

Abstract
Introduction
Upon stimulation, endothelial cells release von Willebrand factor (VWF) in the unusually large (UL) and hyperactive forms that are rapidly cleaved by ADAMTS-13. Mutations in the ADAMTS13 gene result in ULVWF-mediated thrombosis found in patients with familial thrombotic thrombocytopenia purpura (TTP). ADAMTS-13 fits in the consensus of the ADAMTS family metalloproteases, but also contains two unique C- terminal CUB domains. Studying mutations in CUB domains could provide insights into the functional role of these domains.
Methods
Three naturally occurring mutations (C1213Y, W1245del and K1256FS) in the CUB-1 domain found in patients with TTP were expressed in Hela cells. The secretion, stability and VWF-cleaving activity of the mutants under static and flow conditions were examined.
Results
The mutations impaired secretion of ADAMTS-13 to apical surface, but not to extracellular matrix of transfected Hela cells. C1213Y and K1256FS also accelerated, whereas W1245del delayed, extracellular degradation of the mutants. The mutations also resulted in a moderate decrease in cleaving plasma VWF under static conditions. However, the mutated ADAMTS-13 bound to VWF substrate similarly as the wild-type metalloprotease and remained active in cleaving (UL)VWF under flow conditions.
Conclusions
The CUB-1 domain is critical for ADAMTS-13 secretion and stability upon secretion. ADAMTS-13 deficiency found in TTP patients could be resulted from reduced ADAMTS-13 secretion and, in the case of C1213Y and W1245del, accelerated degradation. W1245del is highly resistant to degradation and active in cleaving VWF.
Keywords: TTP, ADAMTS-13, CUB domains, biosynthesis, stability
Introduction
The primary function of ADAMTS-13, a member of the A Disintegrin and Metalloprotease with Thrombospondin type 1 motif (ADAMTS) family metalloprotease, is to cleave unusually large (UL) forms of von Willebrand factor (VWF) multimers that are freshly secreted from activated endothelial cells. The proteolysis occurs on surface of endothelial cells and is a critical step to convert hyperactive ULVWF to smaller forms that are hemostatically active, but no longer prothrombotic. ADAMTS-13 deficiency caused by either mutations in the ADAMTS13 gene (Upshaw Schulman's syndrome) or autoantibodies to the metalloprotease is therefore associated with systemic thrombosis, typically thrombotic thrombocytopenic purpura (TTP). ADAMTS-13 has a domain structure similar to other members of the ADAMTS metalloprotease family, except that it also contains two CUB domains (C1112-E1298 and C1299-T1427, respectively) in its C-terminus (1). “CUB” is an acronym of three proteins, from which the domain was first characterized: complement components C1r/C1s, Uegf (sea urchin fibropellins), and bone morphogenic protein (Bmp1). A structural consensus based on other CUB-containing proteins shows that CUB domain is composed of ten β-strands arranged into two β-sheets that are stabilized by two disulfide bonds (2). The CUB domain has been structurally examined in several species and is believed to be involved in protein-protein interactions. However, the CUB domains in ADAMTS-13 do not precisely fit the structural consensus for disulfide bonds because CUB-1 and CUB-2 contain five and two cysteine residues respectively (1).
The functional significance of CUB domains in ADAMTS-13 has been demonstrated in several studies, but results are not entirely consistent. On one hand, recombinant CUB domains and peptides from the CUB-1 domains bind VWF and partially inhibit the cleavage of ULVWF strings under flow (3), suggesting that these domains may contain the docking sites for ULVWF strings, required for ADAMTS-13 activity under flow conditions. Removal of the CUB domains significantly reduces the binding affinity of the metalloprotease for VWF substrate under static (4) and upon exposure to fluid shear stress (5). An ADAMTS-13 variant lacking CUB domains has moderately lower VWF-cleaving activity that is progressively worsened upon further truncations (6). Consistent with these in vitro data, mutations in CUB domains have been associated with familial TTP (7-11). On the other hand, a truncated ADAMTS-13 lacking the CUB domains (and 7 TSP-1 motifs) cleaves (UL)VWF under static and flow conditions in vitro (12;13). Binding of several ADAMTS-13 truncation variants to VWF is reduced, but not abolished under static and flow conditions (4;5).
