by the fraction of neurons that received an input from that location among all the recorded ones.
Spatial integration distance measures.
We defined the columnar width of the inputs as the difference of between the 10th and 90th percentiles of the input distances relative to the cell along rostral-caudal axis within each layer.
In vivo imaging.
Surgery and animal preparation: mice were given a subcutaneous injection of dexamethasone (5 mg/kg) at least 2 h before surgery to prevent inflammation and edema. Mice were deeply anesthetized using isoflurane (5% induction, 2% maintenance) and given subcutaneous injections of atropine (0.2 mg/kg) and cefazolin (500 mg/kg). Internal body temperature was maintained at 37.5°C using a feedback-controlled heating blanket. The scalp fur was trimmed with scissors and any remaining fur was removed with Nair. The scalp was disinfected with alternating swabs of 70% ethanol and betadine. A patch of skin was removed and the underlying bone cleared of connective tissue with a bone curette. The temporal muscle was detached from the skull and pushed aside, and the skull was thoroughly cleaned and dried. A thin layer of cyanoacrylate glue (VetBond) adhesive was applied to the exposed skull surface and a custom machined titanium headplate (based on the design described by Guo et al., 2014) was affixed to the skull overlying the auditory cortex using VetBond followed by dental acrylic (C&B METABOND). A circular craniotomy (∼3 mm diameter) was made in the center opening of the headplate and the patch of bone was removed. Virus (AAV1.hSyn1.mRuby2.GSG.P2A.GCaMP6s.WPRE.SV40, titer: 3 × 1013; University of Pennsylvania Vector Core) was loaded into beveled glass pipettes and injected slowly into the areas corresponding to auditory cortex in three to five sites (∼30 nl/site; ∼250–300 μm from the surface; ∼2–3 min at each injection site). Pipettes were left in place for at least 5 min after completion of each injection to prevent backflow. After virus injections, a chronic imaging window was implanted. The window consisted of a stack of 2–3 mm diameter coverslips glued with optical adhesive (Norland 71, Edmund Optics) to a 5 mm diameter coverslip; the edges of the window between the glass and the skull were sealed with a silicone elastomer (Kwik-Sil). The edges of the glass and the skull were sealed with dental acrylic. To minimize light reflections, the entire implant except for the imaging window was then coated with black dental cement, created by mixing standard white powder (Dentsply) with iron oxide powder (AlphaChemical, 3:1 ratio; Goldey et al., 2014). Meloxicam (0.5 mg/kg) and a supplemental does of dexamethasone were provided subcutaneously as a postoperative analgesic. Animals were allowed to recover for at least 1 week before the beginning of experiments.
Acoustic stimulation.
Sound stimuli were synthesized using custom software in MATLAB using custom software, passed through a multifunction processor (RX6, TDT), attenuated (PA5, Programmable Attenuator), and delivered via ES1 speaker placed ∼5 cm directly in front of the mouse. The sound system was calibrated between 2.5 and 80 kHz and showed a flat (±3 dB) spectrum over this range. Overall sound pressure level (SPL) at 0 dB attenuation was 90 dB SPL (for tones). Sounds were played at a range of sound levels (40–80 dB SPL, 20 dB steps). Auditory stimuli consisted of sinusoidal amplitude-modulated tones (20 Hz modulation, cosine phase), ranging from 3 to 48 kHz. For wide-field imaging, the frequency resolution of the stimuli was 1 tone/octave; for two-photon imaging, the frequency resolution was 2 tones/octave (0.5 octave spacing). Each of these tonal stimuli was repeated five times with a 6 s interstimulus interval, for a total of either 75 (wide-field) or 135 (two-photon) iterations.
Wide-field imaging.
For wide-field imaging, awake mice were placed into a plastic tube and head restraint system the design of which was similar to that described by Guo et al., (2014). Blue excitation light was provided by an LED (470 nm, Thorlabs) and xenon-arc lamp (Lambda LS, Sutter Instrument) equipped with an excitation filter (470 nm CWL, 40 nm FWHM; Chroma ET470/40×) and directed toward the cranial window. Emitted light was collected through a tandem lens combination (Ratzlaff and Grinvald, 1991) consisting of a 55 mm lens and 85 mm lens affixed to the camera and passed through a long-pass filter (cutoff: 495 nm, Chroma Q495lp) followed by a bandpass emission filter (525 nm CWL, 50 nm FWHM; Chroma HQ525/50). Images were acquired using StreamPix software (NorPix) controlling a CoolSNAP HQ2 CCD camera (Photometrics). After acquiring an image of the surface vasculature, the focal plane was advanced to a depth corresponding to ∼300–400 μm below the brain surface. One trial of stimulation consisted of ∼1–2 s of quiet, followed by sound onset (duration, 1 s; modulation rate, 20 Hz; frequencies, 3–48 kHz, 1 octave spacing; level, 40, 60, 80 dB SPL), followed by 1–2 s of quiet. Each frequency-level combination was randomly repeated five times for a total of 75 iterations. Intertrial interval was 10–15 s. Acquisition of each frame was individually triggered and synchronized with the sound presentation using the Ephus software suite (Suter et al., 2010).
Two-photon imaging.
For two-photon imaging, we used a scanning microscope (Bergamo II series, B248, Thorlabs) coupled to a pulsed femtosecond Ti:Sapphire two-photon laser with dispersion compensation (Vision S, Coherent). The microscope was controlled by ThorImageLS software (Thorlabs). The laser was tuned to λ = 940 nm to simultaneously excite GCaMP6s and mRuby2. Red and green signals were collected through a 16× 0.8 NA microscope objective (Nikon). Emitted photons were directed through 525/50 (green) and 607/70 (red) bandpass filters onto GaAsP photomultiplier tubes. The field-of-view was 370 × 370 μm. Imaging frames of 512 × 512 pixels (pixel size 0.72 μm) were acquired at 30 Hz by bidirectional scanning of an 8 kHz resonant scanner. Beam turnarounds at the edges of the image were blanked with a Pockels cell. The average power for imaging was <70 mW, measured at the sample.
Data analysis.
Wide-field image sequences were analyzed using custom routines written in MATLAB (MathWorks). Images were parsed into trial-based epochs in which each frame sequence represented a single trial consisting of the presentation of a single sound frequency-intensity combination. For each trial, response amplitude (ΔF/F) as a function of time was determined for each pixel using the formula, ΔF/F = (F − F0)/ F0), where F corresponds to the time-varying fluorescence signal at a given pixel, and baseline fluorescence F0 was estimated by averaging the fluorescence values for four frames (∼1 s) before sound onset for a given trial and pixel. For construction of sound-evoked response maps, the amplitude of the ΔF/F pixel response during the 1 s (i.e., ∼4 frames) after stimulus onset was averaged across time and repetitions, yielding an average response magnitude that was assigned to each pixel. Responsive areas in the average response maps were defined on a pixel-by-pixel basis as pixels in which the average brightness of the pixel during the 1 s (i.e., ∼4 frames) after stimulus onset exceeded 2 SD of the pixel brightness during the 1 s preceding stimulus onset across stimulus repetitions.
Two-photon image sequences were corrected for x–y drifts and movement artifacts using either the TurboReg in ImageJ (Thévenaz et al., 1998; Schindelin et al., 2012) or discrete Fourier transform registration implemented in MATLAB (MathWorks) using the mRuby2 labeled neurons. Cell centers were identified manually from the average image of the motion-corrected sequence. Ring-like regions-of-interest (ROI) boundaries were drawn based on the method described by Chen et al. (2013). Overlapping ROI pixels (due to closely juxtaposed neurons) were excluded from analysis. For each labeled neuron, a raw fluorescence signal over time was calculated as the mean fluorescence in each ROI (F) across frames and converted to a relative fluorescence measure (ΔF/F0). F0 was estimated by calculating the average fluorescence during the prestimulus period. Neuropil (NP) subtraction was performed on all soma ROIs. The neuropil ROI was drawn based on the outer boundary of the soma ROI and extended from 1 pixel beyond the soma ROI outer boundary to 15 μm and excluded any pixels assigned to neighboring somata. Thus, the final ΔF/F used for analysis was calculated as ΔF/Fsoma = raw ΔF/Fsoma − (0.9 × ΔF/FNP). Data analysis was performed as described previously (Winkowski and Kanold, 2013). Neurons in which the calcium waveform was significantly modulated by sound presentation were defined by ANOVA (p < 0.01) across baseline (prestimulus) and all sound presentation periods. Percentage responding neurons was defined as the percentage of neurons that exceeded this significance criterion. Frequency tuning curves and frequency response areas were obtained by calculating the mean response (ΔF/F) during the stimulus period for each sound frequency-intensity combination. Best frequency (BF) of a given neuron was defined as the peak (maxΔF/F) of the frequency-tuning curve at the middle sound intensity (i.e., 60 dB SPL). To assess BF variability for each neuron on a local scale (<100 μm), we calculated the interquartile range (IQRBF; in octaves) for all responding neurons within a 100 μm radius around each neuron. To assess tuning curve bandwidth (BW), we first performed linear interpolation of the frequency tuning curve to find the minimum and maximum sound frequencies (Freqmin and Freqmax, respectively) that evoked responses exceeding 60% of the maximum ΔF/F response for each responding neuron. For single-peaked neurons, the log2 (Freqmax/Freqmin) was used as a measure of tuning bandwidth in octaves. Quality factor (Q) was defined as the BF/BW60 for each neuron.
