

Abstract
Purpose
Endoxifen is a tamoxifen metabolite with potent antiestrogenic activity.
Patients and Methods
We performed a phase I study of oral Z-endoxifen to determine its toxicities, maximum tolerated dose (MTD), pharmacokinetics, and clinical activity. Eligibility included endocrine-refractory, estrogen receptor–positive metastatic breast cancer. An accelerated titration schedule was applied until moderate or dose-limiting toxicity occurred, followed by a 3+3 design and expansion at 40, 80, and 100 mg per day. Tumor DNA from serum (circulating cell free [cf); all patients] and biopsies [160 mg/day and expansion]) was sequenced.
Results
Of 41 enrolled patients, 38 were evaluable for MTD determination. Prior endocrine regimens during which progression occurred included aromatase inhibitor (n = 36), fulvestrant (n = 21), and tamoxifen (n = 15). Patients received endoxifen once daily at seven dose levels (20 to 160 mg). Dose escalation ceased at 160 mg per day given lack of MTD and endoxifen concentrations > 1,900 ng/mL. Endoxifen clearance was unaffected by CYP2D6 genotype. One patient (60 mg) had cycle 1 dose-limiting toxicity (pulmonary embolus). Overall clinical benefit rate (stable > 6 months [n = 7] or partial response by RECIST criteria [n = 3]) was 26.3% (95% CI, 13.4% to 43.1%) including prior tamoxifen progression (n = 3). cfDNA mutations were observed in 13 patients (PIK3CA [n = 8], ESR1 [n = 5], TP53 [n = 4], and AKT [n = 1]) with shorter progression-free survival (v those without cfDNA mutations; median, 61 v 132 days; log-rank P = .046). Clinical benefit was observed in those with ESR1 amplification (tumor; 80 mg/day) and ESR1 mutation (cfDNA; 160 mg/day). Comparing tumor biopsies and cfDNA, some mutations (PIK3CA, TP53, and AKT) were undetected by cfDNA, whereas cfDNA mutations (ESR1, TP53, and AKT) were undetected by biopsy.
Conclusion
In endocrine-refractory metastatic breast cancer, Z-endoxifen provides substantial drug exposure unaffected by CYP2D6 metabolism, acceptable toxicity, and promising antitumor activity.
INTRODUCTION
Tamoxifen is a weak antiestrogen that undergoes extensive biotransformation in humans to metabolites 4-hydroxytamoxifen (4HT) and 4-hydroxy N-desmethyl tamoxifen (endoxifen), which have greater antiestrogenic potency than the parent drug.1-5 In humans, 4HT concentrations are low, typically < 5 ng/mL,4,6 whereas endoxifen plasma concentrations are up to 10-fold higher than 4HT, exhibiting substantial variability.4,6,7 CYP2D6 is the main enzyme responsible for the conversion of the primary tamoxifen metabolite, N-desmetyltamoxifen, to endoxifen.4 Patients with low CYP2D6 enzyme activity, as a result of CYP2D6 genetic polymorphisms or the coadministration of potent CYP2D6 inhibitors, exhibit significantly lower endoxifen concentrations when treated with tamoxifen.4
On the basis of early reports demonstrating an association between tamoxifen efficacy and either reduced CYP2D6 metabolism8 or low endoxifen concentrations,9 we hypothesized that oral administration of Z-endoxifen could achieve not only clinically relevant endoxifen concentrations but also antitumor activity possibly superior to that of tamoxifen. Therefore, after confirmation of the substantial bioavailability of Z-endoxifen in mice,10 we conducted a phase I study of Z-endoxifen to determine its toxicity profile, maximum tolerated dose (MTD), pharmacokinetics, pharmacogenetics, and clinical activity in women with estrogen receptor (ER) –positive, hormone-refractory metastatic breast cancer (MBC).
PATIENTS AND METHODS
Formulation
Z-endoxifen hydrochloride was supplied by the Pharmaceutical Management Branch, National Cancer Institute, as 20- and 40-mg capsules.
Eligibility and Enrollment
This study enrolled women age ≥ 18 years with histologically confirmed ER-positive (> 1% nuclear staining) MBC or locally recurrent breast cancer that was either measurable or evaluable. Additional eligibility criteria included: Eastern Cooperative Oncology Group performance status of 0 to 1, adequate blood chemistries (absolute neutrophil count ≥ 1,000/μL; platelet count ≥ 75,000/μL; total bilirubin ≤ 1.5 × institutional upper limit of normal [ULN]; total AST and ALT ≤ 2.5 × ULN [5 × ULN if liver function test elevations resulted from live metastases]; and creatinine ≤ 1.5 × ULN).
Prior Endocrine Therapy
Patients were required to have experienced progression while receiving either tamoxifen (if premenopausal) or an aromatase inhibitor (AI; if postmenopausal) in either the metastatic or adjuvant setting. An unlimited number of endocrine therapy regimens were allowed, including everolimus-based regimens.
Prior Chemotherapy
An unlimited number of prior chemotherapy regimens were allowed in the dose-escalation cohort, with at least one prior chemotherapy regimen required in the adjuvant and/or metastatic setting. Up to two prior chemotherapy regimens were allowed during the expansion phase. Women with human epidermal growth factor receptor 2–positive disease must have experienced progression during at least one prior anti–human epidermal growth factor receptor 2–directed regimen.