In addition to its roles in the VWF-ADAMTS-13 interaction, CUB domains has also been shown to direct ADAMTS-13 secretion primarily to the vascular lumen, not to subendothelium in cultures of endothelial cells and transfected cell lines (14). This polarized secretion is lost when the CUB domains are deleted. Finally, autoantibodies against the CUB domains found in TTP plasma could also accelerate the ADAMTS-13 clearance from the blood circulation (15)
Because of lacking actual structural information, naturally occurring mutations in ADAMTS13 gene found in TTP patients may offer alternative insights into the structure-function relationships of the CUB domains in regulating ADAMTS-13 synthesis, secretion, and VWF-cleaving activity. We, therefore, examined three such mutations that have so far been reported for their effects on biosynthesis and enzymatic activity in vitro.
Methods
Mutations and their clinical phenotypes
Three mutations in the CUB-1 domain have so far been reported in patients with familial TTP (7;9). All were compound heterozygous with CUB-1 mutations located on one allele. The mutation carriers were diagnosed as familial TTP soon after birth or in the neonatal period, with no adult onset case. The clinical and laboratory manifestations include recurrent episodes of hemolytic anemia, thrombocytopenia and severe ADAMTS-13 deficiency (less than 10% VWF-cleaving activity). Among the three mutations (Figure 1), C1213 may potentially disrupt the first putative disulfide bond in the CUB-1 domain; W1245del converts a tryptophan to a stop code, resulting in a new C-terminus containing the first 50-60 residues of CUB-1; K1256FS results in a frame shift so that the CUB-2 domain and the C-terminal 42 amino acids of the CUB-1 domains are replaced with a new C-terminus of 36 residues.
Figure 1.
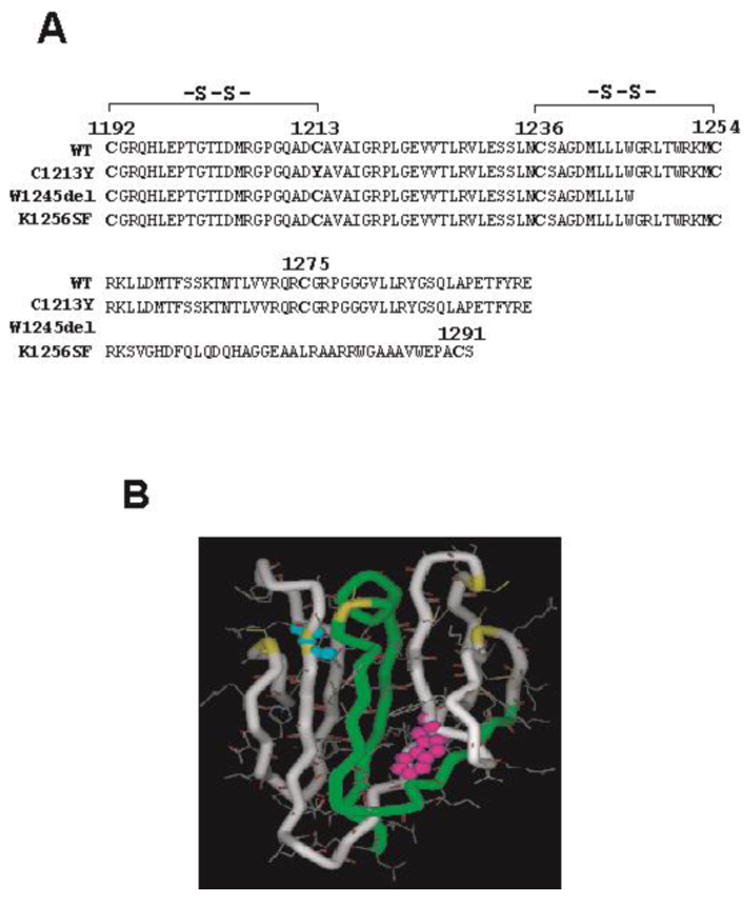
A. Location and consequence of three CUB mutations. B. The potential impacts of mutations in CUB-1 domain structure: C1213Y disrupts the disulfide bond with Cys1192 predicted based on the consensus of the CUB domains from other proteins; W1245del deletes CUB-2 and a part of CUB-1 domains due to a premature stop codon; K1256FS deletes CUB-1 and replaces a part of CUB-1 with a new 36 amino acids C-terminus (including a fifth cysteine in a different location).