Experimental design and statistics.
Cells are grouped according to the hierarchical clustering. Values for each group are plotted as means ± SEM as indicated. For comparison between multi-groups Lilliefors test was first performed to check normality of the distribution. If data passed the test, then ANOVA was used (anova1, MATLAB 2015a). If not, we used a Kruskal–Wallis test (kruskalwallis, MATLAB 2015a). We then used a Tukey–Kramer multiple-comparison test (multicomp, MATLAB 2015a) and obtained p values of pairwise comparison between groups.
Populations of imaged neurons are compared with a rank sum or Student's t test (based on Lilliefors test for normality), and deemed significant if p < 0.05. The p values for the regression fit were obtained using MATLAB function “corr”. Regression coefficients were determined using the “regress” function in MATLAB 2015b.
Results
We use LSPS of caged glutamate (Shepherd et al., 2003; Meng et al., 2014, 2015) to spatially map the functional connectivity of excitatory and inhibitory inputs to A1 L2/3 neurons (n = 68 cells; Fig. 1). We mapped the excitatory and inhibitory inputs to a given neuron by targeting up to 40 × 35 stimulation points spaced 30 μm apart (Fig. 1B). Figure 1C shows maps for three example neurons demonstrating that connectivity maps varied between cells. Although previously we characterized the average connectivity profile in L2/3 cells (Meng et al., 2015), we here aimed to investigate the diversity of connection profiles in populations of L2/3 neurons to potentially uncover distinct subcircuits.
Layer 2/3 cells are heterogeneous with respect to their intracortical inputs
We recorded from 68 neurons and found that connection maps could be diverse. Figure 2 shows overlays of connection maps for excitatory (red) and inhibitory (blue) connections for 42 L2/3 neurons. We first qualitatively characterized the variability of the input maps for each lamina. While all neurons received intralaminar excitatory input from L2/3 the extent and spatial pattern of input varied. Because of large direct responses we could not measure intralaminar inputs originating close (<100 μm) to the somata of L2/3 neurons; however, neurons did show excitatory inputs from more distant locations in L2/3 (up to 460 μm). Many (35/68) neurons received input from distinct “hot spots” in L4 or L5/6 caudal or rostral to the soma (Fig. 2, neurons 3, 5, 8, 11, 12, 13, 27, 30, 31, 37, 38, and 41, black arrows).
Figure 2.
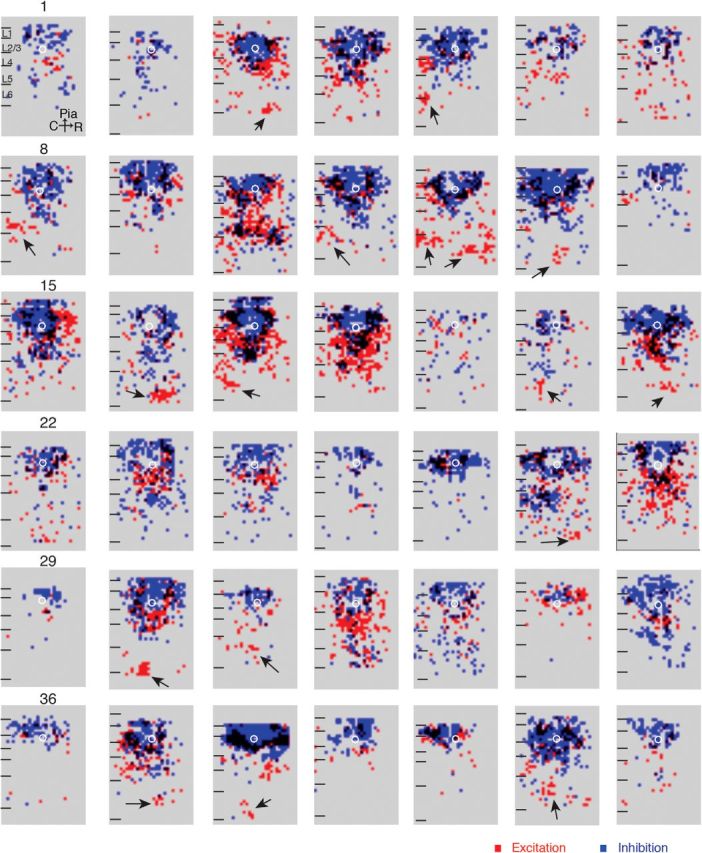
L2/3 cells show diverse connections. Overlays of excitatory (red) and inhibitory (blue) maps for 42 cells. Locations where excitation and inhibition overlap are shown in black. The soma locations of the patched cells are indicated by the white circles.
Interestingly, spatial maps of excitation and inhibition frequently did not overlap with areas that gave rise to inhibitory inputs. Although this is expected around the cell body where excitatory inputs are masked by the direct response, non-overlap for distant inputs suggests patchy non-overlapping translaminar connectivity.
We first characterized what fraction of cells received inputs from each lamina. All L2/3 neurons received input from within L2/3. Most (60/68) L2/3 neurons received excitatory inputs from L4. In 54/60 neurons this input originated from a broad (>150 μm) area, whereas in 8/60 neurons the input originated from a more restricted (<150 μm) area. A fraction (15/68) of L2/3 cells did not receive any excitatory inputs from infragranular layers L5/6. Inhibitory inputs from infragranular layers originated in L5 in 51/68 cells and from L6 in 34/68 cells. Together these results suggest that L2/3 neurons show a wide functional diversity in their laminar input sources.
L2/3 cells segregate into functional classes
So far, we have separately described the spatial diversity of intracortical input patterns from the different layers to each L2/3 cell. We next asked whether a systematic relationship existed between inputs from one layer and inputs from another layer, and whether we can group cells into classes based on laminar connectivity. To identify spatial connection patterns, we converted the spatial input maps into laminar vectors that indicated the fraction of connections a given cell received from each lamina (Fig. 3A). Thus, for a given cell, we obtained two 4-element vectors, one for the amount of excitation and one for amount inhibition that originate from each layer, which characterize the laminar distribution of the respective input. We then performed hierarchical clustering on these vectors (Fig. 3B). The clustering shows that the population of cells can be divided into multiple distinct classes. Qualitative inspection of the clustering results shows that the major group differences are accounted for by differences in the laminar profile of excitatory inputs. To investigate these differences in detail we compared the connection patterns of the major cell groupings.
Layer 2/3 cells separates into two spatially distinct subgroups predominantly based on the amount of L4 input
On a gross level, cells split into two groups (Fig. 3B). To identify the differences in connection patterns between these groups, we constructed average spatial maps of excitatory and inhibitory connection probability of cells in each group by aligning cells and computing the fraction of cells that received input from each location (Fig. 4A,B). Approximately one-third (25/68) of the L2/3 neurons in our sample received relatively stronger inputs from L4 compare to other layers (Group 1; Fig. 2, cells 1, 30, 32), whereas the rest did not (Group 2; Fig. 2, cells 5, 10, 12). Cells that receive excitatory L4 input tended to be localized lower in L2/3 than cells that did not receive L4 input (Fig. 4C). These results are consistent with the reported differences between superficial L2 and deep L3 in rodent (Oviedo et al., 2010); therefore we label Group 1 cells as L3 and Group 2 cells as L2. A quantitative comparison of the fractional inputs showed that L3 cells received most excitatory input from L4 (Fig. 4D1; p < 0.0001). Moreover, L3 cells also received more excitatory inputs from upper L5 (Fig. 4D; p < 0.001).
Figure 4.