Exclusion criteria included: uncontrolled brain metastases or tumors involving the spinal cord or heart; systemic anticancer therapy or radiation therapy within 3 weeks before registration; prior endoxifen treatment, clinically symptomatic cataracts requiring imminent surgery, or bisphosphonate or denosumab use < 90 days before registration; active deep venous thrombosis and/or pulmonary embolus requiring anticoagulant therapy or history of coagulopathy; uncontrolled intercurrent illnesses; and other invasive malignancy diagnosed or recurring < 2 years before registration. Women who were pregnant or breastfeeding were not eligible.
Participating institutions obtained study approval from their institutional review boards and had filed assurances with the Department of Health and Human Services. Written informed consent was required for enrollment.
Study Treatment
Patients received 20, 40, 60, 80, 100, 120, or 160 mg of Z-endoxifen by mouth daily until disease progression, unacceptable adverse events, patient decision to withdraw from the study, or inability to continue treatment. Routine use of colony-stimulating factors was not allowed. Selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors were allowed for the alleviation of vasomotor or estrogen deficiency symptoms.
Management of Toxicity
Appendix (online only).
Patient Evaluations
Appendix.
Statistical Considerations
Dose-escalation phase.
An accelerated titration phase I clinical trial design was chosen as the means to determine a dose to recommend for testing in the phase II clinical trial setting (RP2D). The RP2D was defined as the highest dose tested where at most one of the six patients developed a dose-limiting toxicity (DLT) during the first cycle of treatment. DLT was defined as any of the following adverse events judged to be possibly, probably, or definitely related to Z-endoxifen: any grade ≥ 4 hematologic toxicity, any grade ≥ 3 nonhematologic toxicity, and any grade ≥ 2 toxicity that resulted in < 14 days of treatment during the first treatment cycle.
Dose escalation began with an accelerated phase where two patients were enrolled per dose level, starting at dose level 1. If one or both of these two patients developed a moderate toxicity (any grade ≥ 3 hematologic toxicity or any grade ≥ 2 nonhematologic toxicity except grade 2 vasomotor or estrogen-deficiency symptoms that were possibly, probably, or definitely related to Z-endoxifen during the first cycle of treatment), the accelerated phase ended. Otherwise, the next two patients registered were enrolled at the next dose level. If all the dose levels were exhausted without observation of a moderate toxicity, an additional four patients were to be treated at the highest dose level to establish it as the RP2D. If a moderate toxicity was observed at a given dose level (referred to as Dx), as many as four additional patients were to be enrolled at that dose level. If two or more of the six patients receiving Dx developed a DLT, dose escalation was stopped, and four patients were to be enrolled at the next-lower dose level to confirm it as the RP2D. If at most one of the six patients enrolled at dose-level Dx developed a DLT during the first cycle of treatment, the dose-escalation plan switched to that of a 3+3 phase I clinical trial. No intrapatient dose escalation was allowed.
Expansion phase.
On the basis of the pattern of toxicities observed, tumor response, and endoxifen steady-state concentrations (Csss), three dose levels were chosen for further exploration. Patients were randomly assigned to each of these dose levels using a stratified randomization procedure, with the stratification factors of dominant disease (visceral v other), prior everolimus-containing regimen (yes v no), and hormone resistance (primary v secondary).11 Patients enrolled in the 160 mg per day and expansion cohorts underwent pretreatment tumor biopsies.