The ADAMTS13 cDNA was subcloned into the mammalian expression vector pSecTag-hygromycin (Invitrogen, Carlsbad, CA) that contains the C-terminal His- and Myc-tags. A site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used to introduce mutations: 3638G>A (C1213Y), 3735G>A (W1245del), 3769-3770insT (K1256FS), individually into the wild-type (WT) ADAMTS-13 cDNA. All mutations were verified by direct DNA sequencing.
Transfection and stable cell lines
The ADAMTS-13 cDNA was introduced to Hela cells by a liposome-mediated DNA transfer (Lipofectamine™ 2000; Invitrogen). The transfected cells were maintained in Dulbecco-modified Eagle medium (Invitrogen) supplemented with 10% fetal bovine serum and 4 mM L-glutamine at 37°C with 95% air and 5% CO2. Cells expressing recombinant ADAMTS-13 (rADAMTS-13) were selected in medium containing hygromycin-B (500 μg/ml). For production, confluent cells were incubated in the serum-free Opti-Pro SFM medium (Invitrogen) for 48-72 hrs and rADAMTS-13 was purified from the conditioned medium through a Ni+2 column. WT rADAMTS-13 and mutants were identified by Western blot using a monoclonal ADAMTS-13 antibody as described previously (16).
Analysis of ADAMTS-13 secretion
The conditioned medium of transient transfected Hela cells was collected 72 hours after transfection and probed for expression of WT rADAMTS-13 and mutants by immunoblots. Recombinant proteins secreted from the cells were quantified by densitometry and compared between WT and the mutants. The adherent cells were then washed, lysed and analyzed for intracellular pools of the metalloprotease. In separate experiments, rADAMTS-13 secreted into the extracellular matrix (ECM) was measured to determine secretion polarity. For this, cells were grown to confluent, washed with PBS, and then detached by EDTA (0.5 mM). ECM was dissolved with SDS sample buffer at 37°C and separated by SDS-electrophoresis and probed with an ADAMTS-13 antibody. The amount of rADAMTS-13 and the mutants in the condition medium, cell lysates and ECM were quantified by densitometry on immunoblots.
ADAMTS-13 binding to VWF under static conditions
Interactions of VWF with WT rADAMTS13 and the mutants were measured by a static enzyme-linked immunosorbent assay (ELISA). Plasma VWF (5 μg/well) was immobilized onto wells of microtiter plates for 2 hrs at room temperature. The coated plates were then incubated with 5% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 60 min to block nonspecific binding sites, washed with PBS, and incubated with rADAMTS-13 for an additional 30 min at room temperature. After washing the wells with PBS, bound rADAMTS-13 was detected using a goat anti-ADAMTS-13 antibody made against a synthetic peptide derived from the fourth TSP-1 motif (156, Bethyl Laboratory, Houston, TX) and chemiluminescence.
ADAMTS-13 activity under static and flow conditions
ADAMTS-13 activity was first measured under static conditions using a method modified from that of Tsai et al, with VWF multimers purified from human cryoprecipitate as the substrate (17). Briefly, rADAMTS-13 was diluted (1:5) with low-ionic-strength Tris-saline buffer and then activated with 1 mM of BaCl2 for 5 min. It was mixed with VWF, dialyzed against 1.5 M of urea for 24 hrs at 37°C, and then subjected to 10% SDS-electrophoresis under reducing conditions. The separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane and immunobloted for VWF by a polyclonal anti-VWF antibody (DakoCytomation, Carpinteria, CA).