Dividing L2/3 neurons into two groups: L2, L3. L2/3 neurons subdivide into two groups based on the hierarchical cluster analysis. A, Average maps (aligned to soma, white circle) of connection probability for excitatory connections among Group 1 and 2 cells. Connection probability is encoded according to the pseudocolor scale. White horizontal lines indicate averaged laminar borders and are 100 μm long. B, Average maps (aligned to soma, white circle) of connection probability for inhibitory connections among Group 1 and 2 cells. Connection probability is encoded according to the pseudocolor scale. White horizontal lines indicate averaged laminar borders and are 100 μm long. C, Boxplot of relative cell positions of Group 1 (blue) and Group 2 (red) cells within L2/3. The locations of Group 1 cells are lower than those of Group 2 cells (p = 5.03 × 10−3) thus Group 1 represents L3 and Group 2 represents L2. *p < 0.05, **p < 0.01. D, Layer-specific fractional excitatory (left) and inhibitory (right) charge of L3 (blue) and L2 (red) cells. Data are mean charge ± SEM. Excitation originating from L2/3, L4, and L5 to L2/3 cells shows significant comparing between L3 and L2. L2 has significantly higher percentage excitatory input from L2/3 (p = 7.92 × 10−13) but less percentage from L4 (p = 4.22 × 10−15) and L5 (p = 3.30 × 10−3). Inhibition from L2/3, L4, L5 and L6 does not show significant differences between the groups (L2/3: p = 0.20; L4: p = 0.36; L5: p = 0.60; L6: p = 0.15). E, Bar plot of the distance of 80% of input to each L2/3 cell originating from L2/3, L4, L5, and L6. L3 cells receive input originating from a wider L4 region (p = 0.01); there is no significant difference for L2/3 (p = 0.31), L5 (p = 0.23) and L6 (p = 0.21). F, Boxplot of the direct activation area of L3 and L2 cells. L3 cells (blue) have bigger direct activation area than L2 cells (red; p = 0.028). Triple asterisk indicates a higher significance level.
We next analyzed differences in inhibitory input between L2 and L3. Qualitative inspection of the spatial connection probabilities suggested that L3 cells received more total input from L4 (Fig. 4B). However, comparing the fractional laminar pattern of inhibitory inputs between the groups showed no differences (Fig. 4D2). The difference in the average maps suggests that while the amount of inhibitory L4 might be similar, the columnar pattern of inhibitory inputs from L4 might be different between L2 and L3.
To analyze the spatial extent of the different laminar inputs, we calculated the columnar width that encompassed 80% of inputs from each lamina (Fig. 4E). This analysis revealed that L4 input to L3 cells originated from further across the tonotopic axis (up to 450 μm) than L4 input to L2 cells. Thus, L3 cells integrate ascending information from L4 across a larger range of the tonotopic axis. Together these results already suggest a suborganization of L2/3 into L2 and L3 based on the laminar pattern of both excitatory and inhibitory inputs. To investigate whether these differences could be due to differences in intrinsic features of L2/3 cells we measured the area that resulted in direct responses during LSPS. This area is a measure of the size of the soma and the proximal dendrites. A comparison showed that L3 cells had a larger direct response area than L2 cells (Fig. 4F) suggesting that L3 cells had more proximal dendrites consistent with L3 pyramidal cells having more complex dendritic arbors than L2 cells (Winer, 1984; Ojima et al., 1991). Together these results show that L2/3 in mouse A1 shows a functional sublaminar organization consistent with prior anatomical studies in carnivores.
Layer 2 further separates into two subgroups primarily based on the balance of input from L2/3 and L4
L2 cells fell into two groups (Fig. 3B) based the relative amount of excitatory input these cells receive from within L2/3 and L4 (Fig. 5D; p < 0.0001). Plotting the average connection spatial connection probability maps (Fig. 5A,B) for the three groups showed that the two populations of L2 cells with weak L4 inputs differed in both the amount and spatial extent of excitatory inputs from within L2/3 (Fig. 5C,D). Cells receiving the least L4 input were located more superficially in L2 (Fig. 5C,D); therefore we label these as L2a cells (Fig. 2, cells 38, 40) and the middle group, which did receive some L4 input, as L2b cells (Fig. 2, cells 12, 13). Although L2b cells on average were located between L2a and L3, their position overlapped with these two other classes (Fig. 5C). L2a and L2b cells also differed in their inhibitory inputs (Fig. 5B,D). L2a cells received almost no ascending inhibitory input from L4 and L5, whereas L2b cells did receive some inhibition from L4 and L5 (Fig. 5B,D). Moreover, excitatory L6 inputs to L2b cells seemed to be displaced from the home column of the L2b cells, consistent with prior studies (Oviedo et al., 2010).
Figure 5.

L2 subdivides into two sublayers: L2a, L2b. L2 cells (Fig. 4, Group 2) can be further subdivided into two groups based on the hierarchical cluster analysis. The two subgroups are here renumbered as Group 2 (L2b) and Group 3 (L2a) A, Average maps (aligned to soma, white circle) of connection probability for excitatory connections among Group 1 (Fig. 4A, L3), 2 and 3 cells. Connection probability is encoded according to the pseudocolor scale. White horizontal lines indicate averaged laminar borders and are 100 μm long. B, Average maps (aligned to soma, white circle) of connection probability for inhibitory connections among Group 1, 2, and 3 cells. Connection probability is encoded according to the pseudocolor scale. White horizontal lines indicate averaged laminar borders and are 100 μm long. C, Boxplot of relative cell positions of Group 1 (red), Group 2 (yellow), and Group 3 (blue) cells within L2/3. The locations of Group 1 cells are significantly lower than those of Group 3 cells (p = 1.31 × 10−3). The cell location in Group 2 is between Groups 1 and 3 (multiple comparison: Group 1,2: p = 0.093; Group 2,3: p = 0.067). *p < 0.05, **p < 0.01. Thus Group 1 represents L3, whereas Groups 2 and 3 represent L2b and L2a, respectively. D, Layer-specific fractional excitatory (D1) and inhibitory (D2) charge of L3 (blue), L2b (purple), and L2a (yellow) cells. Data are mean charge ± SEM. The excitation originating from L2/3, L4 shows significance comparing among three groups (multiple comparison: L2/3: L3 vs L2b: p = 9.56 × 10−10; L3 vs L2a: p = 9.56 × 10−10; L2a vs L2b: p = 9.58 × 10−10; L4: L3 vs L2b: p = 9.56 × 10−10; L3 vs L2a: p = 9.56 × 10−10; L2a vs L2b: p = 1.48 × 10−4). L2a has significantly higher percentage of excitatory input from L2/3 but few inputs from other layers. L3 and L2b have more input from L5 than L2a (L3 vs L2a: p = 5.45 × 10−4; L2a vs L2b: p = 2.51 × 10−2). L2b has more input from deep L6 compared with L2a (p = 2.93 × 10−2). Similar to excitation L2a also has significantly higher percentage of inhibitory input from L2/3 (L3 vs L2a: p = 1.20 × 10−3; L2a vs L2b: p = 1.10 × 10−3; L3 vs L2b: p = 0.98) but few inputs from L4 (L3 vs L2a: p = 2.53 × 10−2; L2a vs L2b: p = 7.38 × 10−2; L3 vs L2b: p = 2.01 × 10−3) and L5 (L3 vs L2a: p = 6.57 × 10−3; L2a vs L2b: p = 1.07 × 10−3; L3 vs L2b: p = 0.79) compared with other groups. All three groups have few inputs from deep L6 (L3 vs L2a: p = 0.14; L2a vs L2b: p = 0.47; L3 vs L2b: p = 0.40). E, Barplot of the distance of 80% of input to each L2/3 cell originating from L2/3, L4, L5 and L6. L2a cells receive input originating from narrower tonotopic areas in L4 (L3 vs L2a: p = 7.06 × 10−4; L2a vs L2b: p = 2.11 × 10−2; L3 vs L2b: p = 0.20) and L5 (L3 vs L2a: p = 2.24 × 10−3; L2a vs L2b: p = 1.91 × 10−3; L3 vs L2b: p = 0.99). There is no significant difference of L2/3 and L6 input width among three groups (multiple comparison: L2/3: L3 vs L2b: p = 0.45; L3 vs L2a: p = 0.99; L2a vs L2b p = 0.71; L6: L3 vs L2b: p = 0.83; L3 vs L2a: p = 5.2 × 10−2; L2a vs L2b: p = 0.11). F, Boxplot of the direct activation area of all cells. L3 (blue) cells have bigger direct activation area than L2a cells (yellow; p = 0.018). There is no difference between L3 vs L2b (p = 0.25) and L2a vs L2b (p = 0.21). Triple asterisk indicates a higher significance level.