Data lock occurred on March 5, 2017. Wilcoxon rank sum test was used to assess whether oral clearance differed with respect to CYP2D6 activity score (AS; ≥ 2.0 v ≤ 1.5; score determination summarized in Appendix Table A1, online only). The distribution of progression-free survival (PFS) times was estimated using the Kaplan-Meier method. The median PFS time was defined as the smallest observed PFS time for which the value of the Kaplan-Meier estimate of the PFS function was < 0.50. Log-rank test was used to assess whether PFS differed with respect to AS (≥ 2.0 v ≤ 1.5). Changes in lipid profile after one cycle of treatment were examined in terms of change in total, LDL, and HDL cholesterol.
RESULTS
Study Cohort
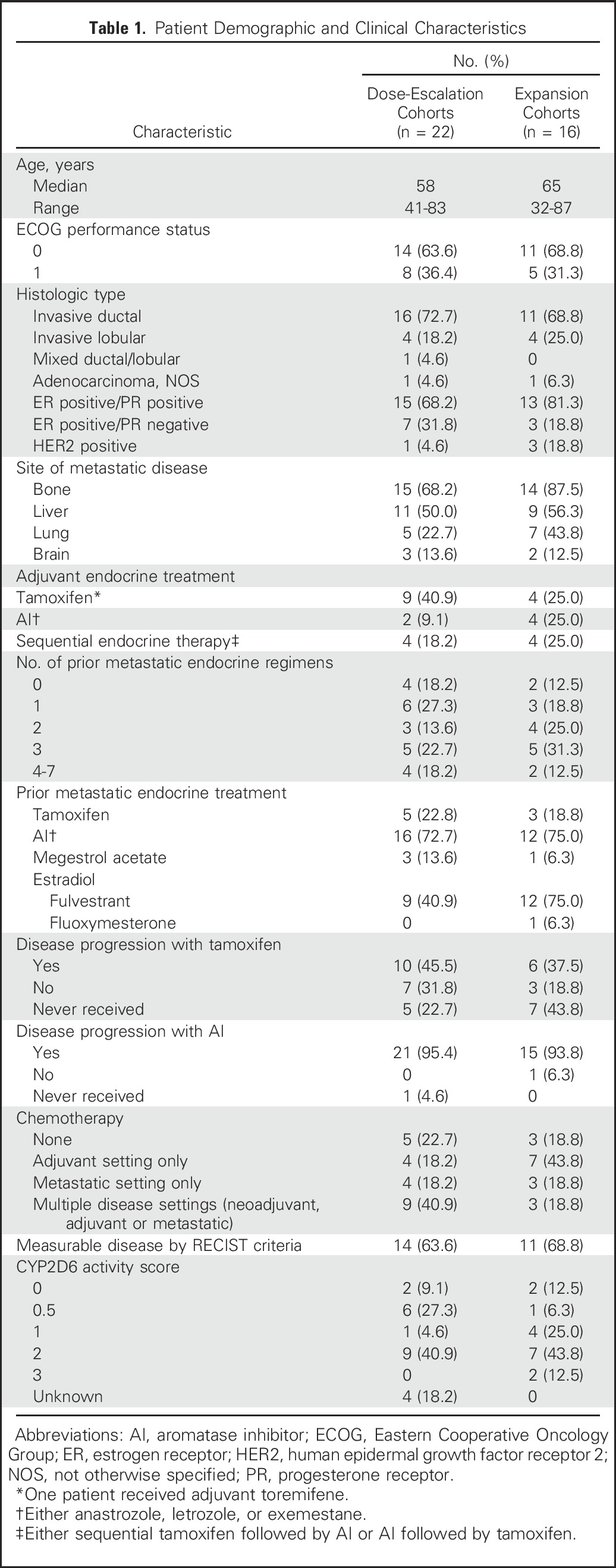
From March 25, 2011, to December 9, 2014, 41 women were enrolled. Three patients were not included in the MTD determination: one (20 mg) discontinued treatment after one 20-mg dose; one (100 mg) sought alternative treatment after 12 100-mg doses; and one (60 mg) skipped 1 week of treatment, restarted of her own volition at a lower dose, and then stopped altogether 1 week later. The demographic and tumor characteristics of the remaining 38 patients are listed in Table 1.
Table 1.
Patient Demographic and Clinical Characteristics
Treatment Course and Toxicities
No moderate toxicities or DLTs were observed among patients enrolled at either the 20- or 40-mg dose level. Accelerated dose escalation stopped at the 60-mg dose level after a grade 3 thromboembolic event (pulmonary embolus). None of the additional five patients at the 60-mg dose level nor any patients enrolled at the subsequent dose levels developed a DLT. Moreover, during the course of the study, only one other severe toxicity was reported (grade 4 hypertriglyceridemia) after five cycles of treatment at the 60-mg dose. No eye toxicity was observed. As such, all of the planned dose levels were exhausted without observation of an MTD. Table 2 lists the number of treatment cycles, moderate or severe toxicities reported, treatment response, PFS time, and overall survival time for each patient by dose level.
Table 2.
Summary of Treatment Cycles, Toxicities, Mutations, Variants, Biopsy Site, Treatment Response, PFS, and OS for Patients by Dose Level

Having exhausted all planned dose levels, the dose levels considered for expansion were based on the observations of substantial endoxifen pharmacokinetic exposure (> 900 ng/mL) without DLT at all dose levels > 80 mg per day, antitumor activity independent of dose level, and prior published data demonstrating that Z-endoxifen Csss achieved at 40 mg per day (500 ng/mL) were associated with in vitro inhibition of estrogen-induced proliferation in ER-positive cells with12 and without ESR1 mutations.5 Dose levels 40, 80, and 100 mg per day were further studied in the expansion cohorts. Sixteen patients were enrolled in the expansion phase, where six were randomly assigned to 40, five to 80, and five to 100 mg per day. None of these patients developed a severe adverse event.
Pharmacokinetics
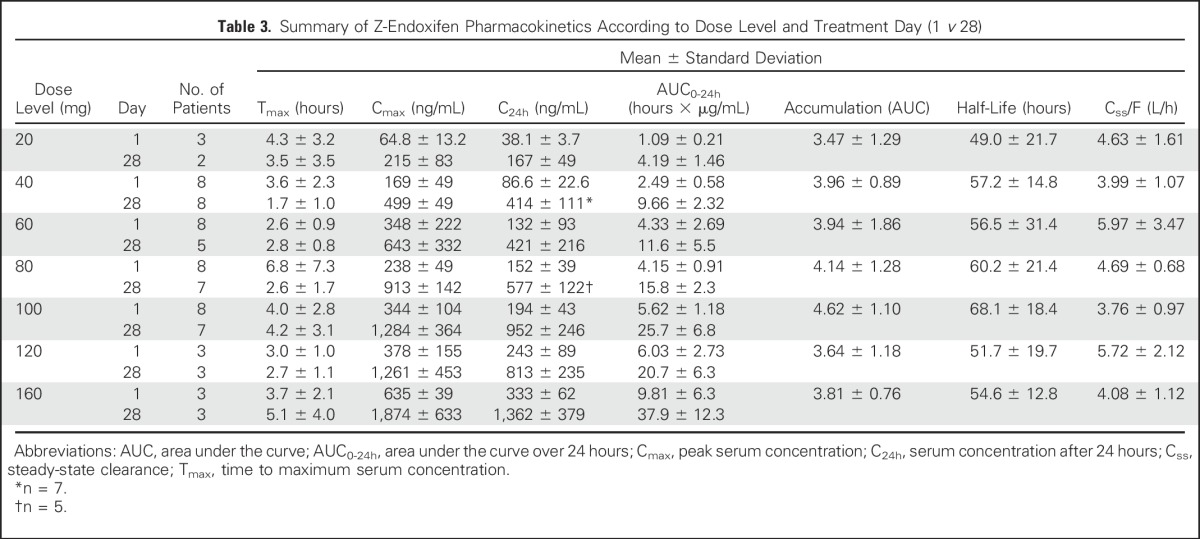
Endoxifen pharmacokinetics were determined in all 41 patients, and the results are summarized in Table 3 and illustrated graphically for patients treated with 40 or 100 mg per day in Appendix Figure A1 (online only). Peak endoxifen concentrations were reached 2 to 4 hours after the day-1 oral dose, and mean values of peak concentration, concentration after 24 hours, and area under the curve over 24 hours increased in proportion to dose (Table 3; Appendix Fig A2, online only). Css was achieved on day 7, with an approximate 3.5-fold accumulation observed on day 28. At the starting dose (20 mg/day) and highest dose level (160 mg/day), endoxifen Css values of 146 ng/mL (390 nM) and 1,950 ng/mL (5,200 nM), respectively, were achieved and maintained throughout the 28-day treatment. Oral clearance did not differ (Wilcoxon P = .3954) between those with a CYP2D6 AS ≥ 2.0 (median, 3.8; interquartile range [IQR], 3.3 to 4.7) and those with a CYP2D6 AS ≤ 1.5 (median, 4.7; IQR, 3.5 to 5.4). Endoxifen Css values remained unchanged after continuous dosing for 8 to 10 months. The mean apparent steady-state clearance was 6.2 L per hour and was not affected by dose increase (Appendix Fig A3, online only).