For the flow assay, human umbilical vein endothelial cells (HUVECs) in 35 mm culture dishes were first stimulated with 25 mM of histamine for 3 minutes and then perfused with Tyrode's buffer containing washed platelets in the presence of WT or mutant ADAMTS-13 through a parallel-plate flow chamber at a flow rate of 0.2 ml/min (2.5 dyne/cm2 shear stress for platelets suspension). Using HUVECs has been approved by the IRB of Baylor College of Medicine on research involving human subjects. Platelets perfused over HUVECs adhered to ULVWF to form long and platelet-decorated strings that were cleaved by rADAMTS-13. For the assay, the VWF cleaving activity is defined as the percent reduction of ULVWF strings in 20 continuous view fields (× 200) in the presence of rADAMTS-13 as compared to its absence (18).
Statistical Analysis
Data are presented as either representative images from multiple experiments or as mean ± SEM of multiple data sets. The Student's t test was used for quantitative data analysis and a p value less than 0.05 was considered to be statistically significant.
Results
CUB-1 mutations impaired ADAMTS-13 secretion
Effects of the CUB mutations on biosynthesis of ADAMTS-13 were evaluated by transiently expressing WT and mutant ADAMTS-13 in Hela cells. We used transient, instead of stable, transfection to eliminate possibility of incorporating different numbers of WT or mutated cDNA into the host genome. Expression and secretion of rADAMTS-13 were measured in the conditioned medium, lysates of transfected cells, and extracellular matrix (ECM). As shown in Figure 2A (and 2D for summary), secretion of all mutants were significantly reduced as compared to WT, counting for 29.0%, 24.4%, and 7.9% of the WT. The doublet bands found in W1245del and K1256FS were likely resulted from differently glycosylated ADAMTS-13.
Figure 2. The CUB-1 mutations impaired ADAMTS-13 secretion.
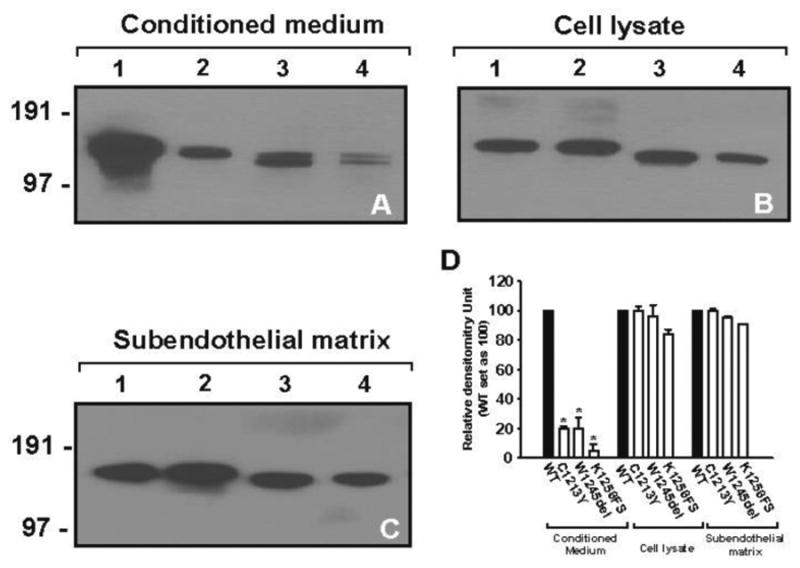
WT and the mutated ADAMTS-13 cDNAs were transiently transfected into Hela cells and expression of the recombinants proteins evaluated in the conditioned media, cell lysates, and ECM by immunoblotting. Secretion of all three mutants was significantly reduced in the conditioned media, but not to ECM or in the intracellular pools, as compared to WT (A-C). The panel D summarized data from the repeated experiments (the Student's t test, *p < 0.01).
There could be four possible reasons for the decreased ADAMTS-13 detected in the culture medium: 1) impaired DNA transcription, 2) intracellular retention of aberrant metalloprotease, 3) disruption of secretion polarity, and 4) proteolytic degradation. We found that the amounts of the mutants in cell lysates were comparable to that of WT (Figure 2B), indicating that the mutants were synthesized at rates comparable to WT ADAMTS-13. We further determined that the CUB mutants were secreted into ECM in the amounts similar to that of WT.