Comparison of the columnar width showed that L2a cells received the most spatially restricted input from L4 and L5 (Fig. 5E). The area of direct response of L2a cells was smaller than that of L3 cells, suggesting that they might correspond to the large and small pyramidal cells found in cat A1 (Mitani et al., 1985; Winer, 1985).
Layer 3 further separates into two subgroups based on the balance of input from L2/3 and L4
Similar to the subdivision of L2 into L2a and L2b, the hierarchical clustering based upon the relative amount of inputs suggested that L3 could also be subdivided (Fig. 3B). Plotting the average connection probability maps for the resulting four groups (Fig. 6A,B) showed that the subdivision of L3 resulted in two classes of L3 cells with the major differences being the relative absence of excitatory input from within L2/3 and L4 (Figs. 3, 6A,D; p < 0.0001). Although there was large overlap in relative position between the two groups of L3 cells (Fig. 6C), L3 cells located deepest in L3 (Fig. 6C, Group 1), termed L3b cells, had the least percentage of input originating from L2/3 but much more inputs from L4 (Figs. 6A,D; 2, cell 28), whereas cells located somewhat more superficially, termed L3a cells, had approximately double the amount of L2/3 input and less input from L4 (Figs. 6D; 2, cells 17, 18). Comparing the inhibitory inputs to L3a and L3b cells showed that L3a cells receive more inhibitory input from L4.
Figure 6.

L3 also subdivides into two sublayers: L3a, L3b. L3 can also be divided into two groups representing L3b (Group 1) and L3a (Group 2). With the prior two subgroups in L2 (Group 3: L2b and Group 4: L2a; same as Fig. 5) we have divided L2/3 cells into 4 groups. A, Average maps (aligned to soma, white circle) of connection probability for excitatory connections among four groups of cells. Connection probability is encoded according to the pseudocolor scale. White horizontal lines indicate averaged laminar borders and are 100 μm long. B, Average maps (aligned to soma, white circle) of connection probability for inhibitory connections among four groups of cells. Connection probability is encoded according to the pseudocolor scale. White horizontal lines indicate averaged laminar borders and are 100 μm long. C, Boxplot of relative cell positions of Group 1 (L3b; green), Group 2 (L3a; light blue), Group 3 (L2b; purple), and Group 4 (L2a; yellow) cells within L2/3. The locations of L2a cells are close to the upper boundary of L2/3 and the locations of L3b cells are close to the lower boundary of L2/3. The cell location of L3a and L2b are in between. *p < 0.05, **p < 0.01. The p values from multiple-comparison test are as follows: L3b vs L3a: p = 0.89; L3b vs L2b: p = 0.27; L3b vs L2a: p = 0.012; L3a vs L2b: p = 0.35; L3a vs L2a: p = 7.89 × 10−3; L2b vs L2a: p = 0.12. D, Layer-specific fractional excitatory (D1) and inhibitory (D2) charge of L3b, L3a, L2b, and L2a cells. Data are mean charge ± SEM. Comparing the excitation originating from L2/3 or L4 between groups shows differences. The main input to L3b cells come from L4, whereas the main input to L2a is within L2/3. L3a and L2b have most input both coming from L2/3 and L4. The p values from multiple-comparison test are as follows: L2/3: L3b vs L3a: p = 6.32 × 10−5; L3b vs L2b: p = 3.77 × 10−9; L3b vs L2a: p = 3.77 × 10−9; L3a vs L2b: p = 3.81 × 10−9; L3a vs L2a: p = 3.77 × 10−9; L2b vs L2a: p = 3.77 × 10−9. L4: L3b vs L3a: p = 8.60 × 10−8; L3b vs L2b: p = 3.77 × 10−9; L3b vs L2a: p = 3.77 × 10−9; L3a vs L2b: p = 3.78 × 10−9; L3a vs L2a: p = 3.77 × 10−9; L2b vs L2a: p = 3.44 × 10−6. L5: L3b vs L3a: p = 0.99; L3b vs L2b: p = 0.76; L3b vs L2a: p = 4.33 × 10−2; L3a vs L2b: p = 0.25; L3a vs L2a: p = 1.63 × 10−3; L2b vs L2a: p = 4.75 × 10−2. L6: L3b vs L3a: p = 0.54; L3b vs L2b: p = 0.14; L3b vs L2a: p = 0.99; L3a vs L2b: p = 0.63; L3a vs L2a: p = 0.41; L2b vs L2a: p = 0.051. Inhibition also shows some difference among four groups. The p values from multiple-comparison tests of inhibition are as follows: L2/3: L3b vs L3a: p = 7.35 × 10−2; L3b vs L2b: p = 0.24; L3b vs L2a: p = 0.69; L3a vs L2b: p = 0.69; L3a vs L2a: p = 2.30 × 10−4; L2a vs L2b: p = 1.34 × 10−3. L4: L3b vs L3a: p = 4.02 × 10−2; L3b vs L2b: p = 0.32; L3b vs L2a: p = 0.99; L3a vs L2b: p = 0.29; L3a vs L2a: p = 5.07 × 10−3; L2a vs L2b: p = 4.94 × 10−3. L5: L3b vs L3a: p = 0.61; L3b vs L2b: p = 0.52; L3b vs L2a: p = 0.47; L3a vs L2b: p = 0.99; L3a vs L2a: p = 6.25 × 10−3; L2a vs L2b: p = 1.98 × 10−3. L6: L3b vs L3a: p = 0.85; L3b vs L2b: p = 0.99; L3b vs L2a: p = 0.78; L3a vs L2b: p = 0.44; L3a vs L2a: p = 0.13; L2a vs L2b: p = 0.64. E, Barplot of the distance of 80% of input to each L2/3 cell originating from L2/3, L4, L5, and L6. The p values from multiple-comparison test are as follows: L2/3: L3b vs L3a: p = 2.60 × 10−3; L3b vs L2b: p = 2.15 × 10−3; L3b vs L2a: p = 5.27 × 10−2; L3a vs L2b: p = 0.99; L3a vs L2a: p = 0.77; L2a vs L2b: p = 0.85. L4: L3b vs L3a: p = 0.28; L3b vs L2b: p = 0.98; L3b vs L2a: p = 0.37; L3a vs L2b: p = 0.10; L3a vs L2a: p = 3.40 × 10−4; L2a vs L2b: p = 3.42 × 10−2. L5: L3b vs L3a: p = 0.51; L3b vs L2b: p = 0.72; L3b vs L2a: p = 0.38; L3a vs L2b: p = 0.92; L3a vs L2a: p = 1.82 × 10−3; L2a vs L2b: p = 3.41 × 10−3. L6: L3b vs L3a: p = 0.011; L3b vs L2b: p = 0.14; L3b vs L2a: p = 0.97; L3a vs L2b: p = 0.31; L3a vs L2a: p = 9.80 × 10−3; L2a vs L2b: p = 0.19. F, Boxplot of the direct activation area of L3b, L3a, L2b, and L2a cells. L3a (light blue) cells have bigger direct activation area than L2a cells (yellow; p = 8.0 × 10−3). There is no difference between L3b, L2b, and L2a (L3b vs L2b: p = 0.93; L3b vs L2a: p = 0.89; L2a vs L2b: p = 0.30). Triple asterisk indicates a higher significance level.
The analysis of the amount of columnar integration revealed further differences between L3a and L3b cells. Excitatory input from L6 to L3b cells originated from a much more restricted tonotopic areas than that to L3a cells (Fig. 6E). Given the large amount of L4 input and small amount of L2/3 input that L3b cells receive this suggest that L3b cells seem to form a faithful relay of L4 inputs. A comparison of the direct-response areas showed a trend for L3b cells to have a smaller direct area suggesting that they might have smaller soma and dendritic trees (Fig. 6F). This also indicates that the decreased input from L2/3 to L3b cells versus L3a cells cannot be attributed to differences in the direct activation area. Thus, these results suggest that L2/3 in mouse A1 can be functionally subdivided into four distinct cell classes based on translaminar input patterns.