Table 3.
Summary of Z-Endoxifen Pharmacokinetics According to Dose Level and Treatment Day (1 v 28)
Antitumor Activity
Antitumor activity, consisting of confirmed partial responses or stable disease > 6 months, was observed at all but the 20- and 120-mg dose levels (Table 2). Among the 25 patients with measurable disease (14 enrolled during dose escalation), three had a partial response on two consecutive evaluations at least 8 weeks apart. Thus, the overall response rate was 12.0% (95% CI, 2.6% to 31.2%). Additionally, five of 25 patients with measurable disease and two of 13 patients with nonmeasurable disease exhibited stable disease for > 6 months. Thus, the clinical benefit rate was 26.3% (95% CI, 13.4% to 43.1%).
Tumor responses and prolonged antitumor activity were observed in patients with prior progression during multiple lines of endocrine therapy. Of the 36 patients with prior progression during treatment with AIs, clinical benefit was observed in those who experienced additional progression during tamoxifen and fulvestrant treatment. Furthermore, of the four patients with prior exemestane/everolimus treatment, three maintained either stable disease (n = 1) or a confirmed partial response (n = 2) lasting > 6 months. The maximum decrease in tumor size according to prior progression with tamoxifen (yes v no) is shown in Figure 1 and to prior exposure to fulvestrant (yes v no) in Appendix Figure A4 (online only). PFS times for all patients are presented in Figure 2 by prior exposure to tamoxifen. Clinical benefit (stable disease ≥ 6 months) was observed in 19% of patients (three of 16) who experienced progression during tamoxifen and 32% (seven of 22) who had no prior tamoxifen treatment or did not experience progression with adjuvant tamoxifen. Figure 3 illustrates the antitumor activity of endoxifen in a patient treated at the 160-mg dose who had previously experienced progression with tamoxifen, anastrozole, fulvestrant, and exemestane/everolimus.
Fig 1.

Maximum decrease in tumor size according to prior tamoxifen treatment. (A) prior progression while taking tamoxifen in the adjuvant or metastatic setting. (B) No prior tamoxifen or no progression while taking tamoxifen in the adjuvant setting.
Fig 2.

Progression-free survival (PFS) times for all patients according to prior tamoxifen exposure. Hashed lines indicate patients who were progression free at time of data lock. Dose level is provided for each patient (mg/day).
Fig 3.

Antitumor activity of Z-endoxifen in a patient with prior progression during four different lines of endocrine therapy, including adjuvant (tamoxifen) and metastatic (anastrozole, fulvestrant, and exemestane plus everolimus) settings: (A) Baseline before starting Z-endoxifen and (B) after 8 cycles of Z-Endoxifen; arrow shows tumor.
At the time of the data lock, two patients were alive without disease progression, 29 were alive with disease progression, and seven had died as a result of disease. The median PFS time was 110 days, with a 1-year PFS rate of 15% (95% CI, 6.2% to 31.4%).
Clinical Benefit According to CYP2D6 Genotype
CYP2D6 activity score was available for 34 patients. PFS among those with AS ≥ 2.0 did not differ from that of those with AS ≤ 1.5 (log-rank P = .8604). Within the subset of 24 patients with prior tamoxifen exposure, the median PFS time was 60 days (n = 11; IQR, 31 to 132 days) among those with AS ≥ 2.0 and 157 days (n = 12; IQR, 72 to 296 days) among those with AS ≤ 1.5.
Effects of Endoxifen on Cholesterol Levels
After one cycle, the median change in total cholesterol was −20 mg/dL (n = 36; range, −49 to 55 mg/dL); the median change in LDL cholesterol was −16.5 mg/dL (n = 36; range, −47 to 93 mg/dL); the median change in HDL cholesterol was −3.5 mg/dL (n = 36; range, −53 to 20 mg/dL); and the median change in triglycerides was −9 mg/dL (n = 36; range, −75 to 73 mg/dL).