CUB-1 mutations affected ADAMTS-13 stability
As in several previous reports including ours, WT rADAMTS-13 in the conditioned medium and purified form degrades overtime during storage, at a rate of approximately 5-30%/week. The main degradation product was approximately 53 kDa containing the C-terminal fragment of ADAMTS-13 as it was recognized by an ADAMTS-13 antibody generated using a peptide derived from the fourth TSP-1 domain (19). It is therefore possible that the degradation is greater for the mutants, leading to lower detection in culture medium. We experimentally examined this possibility and found that the mutants C1213Y and K1256FS were indeed degraded at a significantly greater rate (100% and 76% degradation, respectively, after four weeks in storage, Figure 3). This was particularly significant for the K1256FS mutant because the ratio of mutant detected in medium to that in ECM was much smaller (0.09) as compared to the other two mutants (both were 0.26). In contrast, W1245del was resistance to degradation during the same storage period.
Figure 3. The mutations changed the degradation rates.
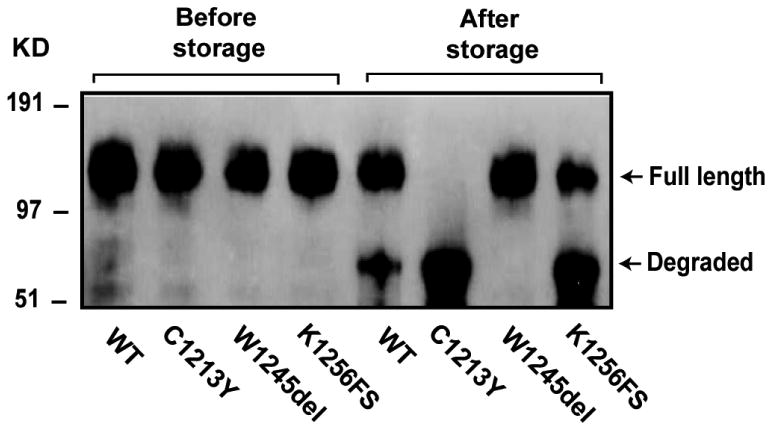
WT and mutated ADAMTS-13 were stored for up to four weeks. Degradation was then measured by the loss of full length metalloprotease and appearance of a degradation product by immunoblotting. The degradation was accelerated and enhanced for C1213Y and K1256FS mutants, but significantly reduced for the W1245del mutant. The figure is a representative of four separate experiments.
The mutants cleaved (UL)VWF under static and flow conditions
As shown in Figure 4A, all the mutants were moderately less active compared to WT for cleaving VWF multimers purified from human plasma cryoprecipitate under static conditions. One concern is that the static assay may not be physiologically relevant because it requires artificial additives such as barium and urea. The VWF-cleaving activity of the mutants was, therefore, measured under flow conditions (18). We found that all three mutants were active in cleaving ULVWF strings at rates comparable to the WT metalloprotease (Figure 4B). Consistent with the activity assays, all three mutants bound immobilized VWF (Figure 5), indicating that the mutations did not affect ADAMTS-13-VWF interaction.
Figure 4. The CUB-1 mutations maintained VWF-cleaving activity.