Layer 2 can be separated into three subgroups based on the amount of L4 and L5 input
So far our hierarchical clustering separated L2 into a small group L2a and a large group L2b. Inspection of the fractional inputs showed that these groups received differing amounts of L4 input (Fig. 3B). Indeed, the hierarchical clustering suggested that L2b could be further subdivided into two groups L2bα and L2bβ (Fig. 3B). Inspection of the average connectivity probability maps (Fig. 7A,B) illustrates the differing inputs between the subgroups. These two subgroups are intermingled within L2/3 (Fig. 7C). A quantification of the laminar inputs (Fig. 7D) showed that the separation within L2b is based on the amount of L4 and L5 input. L2bα (Group 3; Fig. 2, cells 8, 10) received approximately half as much L4 input as L2bβ (Group 4; Fig. 2, cells 3, 5). However, the relative amount of L5 input is reversed with L2bα receiving approximately twice as much as L2bβ (Fig. 7D). Analysis of the amount of columnar integration (Fig. 7E) and direct area (Fig. 7F) showed that L2bα were quite similar with L2bβ (Fig. 7E), suggesting that L2bα and L2bβ cells have similar size of the soma and the proximal dendrites. Even though those two groups of cells have similar cell position and direct area, they are involved into two different types of circuits: L2bα cells receive input primarily from L2/3 and L5, whereas L2bβ cells receive input from L2/3 and L4. Thus, even though L2bα and 2bβ show overlap in depth, they are involved in different circuits.
Figure 7.

L2b further subdivides into two sublayers: L2bα, L2bβ. L2b cells in Figure 5 can be divided into additional two groups L2bα and L2bβ, resulting in five groups of L2/3 cells. The first two subgroups represent L3 (Group 1 = L3b, Group 2 = L3a; same as Fig. 6), whereas L2 consists of three subgroups (Group 3: L2bα, Group 4: L2bβ, Group 5: L2a; same as Fig. 5). A, Average maps (aligned to soma, white circle) of connection probability for excitatory connections among five groups of cells. Connection probability is encoded according to the pseudocolor scale. White horizontal lines indicate averaged laminar borders and are 100 μm long. B, Average maps (aligned to soma, white circle) of connection probability for inhibitory connections among five groups of cells. Connection probability is encoded according to the pseudocolor scale. White horizontal lines indicate averaged laminar borders and are 100 μm long. C, Boxplot of relative cell positions of Group 1 (green), Group 2 (light blue), Group 3 (purple), Group 4 (red), and Group 5 (yellow) cells within L2/3. The locations of L2a cells are close to the upper boundary of L2/3 and the locations of L3b and L3a cells are close to the lower boundary. The cell location in Groups 3 and 4 are in between. *p < 0.05, **p < 0.01. The p values from multiple-comparison tests are as follows: L3b vs L3a: p = 0.95; L3b vs L2bα: p = 0.21; L3b vs L2bβ: p = 0.63; L3b vs L2a: p = 0.02; L3a vs L2bα: p = 0.27; L3a vs L2bβ: p = 0.85; L3a vs L2a: p = 0.012; L2bα vs L2bβ: p = 0.78; L2bα vs L2a: p = 0.66; L2bβ vs L2a: p = 9.68 × 10−2. D, Layer-specific fractional excitatory (D1) and inhibitory (D2) charge of L3b (green), L3a (orange), L2bα (blue), L2bβ (light blue) and L2a (red) cells. Data are mean charge ± SEM. Comparing between each two groups the excitation originating from L2/3 or L4 all shows significance. The main input to L3b cells comes from L4, whereas the main input to L2a is within L2/3. L3a has most input both coming from L2/3 and L4. L2bα has input primarily coming from L2/3 and L5. The p values from Multi-comparison test are as follows: L2/3: L3b vs L3a: p = 2.34 × 10−5; L3b vs L2bα: p = 9.92 × 10−9; L3b vs L2bβ: p = 9.92 × 10−9; L3b vs L2a: p = 9.92 × 10−9; L3a vs L2bα: p = 9.92 × 10−9; L3a vs L2bβ: p = 9.79 × 10−8; L3a vs L2a: p = 9.92 × 10−9; L2bα vs L2bβ: p = 6.62 × 10−3; L2bα vs L2a: p = 1.06 × 10−8; L2bβ vs L2a: p = 9.92 × 10−9. L4: L3b vs L3a: p = 1.04 × 10−8; L3b vs L2bα: p = 9.92 × 10−9; L3b vs L2bβ: p = 9.92 × 10−9; L3b vs L2a: p = 9.92 × 10−9; L3a vs L2bα: p = 9.92 × 10−9; L3a vs L2bβ: p = 3.39 × 10−8; L3a vs L2a: p = 9.92 × 10−9; L2bα vs L2bβ: p = 4.39 × 10−6; L2bα vs L2a: p = 3.91 × 10−2; L2bβ vs L2a: p = 1.04 × 10−8. L5: L3b vs L3a: p = 1; L3b vs L2bα: p = 0.96; L3b vs L2bβ: p = 0.21; L3b vs L2a: p = 3.41 × 10−2; L3a vs L2bα: p = 0.97; L3a vs L2bβ: p = 7.83 × 10−3; L3a vs L2a: p = 7.83 × 10−4; L2bα vs L2bβ: p = 3.44 × 10−3; L2bα vs L2a: p = 3.62 × 10−4; L2bβ vs L2a: p = 0.66. L6: L3b vs L3a: p = 0.65; L3b vs L2bα: p = 0.82; L3b vs L2bβ: p = 5.2 × 10−2; L3b vs L2a: p = 1; L3a vs L2bα: p = 1; L3a vs L2bβ: p = 0.25; L3a vs L2a: p = 0.51; L2bα vs L2bβ: p = 0.21; L2bα vs L2a: p = 0.74; L2bβ vs L2a: p = 0.01. Inhibition also shows differences among the groups. L2a has most input from L2/3 and little originating from other layers compared with L3a, L2bα, and L2bβ; The p values from multiple-comparison tests of inhibition are as follows: L2/3: L3b vs L3a: p = 0.11; L3b vs L2bα: p = 0.25; L3b vs L2bβ: p = 0.53; L3b vs L2a: p = 0.81; L3a vs L2bα: p = 1; L3a vs L2bβ: p = 0.66; L3a vs L2a: p = 3.98 × 10−4; L2bα vs L2bβ: p = 0.93; L2bα vs L2a: p = 3.50 × 10−3; L2bβ vs L2a: p = 0.012. L4: L3b vs L3a: p = 6.43 × 10−2; L3b vs L2bα: p = 9.92 × 10−9; L3b vs L2bβ: p = 9.92 × 10−9; L3b vs L2a: p = 9.92 × 10−9; L3a vs L2bα: p = 0.61; L3a vs L2bβ: p = 0.58; L3a vs L2a: p = 8.77 × 10−3; L2bα vs L2bβ: p = 1; L2bα vs L2a: p = 0.35; L2bβ vs L2a: p = 0.17. L5: L3b vs L3a: p = 0.72; L3b vs L2bα: p = 0.28; L3b vs L2bβ: p = 0.92; L3b vs L2a: p = 0.58; L3a vs L2bα: p = 0.80; L3a vs L2bβ: p = 0.97; L3a vs L2a: p = 8.61 × 10−3; L2bα vs L2bβ: p = 0.44; L2bα vs L2a: p = 9.16 × 10−4; L2bβ vs L2a: p = 0.034. L6: L3b vs L3a: p = 0.93; L3b vs L2bα: p = 1; L3b vs L2bβ: p = 1; L3b vs L2a: p = 0.88; L3a vs L2bα: p = 0.86; L3a vs L2bβ: p = 0.57; L3a vs L2a: p = 0.20; L2bα vs L2bβ: p = 1; L2bα vs L2a: p = 0.76; L2bβ vs L2a: p = 0.87. E, Barplot of the distance of 80% of input to each L2/3 cell originating from L2/3, L4, L5, and L6. The p values from multiple-comparison test are as follows: L2/3: L3b vs L3a: p = 4.59 × 10−3; L3b vs L2bα: p = 9.62 × 10−3; L3b vs L2bβ: p = 7.53 × 10−3; L3b vs L2a: p = 8.32 × 10−2; L3a vs L2bα: p = 1; L3a vs L2bβ: p = 1; L3a vs L2a: p = 0.88; L2bα vs L2bβ: p = 1; L2bα vs L2a: p = 0.92; L2bβ vs L2a: p = 0.95. L4: L3b vs L3a: p = 0.38; L3b vs L2bα: p = 1; L3b vs L2bβ: p = 0.93; L3b vs L2a: p = 0.48; L3a vs L2bα: p = 6.24 × 10−2; L3a vs L2bβ: p = 0.56; L3a vs L2a: p = 5.06 × 10−4; L2bα vs L2bβ: p = 0.63; L2bα vs L2a: p = 0.44; L2bβ vs L2a: p = 0.022. L5: L3b vs L3a: p = 0.63; L3b vs L2bα: p = 0.99; L3b vs L2bβ: p = 0.67; L3b vs L2a: p = 0.49; L3a vs L2bα: p = 0.77; L3a vs L2bβ: p = 1; L3a vs L2a: p = 2.91 × 10−3; L2bα vs L2bβ: p = 0.81; L2bα vs L2a: p = 0.95; L2bβ vs L2a: p = 3.43 × 10−3. L6: L3b vs L3a: p = 9.89 × 10−3; L3b vs L2bα: p = 0.29; L3b vs L2bβ: p = 0.24; L3b vs L2a: p = 1; L3a vs L2bα: p = 0.43; L3a vs L2bβ: p = 0.30; L3a vs L2a: p = 3.90 × 10−3; L2bα vs L2bβ: p = 1; L2bα vs L2a: p = 0.29; L2bβ vs L2a: p = 0.22. F, Boxplot of the direct activation area of the cell groups. L3a cells have bigger direct activation area than group L2bα and L2a cells (L3a vs L2bα: p = 0.037; L3a vs L2a: p = 0.011). There is no difference between the other groups (L3b vs L3a: p = 0.27; L3b vs L2bα: p = 0.99; L3b vs L2bβ: p = 0.80; L3b vs L2a: p = 0.95; L3a vs L2bβ: p = 0.67; L2bα vs L2bβ: p = 0.41; L2bα vs L2a: p = 0.97). Triple asterisk indicates a higher significance level.