Tumor and Cell-Free DNA Sequencing
Circulating cell-free DNA (cfDNA) was available for analysis in 36 patients. cfDNA mutations were observed in 13 (36.1%), including ESR1 (Y537N or D538G; n = 5), PIK3CA (H1047R or E542K; n = 8), TP53 (K132R, R248Q, R267Q, or H179Y; n = 4), AKT (Q79K; n = 1), and KRAS (G12D; n = 1). Of the five patients with cfDNA ESR1 mutations, one exhibited clinical benefit at the highest dose level (160 mg/day). PFS was shorter in those with versus those without detectable cfDNA alterations (median, 61 v 132 days; log-rank P = .046).
Fourteen patients had simultaneous collection of a fresh tumor biopsy13 and serum for DNA mutation detection; their PFS and overall survival are listed in Table 2 according to the mutation data. All tumor-sequencing data for these 14 patients are included in Appendix Figure A5 (online only). Of note, substantial discordance in detection rates was observed comparing these two modalities. For example, of the eight PIK3CA mutations, four were detected in both tumor and serum, whereas four were detected in the tumor but not serum. Of the two ESR1 cfDNA mutations detected, neither was found in the tumor (verified by targeted next-generation sequencing of the tumor samples). Of the four TP53 mutations detected, one was detected in both tumor and serum, one was detected in the tumor but not the serum, and two were detected in serum but not tumor. Of the two AKT1 mutations detected, one was detected in the tumor but not serum, and the other detected in serum but not tumor.
DISCUSSION
In this first-in-human study of Z-endoxifen in women with hormone-refractory MBC, Z-endoxifen resulted in therapeutic endoxifen concentrations, minimal toxicity, and substantial antitumor activity in women with endocrine-refractory breast cancer who had experienced progression with AIs, fulvestrant, tamoxifen, or exemestane and everolimus. On the basis of these data, a randomized phase II clinical trial (A011203; ClinicalTrials.gov identifier: NCT02311933) is ongoing comparing endoxifen (80 mg/day) with tamoxifen (20 mg/day) in women experiencing progression during prior AI therapy.
Prior attempts to improve the risk/benefit ratio of selective ER modulators (SERMs) have included tamoxifen dose escalation14,15 and development of alternative SERMs, such as raloxifene,16,17 droloxifene,18 arzoxifene,19 and toremifene,20,21 with none of these approaches resulting in superiority compared with the 20 mg per day tamoxifen dose. The genesis for Z- endoxifen drug development was based on in vitro observations that Z-endoxifen more potently inhibited tumor growth compared with tamoxifen5 and pharmacogenetic analyses of tamoxifen trials demonstrating an association between efficacy and reduced CYP2D6 metabolism8,22 or low endoxifen concentrations.9
The optimal dose of Z-endoxifen is unknown. Maximum inhibition of estrogen-induced stimulation and ER transcription is achieved with endoxifen concentrations ranging between 100 and 1,000 nm,5 with higher Z-endoxifen concentrations necessary when estradiol concentrations mimicking the premenopausal and postmenopausal settings are used to stimulate cell growth23,24 or in cells with ESR1 mutations.12 In this study, Z-endoxifen Csss of 499, 913, and 1,362 ng/mL were observed at the 40, 80, and 160 mg per day dose levels, respectively, with no differences in either toxicity or antitumor activity with respect to dose level.
cfDNA mutations were observed in 36% of patients, with shorter PFS in those with versus without detectable cfDNA alterations (median, 61 v 132 days; log-rank P = .046). This observation might simply reflect a higher tumor burden for patients with detectable cfDNA alterations. Of the five patients with cfDNA ESR1 mutations, one exhibited clinical benefit at the highest dose level (160 mg/day). Of note, clinical benefit was observed in one patient with tumor ESR1 amplification (80 mg/day). Evaluation of the 80 mg per day dose of Z-endoxifen was chosen for the randomized phase II trial of endoxifen and tamoxifen (A011203), and evaluation of ESR1 alterations in plasma and fresh tumor is planned in both arms.
CYP2D6 is not known to metabolize endoxifen25 and was not associated with either Z-endoxifen oral clearance or PFS. However, in patients who experience progression with tamoxifen, Z-endoxifen may still exhibit antitumor activity, especially in patients unable to achieve therapeutic Z-endoxifen concentrations while receiving tamoxifen. Our data provide preliminary support for this hypothesis, because patients with prior tamoxifen exposure and with CYP2D6 AS ≥ 2.0 generally had rapid clinical progression (median PFS, 60 days) in contrast to patients with reduced CYP2D6 metabolism (AS ≤ 1.5), in whom longer median PFS was observed. The relationship between CYP2D6 genotype, endoxifen exposure, and the antitumor benefit of both drugs will be evaluated in the randomized phase II trial of Z-endoxifen and tamoxifen (A011203).
A beneficial effect of SERMs is reduction in cholesterol. Our preliminary data suggest that endoxifen may reduce both total and LDL cholesterol levels. Adverse effects of SERMs include thromboembolism, uterine cancer, and a higher risk of cataracts.26 Prior studies evaluating high-dose tamoxifen have demonstrated retinal toxicity.27 Additionally, the combination of high-dose tamoxifen (with tamoxifen Csss of 4 to 8 μmol/L) and vinblastine resulted in substantial neurotoxicity, including tremor, hyperreflexia, dysmetria, unsteady gait, and dizziness.28 In our current study, no such neurotoxicity was observed, despite endoxifen concentrations > 5 μmol/L achieved at the highest dose level (160 mg/day). Furthermore, dilated eye examinations demonstrated no eye toxicity, even in patients remaining on study for as long as 6 months. One patient experienced thromboembolism at the 60 mg per day dose level. Long-term studies will be necessary to fully evaluate the adverse effect profile of Z-endoxifen.