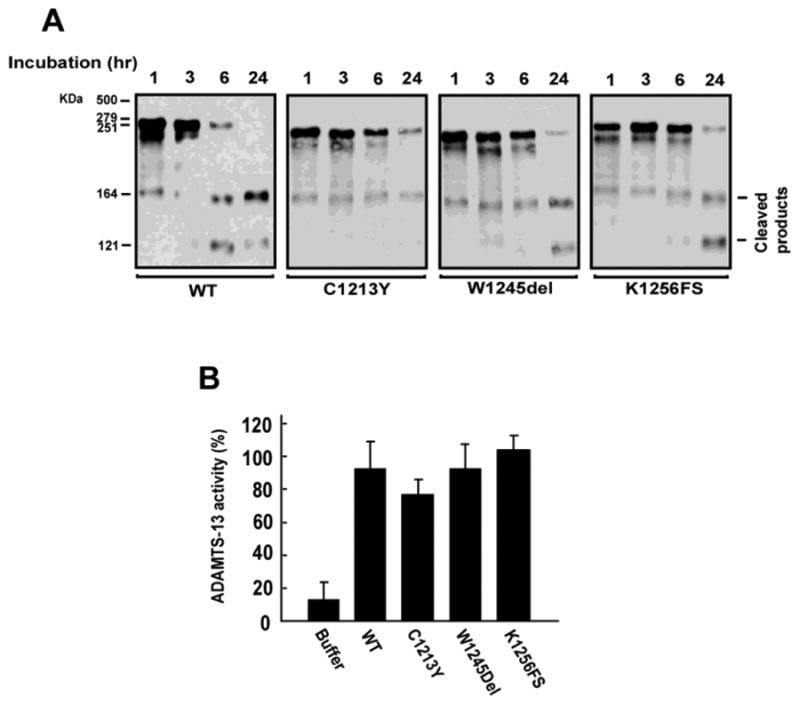
The recombinant WT and mutants were tested for their (UL)VWF cleaving activity. (A) The static activity assay measured the cleavage of VWF multimers purified from plasma cryoprecipitate in the presence of barium and urea at different time points (1, 3, 6 and 24 hours). The VWF-cleaving activities of the mutants were moderately lower (80-90%) as compared to WT under static conditions (Figure 4A is a representative of four separate experiments). (B) The flow assay measured the cleavage of ULVWF-platelet strings formed on endothelial cells under flow conditions (2.5dyn/cm2 of calculated wall shear stress). There was no significant difference between the WT and mutants (the Student's t test, n = 3).
Figure 5. The CUB-1 mutations did not impair VWF binding.
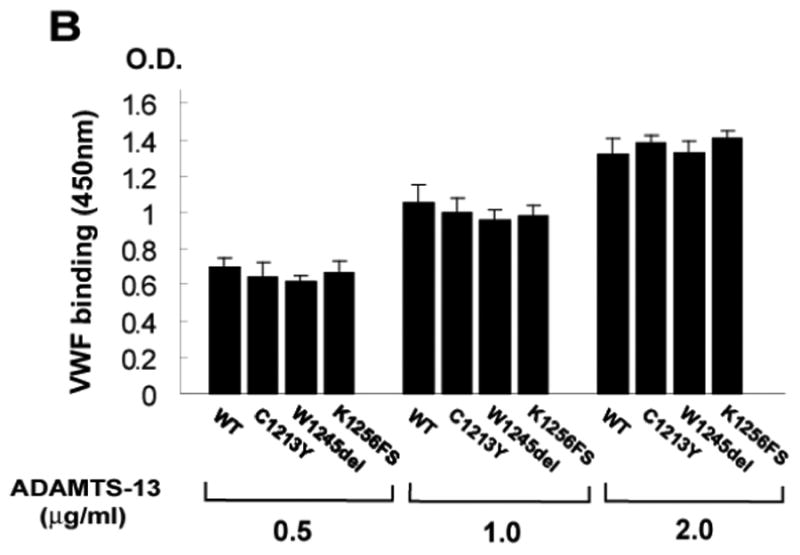
The binding of the CUB-1 mutants at various concentrations (0.5, 1.0, 2.0 μg/ml) to the purified VWF multimers was measured under static conditions. Consistent with the activity measurements, all three mutants bound immobilized VWF at levels similar to each other and to WT (ANOVA test, n = 4, p > 0.05 among WT and mutants).
Discussion
We have shown that three naturally occurring mutations in the CUB-1 domain severely impair secretion of ADAMTS-13 into the conditioned medium, but not extracellular matrix of transfected Hela cells (Figure 2). These data are consistent with an early notion that the CUB domains are critical for directing ADAMTS-13 secretion to the luminal surface of the metalloprotease producing cells (14). The reason for this selective impairment to ADAMTS-13 secretion remains to be further investigated, but may be caused by disrupting a CUB association with lipid rafts (14). The impairment to the targeted secretion may be particularly important for ADAMTS-13 activity associated with endothelial cells (14;20) and platelets (19;23), but less relevant to that produced by hepatic stellate cells (21;22).