Although the hierarchical clustering suggested that further subdivisions are possible based on relative laminar balances of excitatory and inhibitory inputs, the small number of cells in each of the finely subdivided groups does not allow us to reliably calculate spatial probability maps. One of the main differences between L2 and L3 and the distinct sublamina was both the cell position within L4 and the amount of L4 input. Plotting the relative amount of L4 input as a function of relative cell position showed that cell position and the amount of L4 input covaried (Fig. 8A). Thus, these results show that L2/3 of A1 shows functional and spatial sublamination based on the relative amount of L4 input.
Figure 8.

Amount of L4 inputs varies by position in L2/3. A, Scatter plot of percentage of L4 excitatory input vs cell position. The cells that receive more L4 excitatory input tend to be closer to L2/3 lower boundary. B, Graphic summary for different cell types based on the functional excitatory and inhibitory connections. Cells could be approximately divided into two big groups: the one group named L2 are close to the upper boundary of L2/3 and the cells in this group receive primary input from within L2/3. The other group named L3 is close to the lower boundary of L2/3. The cells in this group receive more input from L4 and L5. Then we further divide the L2 group into L2a and L2b. L2a cells almost only receive input from within L2/3. L2b cells also receive input originating from L4 and L5/6, even though the main input for L2b still comes from L2/3. At the end, we further divide L3 cells into L3a and L3b. L3b cells are much closer to L2/3 lower boundary. The primary input to L3b cells come from L4 not L2/3 and it is approximately double the amount of input originating from L4 to L3a cells.
Thus, together our clustering results suggest that L2/3 in mouse A1 can be functionally subdivided into at least five distinct cell classes based on translaminar input patterns (Fig. 8B).
In vivo two-photon imaging shows sublaminar differences in bandwidth but not tuning heterogeneity within L2/3
So far, we identified sublaminar differences in the laminar and columnar circuits innervating L2/3 cells in a slice preparation. Since these circuits transmit sensory information, we hypothesized that sound-evoked response features in the sublaminae of L2/3 in vivo should also be different. Prior laminar recordings in cat auditory cortex have shown that L2 cells integrate over a larger frequency range than L3 cells (Atencio and Schreiner, 2010). In contrast, recordings from mouse have suggested that L2 cells are more strongly driven by tones than L3 cells (Oviedo et al., 2010). To reconcile these results, we performed in vivo two-photon imaging experiments in A1 of awake mice (n = 8 mice; Fig. 9A) and imaged at depths ranging from 147 to 244 μm. We injected the AAV-mRuby2-gCaMP6s (Rose et al., 2016) vector encoding the sensitive, genetically-encoded calcium indicator GCaMP6 (Chen et al., 2013) into A1 and installed a chronic window (Fig. 9B,C). To identify A1 and differentiate major subdivisions of the auditory cortex, we imaged sound-evoked activity through wide-field imaging of evoked calcium signals reported by GCaMP6s. Amplitude-modulated tones of varying frequencies and intensity combination caused short-latency activation of auditory cortex (Fig. 9B) consistent with prior studies (Issa et al., 2014; Baba et al., 2016) and allowed us to identify large-scale tonotopic organization and delineate major subdivisions of the auditory cortex including A1, AAF, and A2 by the presence of strong responses to tonal stimuli and the presence (or absence) of tonotopic gradients.
Figure 9.

Imaging of sound evoked activity in L2/3 neurons of auditory cortex in awake mice reveals sublaminar differences in frequency integration. A, Image of awake mouse under two-photon microscope. B, Left, Wide-field epifluorescence image of cranial view showing expression of GCaMP6. Scale Bar, 1 mm. Right, Composite frequency map of sound-evoked activation areas for midlevel sound intensity (60 dB SPL). Tonotopic and non-tonotopic gradients indicate location of A1 and A2, respectively. C, Full resolution field-of-view captured with two-photon imaging showing cellular expression of AAV-mRuby2-GCamp6s in A1. Scale bar, 100 μm. D, Overview of frequency response areas for four responsive neurons in a single field-of-view. E, Evoked intracellular calcium responses for two pyramidal neurons in A1 to 9 different frequencies (column) and three sound levels (row). Each gray line indicates the response of the neurons to a single sound presentation; black line indicates the average across stimulus presentations. Five repeats per condition. Red vertical line indicates sound onset. F, Scatterplot showing percentage of responding neurons as a function of L2/3 depth (n = 8 FOV). Black line shows linear regression. p = 0.188. G, Scatterplot showing BW60 as a function of imaging plane depth (n = 576 neurons). Red line shows linear regression. p = 0.016. H, Boxplot showing BW60 for L2 and L3 neurons. p = 0.016, ANOVA. I, Scatterplot showing Q60 factor as a function of cortical depth (n = 576 neurons). Red line shows linear regression. p = 0.04. J, Scatterplot showing IQRBF100 as a function of depth (n = 576 neurons). Black line shows linear regression. p > 0.05. Triple asterisk indicates a higher significance level.
Only a fraction of individual A1 neurons in a given imaging field were responsive to sound stimuli and showed frequency selectivity (Fig. 9D,E). L2/3 neurons within an imaging field exhibited diverse tuning characteristics, with some neurons showing clear frequency-selective frequency-response-areas (FRAs), whereas others appeared less selective (Fig. 9D). Plotting the fraction of responsive cells as a function of depth showed no significant differences across imaging depths within L2/3 (Fig. 9F). We next calculated the bandwidth of the FRA at midlevel (60 dB SPL) and found that bandwidth varied with L2/3 depth (Fig. 9G–I). Cells in superficial L2/3, i.e., L2, showed broader tuning than cells in L3 (Fig. 9G–I) consistent with studies in cat A1 (Atencio and Schreiner, 2010).
Prior in vivo two-photon imaging in anesthetized animals using synthetic dyes (Bandyopadhyay et al., 2010; Rothschild et al., 2010; Winkowski and Kanold, 2013) and in vivo patch-clamp (Maor et al., 2016) revealed that neighboring L2/3 neurons show diverse frequency preference. In contrast, in vivo imaging of sparse populations in acute, awake preparations with the moderately sensitive indicator GCaMP3 indicated a higher degree of similarly-responding neurons locally (Issa et al., 2014). To quantify the diversity in local frequency selectivity, we calculated the IQR of preferred frequency (BF, in octaves) of neighboring neurons within 100 μm of the central neuron (Winkowski and Kanold, 2013). Given the frequency range of our stimuli, IQRBF100 could range from 0, indicating no variability, to 5, indicating a broad distribution of BF locally. We found that within a local spatial area around each neuron (radius of 100 μm) neighboring L2/3 neurons showed a range of tuning characteristics and that IQRBF100 did not vary with depth (Fig. 9J). Together these results show that within mouse A1 there is a functional sublaminar organization with more superficial cells responding to a broader range of sounds than cells deeper in L2/3.