In summary, the direct administration of Z-endoxifen provides substantial drug exposure unaffected by CYP2D6 metabolism, acceptable toxicity, and promising antitumor activity. The ongoing randomized phase II trial (A011203) will provide further insight into the antitumor activity of Z-endoxifen in a direct comparison with tamoxifen.
Appendix
Management of Toxicity
Adverse events were documented using Common Terminology Criteria for Adverse Events (version 4) before each 4-week cycle of treatment. Z-endoxifen was to be held until emergent toxicities resolved to severity ≤ grade 1 with a reduction of one dose level when treatment was resumed for grade ≥ 2 eye toxicity, any grade ≥ 3 nonhematologic adverse event, grade ≥ 3 neutrophil count decrease, or grade ≥ 3 platelet count decrease. If symptoms did not resolve to ≤ grade 1 within 14 days, Z-endoxifen was discontinued.
Patient Evaluations
Within 14 days before study registration, before each treatment cycle, and at treatment discontinuation, patients underwent a complete physical examination, blood chemistries, and toxicity assessments. Imaging studies for disease status were performed within 28 days before registration and at the end of every other monthly treatment cycle. After registration but before the start of treatment and at the end of the second cycle, patients also underwent a dilated eye examination and collection of blood and tumor samples. Additionally, any patient still receiving therapy after six cycles underwent a follow-up eye examination. For the expansion cohorts, patients underwent a pretreatment biopsy.
Pharmacokinetics
Whole blood was collected on day 1 before study drug administration and after drug administration at 0.5, 1, 2, 3, 4, 6, and 8 hours; day 2 at 24 hours after day 1 drug administration (no Z-endoxifen to be taken on day 2); day 3 at 48 hours after day 1 drug administration (Z-endoxifen to resume after blood drawn); days 7, 14, and 28 before drug administration; and day 28 after drug administration at 0.5, 1, 2, 3, 4, 6, 8, and 24 hours.
Plasma concentrations of Z-endoxifen were determined using a validated high-performance liquid chromatography assay with fluorescence detection (Lee KH, et al: Analyt Technol Biomed Life Sci 791:245-253, 2003). The plasma concentration-time data were analyzed by noncompartmental analysis using the program Winnonlin Pro (Pharsight, Mountainview, CA) to obtain estimates of the pharmacokinetic parameters: oral clearance (Cl/F), area under the curve from zero to time t, area under the curve from zero to infinity, peak serum concentration, terminal half-life, and time of peak drug concentration.
Pharmacogenetics
CYP2D6 genotype was derived from a peripheral-blood specimen. Genotyping was performed in the Clinical Laboratory Improvement Amendments–certified Mayo Clinic genotyping facility using the Luminex platform. When needed, TaqMan assay and Sanger sequencing were additionally performed. The CYP2D6 activity score (AS) was determined for each patient according to the method introduced by Gaedigk et al (Clin Pharmacol Ther 83:234-242, 2008). Each allele is assigned values as outlined in Appendix Table A1, and the AS is the sum of the values assigned to each allele. Patients were then classified as having extensive or increased CYP2D6 metabolism if AS ≥ 2.0 or reduced CYP2D6 metabolism if AS ≤ 1.5.
Tumor DNA Sequencing
Comprehensive genomic profiling (FoundationOne, Cambridge, MA) testing was performed using tumor biopsies for patients enrolled in the 160 mg per day and expansion cohorts as described previously.13 In brief, DNA was extracted from 40 mm of formalin-fixed, paraffin-embedded sections, and comprehensive genomic profiling was performed on hybridization-captured, adaptor ligation–based libraries to a mean coverage depth of 599 X for up to 405 cancer-related genes plus introns from up to 31 genes frequently rearranged in cancer. Sequence data were analyzed for clinically relevant classes of genomic alterations, including base-pair substitutions, insertions/deletions, copy-number alterations, and rearrangements.
ESR1 DNA Sequencing Confirmation
For patients in the 160 mg per day and expansion cohorts, DNA derived from tumor biopsies was subjected to targeted next-generation sequencing testing to validate the ESR1 mutation findings from the FoundationOne sequencing. Hotspot regions of ESR1 exons 7 and 8 were amplified using 10 ng of patient sample DNA, with KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Wilmington, MA) and custom-designed primers targeting the gene-specific regions. The primers included a 5′ oligo tail containing the sequencing primer region of the Illumina next-generation sequencing adapter sequence (San Diego, CA). Polymerase chain reaction (PCR) products were purified using Agencourt AMPure (Beckman Coulter, Brea, CA), and the eluted PCR product was used to perform a second PCR reaction containing index primers to incorporate a sample-specific index sequence and the remaining Illumina adapter sequence into the initial PCR amplicons. After final AMPure purification, samples were sequenced on a MiSeq instrument (Illumina), and data were analyzed using a custom bioinformatics pipeline to detect genomic alterations with ≥ 5% mutant allele frequency.
Cell-Free DNA Sequencing
Whole-blood samples were collected prospectively for projected biomarker studies at baseline and before the initiation of cycle 2. Serum was stored at −80°C until analyzed. Cell-free DNA (cfDNA) was extracted from the baseline serum samples using the Qiagen QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany) with final concentrations of 1.49 to 54.51 ng/uL. cfDNA was interrogated with the ClearID Breast Cancer Sequencing Test (Cynvenio Biosystems, Westlake Village, CA), which includes 621 amplicons in 27 genes as previously described (Song PY, et al: J Clin Oncol 35, 2017 [abstr 1091]). Libraries were constructed with 10 ng of WBC-derived genomic DNA or 10 uL of purified cfDNA (containing a minimum of 15 ng of DNA) using amplicon-based resequencing on an Ion S5 XL System (Thermo Fisher Scientific, Waltham, MA). Primary FASTQ sequences were aligned to National Center for Biotechnology Information GRCh37, and mutations present at > 1% representation from > 2,000 read coverage in a case-controlled analysis were called.