In addition to defective secretion, the mutants have also changed the rate of extracellular degradation. ADAMTS-13 is a relatively stable metalloprotease with a half-life of 2-3 days in vivo (24). However, it has long been observed that WT ADAMTS-13 is progressively degraded during storage, even at -80°C (3;13). As shown in this study, the mutations result in remarkably different rates of extracellular degradation during storage as compared to WT. Two mutants of C1213Y and K1256FS, one converting a C1213 to a tyrosine and the other replacing the CUB domains with a new shorter sequence, are degraded at significantly greater rates than WT. In contrast, W1245del, which remove CUB-2 and a part of CUB-1, is largely resistant to degradation (Figure 3). One explanation for the diverged rates of degradation is that the two mutants with accelerated rates of degradation contain the fifth cysteine residue (it is in a different position in K1256FS due to a frame shift), whereas the degradation resistant W1245del does not (Figure 1). The fifth cysteine in CUB-1 domain is likely to be in thiol form and may regulate the stability of the metalloprotease.
All three mutants may have moderately reduced the VWF-cleaving activity under static conditions and retain their VWF-cleaving activity under flow conditions (Figure 4). Consistent with the activity measurements, they bind to VWF substrate at levels comparable to WT (Figure 5). The data may not be sufficient to determine if CUB-1 contains a VWF binding site because none of the mutants removes the CUB domain entirely. It is possible that the VWF-binding site is located in the N-terminal half of the CUB-1 domain (C1192 to W1245). The suggestion is consistent with our early observation that synthetic peptides derived from the first half of the CUB-1 domain block ADAMTS-13 cleavage of ULVWF strings under flow conditions (3).
In summary, we have demonstrated that naturally occurring mutations in the CUB-1 domain result in normal rates of synthesis, but lower rates of secretion to the luminal surface of transfected Hela cells. They also have distinct sensitivities to extracellular degradation: W1245del is resistant, but C1245Y and K1256FS are much more sensitive to degradation as compared to the WT metalloprotease. Despite of their reduced rates of secretion and diverged rates of degradation, all three mutants maintain the VWF binding site and VWF-cleaving activity. The results indicate that ADAMTS-13 deficiency found in patients carrying these mutations is likely to be caused by reduced secretion of the metalloprotease to circulation and, in the case of C1213Y and K1256FS, increased extracellular degradation. The data demonstrate not only that the CUB domains are critical for ADAMTS-13 structural stability, but also that rADAMTS-13 can be structurally modified to engineer a variant that is highly resistant to degradation, while maintaining or even enhancing VWF-cleaving activity. Such a modification could have a greater potential for therapeutic use.
Footnotes
Disclosure of Conflict of Interests: The authors have no conflict of interest to disclose.
This work was supported by NIH grant HL71895.
References
- 1.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276:41059–41063. doi: 10.1074/jbc.C100515200. [DOI] [PubMed] [Google Scholar]
- 2.Romero A, Romao MJ, Varela PF, Kolln I, Dias JM, Carvalho AL, Sanz L, Topfer-Petersen E, Calvete JJ. The crystal structures of two spermadhesins reveal the CUB domain fold. Nat Struct Biol. 1997;4:783–788. doi: 10.1038/nsb1097-783. [DOI] [PubMed] [Google Scholar]
- 3.Tao Z, Peng Y, Nolasco L, Cal S, Lopez-Otin C, Li R, Moake JL, Lopez JA, Dong JF. Recombinant CUB-1 domain polypeptide inhibits the cleavage of ULVWF strings by ADAMTS13 under flow conditions. Blood. 2005;106:4139–4145. doi: 10.1182/blood-2005-05-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majerus EM, Anderson PJ, Sadler JE. Binding of ADAMTS13 to von Willebrand factor. J Biol Chem. 2005;280:21773–21778. doi: 10.1074/jbc.M502529200. [DOI] [PubMed] [Google Scholar]