Discussion
We here used LSPS to study the spatial organization of excitatory and inhibitory intracortical inputs to L2/3 neurons in A1. Using hierarchical clustering we identify multiple parallel circuits to layer 2/3 neurons in A1. Cells divide into two major groups, corresponding to L2 and L3, based on their ascending input from L4. Each group can be further subdivided into functional subgroups, L2a and L2b, L3a and L3b, respectively, and these subgroups are located at different depths within L2/3. Thus, even though the supragranular layers of mouse auditory cortex do not show obviously cytoarchitectonic sublaminar organization, a functional analysis of connectivity shows clear laminar differences. Together, these results show that the supragranular layer of mouse auditory cortex contains multiple cell populations defined by distinct translaminar integration patterns. While prior LSPS studies divided L2/3 into 2 groups based on mapping excitatory inputs and laminar grouping (Oviedo et al., 2010), here we used an unbiased clustering approach on excitatory as well as inhibitory inputs in a large number of neurons to detect a further sublamination. Moreover, in contrast to prior results showing stronger sound-evoked responses in L2 (Oviedo et al., 2010), our in vivo imaging shows that cells in all sublayers respond to sound and that the frequency integration varies with laminar position.
Under our recording conditions LSPS has a spatial resolution of ∼80–100 μm (Meng et al., 2015), thus our technique cannot resolve differences on this scale. Nevertheless, we identify sublaminar differences in connectivity. We identified five distinct sublaminar cell groups, but our data suggest the presence of even more groups, potentially based on their integration across the tonotopic axis (e.g., rostral vs caudal).
We find that on the largest scale cells fall into classes based on the presence or absence of L4 input, and that cells contained in these classes are located either deeper or more superficially. This grouping is consistent with prior studies identifying differences in the L4 input to L2/3 by grouping cells into L2 or L3 based on recording depth (Oviedo et al., 2010). We were here able to further identify subgroups within these larger classes and these subgroups varied in the spatial integration of L4 input along the tonotopic axis.
We found a population of cells (L3b) in our sample that received few inputs from L2/3 and did not receive inputs from deeper cortical layers, but were predominantly innervated by L4. Thus, these cells would be expected to show similar frequency tuning and representation as the underlying L4 neurons (Fig. 8B). Given that the topographic maps of frequency are heterogeneous in L2/3 but still aligned with L4 (Winkowski and Kanold, 2013), these cells could “anchor” the topographic map in L2/3. In contrast, the other L2/3 neurons, which receive a smaller fraction of L4 input, would be dominated by intercortical inputs which can show diverse tuning (Chen et al., 2011). Moreover, the dominant L4 input to L3b cells suggests that these cells will be strongly driven by auditory stimuli. The presence of these distinct populations of L2/3 cells could reconcile seemingly conflicting studies of the frequency organization of A1. Multiple studies using in vivo two-photon Ca2+ imaging with sensitive indicators in densely labeled L2/3 detected heterogeneous frequency organization of L2/3 (Bandyopadhyay et al., 2010; Rothschild et al., 2010; Winkowski and Kanold, 2013; Kanold et al., 2014), which were corroborated by sensitive in vivo patch-clamp recordings (Maor et al., 2016). In contrast, imaging sparse populations with less sensitive genetically encoded indicators (GCaMP3) detected a more homogeneous frequency organization in L2/3 (Issa et al., 2014). Our results here suggest that these less sensitive indicators might be biased toward L3b cells which are expected to respond strongly to sound.
Prior LSPS studies of A1 using only excitatory inputs revealed that L2/3 neurons receive biased inputs from within L2/3 and from L5/L6 (Oviedo et al., 2010). A fraction of cells in our sample also received inputs from L5/6 (L3a, L2b) but we only detect a spatial bias in a subset of L2/3 neurons. Prior studies only detected these biases in a sample of cells at the lower margin of L2/3 (Oviedo et al., 2010), whereas the deepest cells (L3b) in our sample received only few L5/6 inputs. Thus, these cells likely represent our L3a cells. Because L5A and L6 neurons receive inputs from a wide range of frequencies (Zhou et al., 2010; Sun et al., 2013) the feedback input to L3a would be expected to broaden or change the tuning preferences of the L3a neuron from the frequency they inherited from L4. Across our recorded population, the spatial location of the L5/6 input with respect to the tonotopic position of the L2/3 neuron can be on the high- or low-frequency side, respectively (rostral or caudal; Fig. 2) suggesting that the frequency center of the wideband input can differ. A peculiar feature of the spatial maps of excitation and inhibition was that areas that gave rise to excitatory input frequently did not overlap with areas that gave rise to inhibitory inputs (Fig. 2). This patchy non-overlapping translaminar excitatory and inhibitory connectivity suggests that excitation and inhibition might have different frequency preferences. This is reminiscent of the fragmented receptive fields that can be found in A1 when probed with tones or more complex stimuli (Schreiner et al., 2000; Depireux et al., 2001), and such receptive field topology could give rise to sensitivity to changing frequency composition such as frequency-modulated sweeps (Poon and Yu, 2000).
Our in vivo imaging reveals that on the basis of single cells, neurons in different sublaminae of L2/3 show different amount of across-frequency integration, although the general heterogeneity of best frequency is similar in each sublayer. This is consistent with in vivo recordings in cat, which have suggested laminar differences in spectrotemporal receptive fields and correlation patterns (Atencio et al., 2009; Atencio and Schreiner, 2010). The increased frequency integration in more superficial layers is consistent with our in vitro results showing a wider connectivity across the tonotopic axis within L2/3 and a higher fraction of inputs from supragranular layers versus L4 in these cells.
We here show a change of intracortical connectivity to L2/3 neurons with depth in L2/3. A depth-dependent change in connectivity in L2/3 has also been observed in primary somatosensory cortex (S1; Shepherd and Svoboda, 2005; Bureau et al., 2006; Staiger et al., 2015). These studies suggested that deeper L2/3 cells received the strongest L4 input consistent with our results. Moreover, more superficial L2/3 cells in S1 were found to receive more L5a inputs, consistent with our findings of L5 input in L3a and L2b. These similarities indicate that subcircuits within L2/3 of A1 and S1 might serve similar functions with respect to the integration of lemniscal and non-lemniscal information. The intermingled nature of the middle groups of L2/3 cells suggest that within one sublamina multiple types of cells can be present potentially differentially integrating lemniscal and non-lemniscal inputs.
Together our results show that despite the lack of obvious cytoarchitectonic differences, supragranular mouse A1 shows a functional subdivision based on the laminar input pattern reminiscent of anatomical subdivisions in carnivore. Thus, multiple parallel circuits exist in supragranular mouse A1.
Footnotes
This work was supported by NIH R01EY022720 (H.K.L. and P.O.K.), NIH R01DC009607 (P.O.K.), NIH U01NS090569 (P.O.K.), and NIH R01GM056481 (J.P.Y.K.). We thank Dr. Eike Budinger for comments on the paper.
The authors declare no competing financial interests.
