
Fig. 4. Hybrid model of the full-length CXCL12:CXCR4 complex and experimental validation.
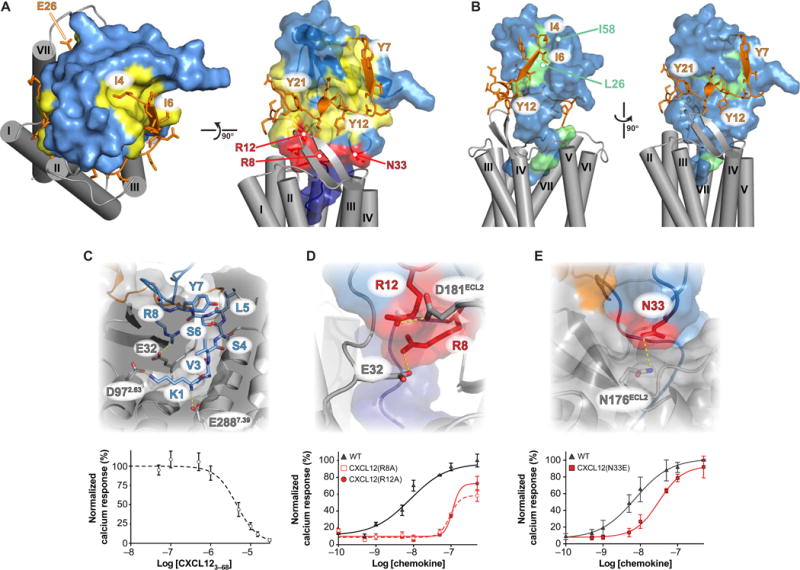
(A) Combining the LM:CXCR41–38 NMR structure and the CXCR4 crystal structure enabled modeling of the intact 1:1 signaling complex. Model generation and coordinates are located in data file S1. CXCL12 is colored light blue, with site 1, 1.5, and 2 contacts shown in yellow, red, and dark blue, respectively. CXCR4 residues 4 to 28 are colored orange, and the TM region is shaded in gray. (B) CXCL12 methyl groups that exhibited NMR intensity reductions of at least 10% from CXCR4-mediated TCS (16) are highlighted in green. (C) The N-terminal residues of CXCL12 occupy the orthosteric pocket, where salt bridges from the Lys1 α-amine and ε-amino groups to Glu288 and Asp97 of CXCR4 contribute substantially to the binding energy. N-terminal truncation of the first two residues abolishes the Ca2+ flux agonist activity of CXCL12 (fig. S8B), and the CXCL123–68 protein competes only weakly with 10 nM WT CXCL12 [IC50 = 4.5 ± 0.9 μM (SD)]. Data are means ± SD of four replicates from two experiments. (D) Arg8 and Arg12 of CXCL12 form salt bridges with Glu32 and Asp181 of CXCR4, respectively. As predicted, mutagenesis reduced the Ca2+ flux response from 7.3 ± 2.2 nM for WT to 110 ± 11 nM for CXCL12(R8A) or 95 ± 10 nM for CXCL12(R12A). Data are means ± SD of four replicates from two experiments. (E) Our model suggests that CXCL12 Asn33 contributes to binding and signaling but is not predicted to be a component of either site 1 or site 2. A fourfold change in the magnitude of Ca2+ flux [Kd = 21 ±5 nM versus 5.2 ± 2nM (SD)] confirms the contributions of Asn33 to receptor activation. Data are means ±SD of four experiments.