Table A1.
Values Assigned to Given CYP2D6 Allele to Determine Activity Score

Fig A1.

Z-endoxifen peak serum concentration (40 or 100 mg/day) on days 1 and 28.
Fig A2.

Z-endoxifen area under the curve versus dose.
Fig A3.

Z-endoxifen steady-state clearance versus dose.
Fig A4.

Maximum decrease in tumor size according to prior fulvestrant treatment: (A) Prior progression on fulvestrant or (B) no prior fulvestrant.
Fig A5.

Progression-free survival (PFS) times according to tumor and plasma DNA mutations for patients treated at 160 mg per day as well as those in expansion cohorts (40, 80, and 100 mg/day). (*) Detected by circulating tumor DNA.
Footnotes
Supported in part by Specialized Programs of Research Excellence Grants No. P50CA 116201 (M.P.G., V.J.S., J.M.R., D.W.N., M.A.M., J.R.H., M.M.A., D.V., J.N.I.), R01 CA133049-01 (M.P.G., V.J.S.), and U01 CA69912 (M.P.G., C.E.) from the Mayo Clinic; by the Regis Foundation (M.P.G.), Prospect Creek Foundation (M.P.G., J.R.H.), and George M. Eisenberg Foundation for Chanties (M.P.G., J.R.H., J.N.I.); and by the Division of Cancer Treatment and Diagnosis and Center for Cancer Research, National Cancer Institute.
Clinical trial information: NCT01327781.
See accompanying Editorial on page 3378
AUTHOR CONTRIBUTIONS
Conception and design: Matthew P. Goetz, Vera J. Suman, Joel M. Reid, Charles Erlichman, John R. Hawse, James H. Doroshow, Jerry M. Collins, Howard Streicher, Matthew M. Ames, James N. Ingle
Financial support: Matthew P. Goetz
Provision of study materials or patients: Matthew P. Goetz
Collection and assembly of data: Matthew P. Goetz, Joel M. Reid, Don W. Northfelt, Michael A. Mahr, Mary Kuffel, Sarah A. Buhrow, Stephanie L. Safgren, Renee M. McGovern, Dan Visscher, Zachary R. Chalmers, Garrett Frampton, Benjamin R. Kipp, Minetta C. Liu
Data analysis and interpretation: Matthew P. Goetz, Vera J. Suman, Joel M. Reid, Don W. Northfelt, Andrew T. Ralya, Mary Kuffel, Sarah A. Buhrow, John Black, Travis Dockter, Tufia Haddad, Alex A. Adjei, Dan Visscher, Zachary R. Chalmers, Garrett Frampton, Minetta C. Liu, Matthew M. Ames, James N. Ingle
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
First-in-Human Phase I Study of the Tamoxifen Metabolite Z-Endoxifen in Women With Endocrine-Refractory Metastatic Breast Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Matthew P. Goetz
Consulting or Advisory Role: Eli Lilly, bioTheranostics, RNA Diagnostics
Research Funding: Eli Lilly, Pfizer
Vera J. Suman
No relationship to disclose
Joel M. Reid
No relationship to disclose
Don W. Northfelt
No relationship to disclose
Michael A. Mahr
No relationship to disclose
Andrew T. Ralya
Employment: QuintilesIMS, Quintiles
Mary Kuffel
No relationship to disclose
Sarah A. Buhrow
No relationship to disclose
Stephanie L. Safgren
No relationship to disclose
Renee M. McGovern
No relationship to disclose
John Black
Leadership: OneOme
Stock or Other Ownership: OneOme, Assurex Health
Patents, Royalties, Other Intellectual Property: OneOme, Assurex Health
Travis Dockter
No relationship to disclose
Tufia Haddad
Research Funding: Takeda Oncology (Inst)
Charles Erlichman
No relationship to disclose
Alex A. Adjei
No relationship to disclose
Dan Visscher
Patents, Royalties, Other Intellectual Property: Mayo Clinic (Inst)
Zachary R. Chalmers
Employment: Foundation Medicine
Stock or Other Ownership: Foundation Medicine
Travel, Accommodations, Expenses: Foundation Medicine
Garrett Frampton
Employment: Foundation Medicine
Stock or Other Ownership: Foundation Medicine
Benjamin R. Kipp
Research Funding: Abbott Molecular
Patents, Royalties, Other Intellectual Property: Abbott Molecular
Minetta C. Liu
Research Funding: Eisai (Inst), Seattle Genetics (Inst), Celgene (Inst), Veridex (Inst), Novartis (Inst), Genentech (Inst), GRAIL (Inst), Merck (Inst)
Travel, Accommodations, Expenses: GRAIL, Merck, Celgene
John R. Hawse
No relationship to disclose
James H. Doroshow
No relationship to disclose
Jerry M. Collins
No relationship to disclose
Howard Streicher
No relationship to disclose
Matthew M. Ames
No relationship to disclose
James N. Ingle
No relationship to disclose
REFERENCES
- 1.Jordan VC, Collins MM, Rowsby L, et al. : A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J Endocrinol 75:305-316, 1977 [DOI] [PubMed] [Google Scholar]
- 2.Allen KE, Clark ER, Jordan VC: Evidence for the metabolic activation of non-steroidal antioestrogens: A study of structure-activity relationships. Br J Pharmacol 71:83-91, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borgna JL, Rochefort H: Hydroxylated metabolites of tamoxifen are formed in vivo and bound to estrogen receptor in target tissues. J Biol Chem 256:859-868, 1981 [PubMed] [Google Scholar]