References
- Atencio CA, Schreiner CE (2010) Columnar connectivity and laminar processing in cat primary auditory cortex. PLoS One 5:e9521. 10.1371/journal.pone.0009521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atencio CA, Sharpee TO, Schreiner CE (2009) Hierarchical computation in the canonical auditory cortical circuit. Proc Natl Acad Sci U S A 106:21894–21899. 10.1073/pnas.0908383106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba H, Tsukano H, Hishida R, Takahashi K, Horii A, Takahashi S, Shibuki K (2016) Auditory cortical field coding long-lasting tonal offsets in mice. Sci Rep 6:34421. 10.1038/srep34421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Shamma SA, Kanold PO (2010) Dichotomy of functional organization in the mouse auditory cortex. Nat Neurosci 13:361–368. 10.1038/nn.2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I, von Saint Paul F, Svoboda K (2006) Interdigitated paralemniscal and lemniscal pathways in the mouse barrel cortex. PLoS Biol 4:e382. 10.1371/journal.pbio.0040382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300. 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Leischner U, Rochefort NL, Nelken I, Konnerth A (2011) Functional mapping of single spines in cortical neurons in vivo. Nature 475:501–505. 10.1038/nature10193 [DOI] [PubMed] [Google Scholar]
- Code RA, Winer JA (1985) Commissural neurons in layer III of cat primary auditory cortex (AI): pyramidal and non-pyramidal cell input. J Comp Neurol 242:485–510. 10.1002/cne.902420404 [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Rose HJ, Metherate R (2002) Auditory thalamocortical synaptic transmission in vitro. J Neurophysiol 87:361–384. 10.1152/jn.00549.2001 [DOI] [PubMed] [Google Scholar]
- Depireux DA, Simon JZ, Klein DJ, Shamma SA (2001) Spectro-temporal response field characterization with dynamic ripples in ferret primary auditory cortex. J Neurophysiol 85:1220–1234. [DOI] [PubMed] [Google Scholar]
- Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, Andermann ML (2014) Removable cranial windows for long-term imaging in awake mice. Nat Protoc 9:2515–2538. 10.1038/nprot.2014.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZV, Hires SA, Li N, O'Connor DH, Komiyama T, Ophir E, Huber D, Bonardi C, Morandell K, Gutnisky D, Peron S, Xu NL, Cox J, Svoboda K (2014) Procedures for behavioral experiments in head-fixed mice. PLoS One 9:e88678. 10.1371/journal.pone.0088678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JB, Haeffele BD, Agarwal A, Bergles DE, Young ED, Yue DT (2014) Multiscale optical Ca2+ imaging of tonal organization in mouse auditory cortex. Neuron 83:944–959. 10.1016/j.neuron.2014.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanold PO, Nelken I, Polley DB (2014) Local versus global scales of organization in auditory cortex. Trends Neurosci 37:502–510. 10.1016/j.tins.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao JPY. (2006) Caged molecules: principles and practical considerations. In: Current protocols in neuroscience (Gerfen C, Holmes A, Rogawski M, Sibley D, Skolnick P, Wray S, eds). Hoboken, NJ: Wiley. [DOI] [PubMed] [Google Scholar]
- Maor I, Shalev A, Mizrahi A (2016) Distinct spatiotemporal response properties of excitatory versus inhibitory neurons in the mouse auditory cortex. Cereb Cortex 26:4242–4252. 10.1093/cercor/bhw266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Kao JP, Kanold PO (2014) Differential signaling to subplate neurons by spatially specific silent synapses in developing auditory cortex. J Neurosci 34:8855–8864. 10.1523/JNEUROSCI.0233-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Kao JP, Lee HK, Kanold PO (2015) Visual deprivation causes refinement of intracortical circuits in the auditory cortex. Cell Rep 12:955–964. 10.1016/j.celrep.2015.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani A, Shimokouchi M, Itoh K, Nomura S, Kudo M, Mizuno N (1985) Morphology and laminar organization of electrophysiologically identified neurons in the primary auditory cortex in the cat. J Comp Neurol 235:430–447. 10.1002/cne.902350403 [DOI] [PubMed] [Google Scholar]
- Muralidharan S, Dirda ND, Katz EJ, Tang CM, Bandyopadhyay S, Kanold PO, Kao JP (2016) Ncm, a photolabile group for preparation of caged molecules: synthesis and biological application. PLoS One 11:e0163937. 10.1371/journal.pone.0163937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojima H, Honda CN, Jones EG (1991) Patterns of axon collateralization of identified supragranular pyramidal neurons in the cat auditory cortex. Cereb Cortex 1:80–94. 10.1093/cercor/1.1.80 [DOI] [PubMed] [Google Scholar]
- Oviedo HV, Bureau I, Svoboda K, Zador AM (2010) The functional asymmetry of auditory cortex is reflected in the organization of local cortical circuits. Nat Neurosci 13:1413–1420. 10.1038/nn.2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon PW, Yu PP (2000) Spectro-temporal receptive fields of midbrain auditory neurons in the rat obtained with frequency modulated stimulation. Neurosci Lett 289:9–12. 10.1016/S0304-3940(00)01251-9 [DOI] [PubMed] [Google Scholar]
- Ratzlaff EH, Grinvald A (1991) A tandem-lens epifluorescence macroscope: hundred-fold brightness advantage for wide-field imaging. J Neurosci Methods 36:127–137. 10.1016/0165-0270(91)90038-2 [DOI] [PubMed] [Google Scholar]
- Rose T, Jaepel J, Hübener M, Bonhoeffer T (2016) Cell-specific restoration of stimulus preference after monocular deprivation in the visual cortex. Science 352:1319–1322. 10.1126/science.aad3358 [DOI] [PubMed] [Google Scholar]
- Rothschild G, Nelken I, Mizrahi A (2010) Functional organization and population dynamics in the mouse primary auditory cortex. Nat Neurosci 13:353–360. 10.1038/nn.2484 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner CE, Read HL, Sutter ML (2000) Modular organization of frequency integration in primary auditory cortex. Annu Rev Neurosci 23:501–529. 10.1146/annurev.neuro.23.1.501 [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Svoboda K (2005) Laminar and columnar organization of ascending excitatory projections to layer 2/3 pyramidal neurons in rat barrel cortex. J Neurosci 25:5670–5679. 10.1523/JNEUROSCI.1173-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM, Pologruto TA, Svoboda K (2003) Circuit analysis of experience-dependent plasticity in the developing rat barrel cortex. Neuron 38:277–289. 10.1016/S0896-6273(03)00152-1 [DOI] [PubMed] [Google Scholar]
- Sorensen SA, Bernard A, Menon V, Royall JJ, Glattfelder KJ, Desta T, Hirokawa K, Mortrud M, Miller JA, Zeng H, Hohmann JG, Jones AR, Lein ES (2015) Correlated gene expression and target specificity demonstrate excitatory projection neuron diversity. Cereb Cortex 25:433–449. 10.1093/cercor/bht243 [DOI] [PubMed] [Google Scholar]
- Staiger JF, Bojak I, Miceli S, Schubert D (2015) A gradual depth-dependent change in connectivity features of supragranular pyramidal cells in rat barrel cortex. Brain Struct Funct 220:1317–1337. 10.1007/s00429-014-0726-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YJ, Kim YJ, Ibrahim LA, Tao HW, Zhang LI (2013) Synaptic mechanisms underlying functional dichotomy between intrinsic-bursting and regular-spiking neurons in auditory cortical layer 5. J Neurosci 33:5326–5339. 10.1523/JNEUROSCI.4810-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter BA, O'Connor T, Iyer V, Petreanu LT, Hooks BM, Kiritani T, Svoboda K, Shepherd GM (2010) Ephus: multipurpose data acquisition software for neuroscience experiments. Front Neural Circuits 4:100. 10.3389/fncir.2010.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao C, Zhang G, Zhou C, Wang L, Yan S, Tao HW, Zhang LI, Zhou Y, Xiong Y (2017) Diversity in excitation-inhibition mismatch underlies local functional heterogeneity in the rat auditory cortex. Cell Rep 19:521–531. 10.1016/j.celrep.2017.03.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE, Unser M (1998) A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process 7:27–41. 10.1109/83.650848 [DOI] [PubMed] [Google Scholar]
- Watkins PV, Kao JP, Kanold PO (2014) Spatial pattern of intra-laminar connectivity in supragranular mouse auditory cortex. Front Neural Circuits 8:15. 10.3389/fncir.2014.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer JA. (1984) The pyramidal neurons in layer III of cat primary auditory cortex (AI). J Comp Neurol 229:476–496. 10.1002/cne.902290404 [DOI] [PubMed] [Google Scholar]
- Winer JA. (1985) Structure of layer II in cat primary auditory cortex (AI). J Comp Neurol 238:10–37. 10.1002/cne.902380103 [DOI] [PubMed] [Google Scholar]
- Winguth SD, Winer JA (1986) Corticocortical connections of cat primary auditory cortex (AI): laminar organization and identification of supragranular neurons projection to area AII. J Comp Neurol 248:36–56. 10.1002/cne.902480104 [DOI] [PubMed] [Google Scholar]
- Winkowski DE, Kanold PO (2013) Laminar transformation of frequency organization in auditory cortex. J Neurosci 33:1498–1508. 10.1523/JNEUROSCI.3101-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Kao JP, Kanold PO (2009) Functional excitatory microcircuits in neonatal cortex connect thalamus and layer 4. J Neurosci 29:15479–15488. 10.1523/JNEUROSCI.4471-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu BH, Wu GK, Kim YJ, Xiao Z, Tao HW, Zhang LI (2010) Preceding inhibition silences layer 6 neurons in auditory cortex. Neuron 65:706–717. 10.1016/j.neuron.2010.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]