- 4.Stearns V, Johnson MD, Rae JM, et al. : Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst 95:1758-1764, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Hawse JR, Subramaniam M, et al. : The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor alpha for degradation in breast cancer cells. Cancer Res 69:1722-1727, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Mürdter TE, Schroth W, Bacchus-Gerybadze L, et al. : Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin Pharmacol Ther 89:708-717, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Safgren SL, Suman VJ, Kosel ML, et al. : Evaluation of CYP2D6 enzyme activity using a 13C-dextromethorphan breath test in women receiving adjuvant tamoxifen. Pharmacogenet Genomics 25:157-163, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goetz MP, Rae JM, Suman VJ, et al. : Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 23:9312-9318, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Madlensky L, Natarajan L, Tchu S, et al. : Tamoxifen metabolite concentrations, CYP2D6 genotype, and breast cancer outcomes. Clin Pharmacol Ther 89:718-725, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reid JM, Goetz MP, Buhrow SA, et al. : Pharmacokinetics of endoxifen and tamoxifen in female mice: Implications for comparative in vivo activity studies. Cancer Chemother Pharmacol 74:1271-1278, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardoso F, Costa A, Norton L, et al. : ESO-ESMO 2nd international consensus guidelines for advanced breast cancer (ABC2). Ann Oncol 25:1871-1888, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson DR, Wu YM, Vats P, et al. : Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 45:1446-1451, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frampton GM, Fichtenholtz A, Otto GA, et al. : Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31:1023-1031, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tormey DC, Lippman ME, Edwards BK, et al. : Evaluation of tamoxifen doses with and without fluoxymesterone in advanced breast cancer. Ann Intern Med 98:139-144, 1983 [DOI] [PubMed] [Google Scholar]
- 15.Goldhirsch A, Joss RA, Leuenberger U, et al. : An evaluation of tamoxifen dose escalation in advanced breast cancer. Am J Clin Oncol 5:501-503, 1982 [PubMed] [Google Scholar]
- 16.Buzdar AU, Marcus C, Holmes F, et al. : Phase II evaluation of Ly156758 in metastatic breast cancer. Oncology 45:344-345, 1988 [DOI] [PubMed] [Google Scholar]
- 17.Gradishar W, Glusman J, Lu Y, et al. : Effects of high dose raloxifene in selected patients with advanced breast carcinoma. Cancer 88:2047-2053, 2000 [DOI] [PubMed] [Google Scholar]
- 18.Buzdar A, Hayes D, El-Khoudary A, et al. : Phase III randomized trial of droloxifene and tamoxifen as first-line endocrine treatment of ER/PgR-positive advanced breast cancer. Breast Cancer Res Treat 73:161-175, 2002 [DOI] [PubMed] [Google Scholar]
- 19.Deshmane V, Krishnamurthy S, Melemed AS, et al. : Phase III double-blind trial of arzoxifene compared with tamoxifen for locally advanced or metastatic breast cancer. J Clin Oncol 25:4967-4973, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Hayes DF, Van Zyl JA, Hacking A, et al. : Randomized comparison of tamoxifen and two separate doses of toremifene in postmenopausal patients with metastatic breast cancer. J Clin Oncol 13:2556-2566, 1995 [DOI] [PubMed] [Google Scholar]
- 21.Pagani O, Gelber S, Price K, et al. : Toremifene and tamoxifen are equally effective for early-stage breast cancer: First results of International Breast Cancer Study Group Trials 12-93 and 14-93. Ann Oncol 15:1749-1759, 2004 [DOI] [PubMed] [Google Scholar]
- 22. Goetz MP, Suman VJ, Hoskin TL, et al: CYP2D6 metabolism and patient outcome in the Austrian Breast and Colorectal Cancer Study Group trial (ABCSG) 8. Clin Cancer Res 19:500-507, 2013. [DOI] [PMC free article] [PubMed]
- 23.Maximov PY, McDaniel RE, Fernandes DJ, et al. : Simulation with cells in vitro of tamoxifen treatment in premenopausal breast cancer patients with different CYP2D6 genotypes. Br J Pharmacol 171:5624-5635, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maximov PY, McDaniel RE, Fernandes DJ, et al. : Pharmacological relevance of endoxifen in a laboratory simulation of breast cancer in postmenopausal patients. J Natl Cancer Inst 106:dju283, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desta Z, Ward BA, Soukhova NV, et al. : Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: Prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther 310:1062-1075, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Fisher B, Costantino JP, Wickerham DL, et al. : Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J Natl Cancer Inst 90:1371-1388, 1998 [DOI] [PubMed] [Google Scholar]
- 27.Kaiser-Kupfer MI, Lippman ME: Tamoxifen retinopathy. Cancer Treat Rep 62:315-320, 1978 [PubMed] [Google Scholar]
- 28.Trump DL, Smith DC, Ellis PG, et al. : High-dose oral tamoxifen, a potential multidrug-resistance-reversal agent: Phase I trial in combination with vinblastine. J Natl Cancer Inst 84:1811-1816, 1992 [DOI] [PubMed] [Google Scholar]