- 5.Zhang P, Pan W, Rux AH, Sachais BS, Zheng XL. The cooperative activity between the carboxyl-terminal TSP1 repeats and the CUB domains of ADAMTS13 is crucial for recognition of von Willebrand factor under flow. Blood. 2007;110:1887–1894. doi: 10.1182/blood-2007-04-083329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou W, Dong L, Ginsburg D, Bouhassira EE, Tsai HM. Enzymatically active ADAMTS13 variants are not inhibited by anti-ADAMTS13 autoantibodies: a novel therapeutic strategy? J Biol Chem. 2005;280:39934–39941. doi: 10.1074/jbc.M504919200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Licht C, Stapenhorst L, Simon T, Budde U, Schneppenheim R, Hoppe B. Two novel ADAMTS13 gene mutations in thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome (TTP/HUS) Kidney Int. 2004;66:955–958. doi: 10.1111/j.1523-1755.2004.00841.x. [DOI] [PubMed] [Google Scholar]
- 8.Pimanda JE, Maekawa A, Wind T, Paxton J, Chesterman CN, Hogg PJ. Congenital thrombotic thrombocytopenic purpura in association with a mutation in the second CUB domain of ADAMTS13. Blood. 2004;103:627–629. doi: 10.1182/blood-2003-04-1346. [DOI] [PubMed] [Google Scholar]
- 9.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 10.Assink K, Schiphorst R, Allford S, Karpman D, Etzioni A, Brichard B, van de KN, Monnens L, van den Heuvel L. Mutation analysis and clinical implications of von Willebrand factor-cleaving protease deficiency. Kidney Int. 2003;63:1995–1999. doi: 10.1046/j.1523-1755.63.6s.1.x. [DOI] [PubMed] [Google Scholar]
- 11.Riksen NP, Luken BM, Klasen IS, Voorberg J, Crama N, van Deuren M. Antibodies against the CUB1-2 domains of ADAMTS13 in a patient with benign monoclonal gammopathy: no causal relationship. Haematologica. 2007;92:e74–e76. doi: 10.3324/haematol.11475. [DOI] [PubMed] [Google Scholar]
- 12.Tao Z, Wang Y, Choi H, Bernardo A, Nishio K, Sadler JE, Lopez JA, Dong JF. Cleavage of ultra-large multimers of Von Willebrand factor by C-terminal truncated mutants of ADAMTS-13 under flow. Blood. 2005;106:141–143. doi: 10.1182/blood-2004-11-4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng X, Nishio K, Majerus EM, Sadler JE. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J Biol Chem. 2003;278:30136–30141. doi: 10.1074/jbc.M305331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shang D, Zheng XW, Niiya M, Zheng XL. Apical sorting of ADAMTS13 in vascular endothelial cells and Madin-Darby canine kidney cells depends on the CUB domains and their association with lipid rafts. Blood. 2006;108:2207–2215. doi: 10.1182/blood-2006-02-002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shelat SG, Smith P, Ai J, Zheng XL. Inhibitory autoantibodies against ADAMTS-13 in patients with thrombotic thrombocytopenic purpura bind ADAMTS-13 protease and may accelerate its clearance in vivo. J Thromb Haemost. 2006;4:1707–1717. doi: 10.1111/j.1538-7836.2006.02025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tao Z, Anthony K, Peng Y, Choi H, Nolasco L, Rice L, Moake JL, Dong JF. Novel ADAMTS-13 mutations in an adult with delayed onset thrombotic thrombocytopenic purpura. J Thromb Haemost. 2006;4:1931–1935. doi: 10.1111/j.1538-7836.2006.02098.x. [DOI] [PubMed] [Google Scholar]
- 17.Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87:4235–4244. [PubMed] [Google Scholar]
- 18.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, Lopez JA. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 19.Liu L, Choi H, Bernardo A, Bergeron AL, Nolasco L, Ruan C, Moake JL, Dong JF. Platelet-derived VWF-cleaving metalloprotease ADAMTS-13. J Thromb Haemost. 2005;3:2536–2544. doi: 10.1111/j.1538-7836.2005.01561.x. [DOI] [PubMed] [Google Scholar]
- 20.Turner N, Nolasco L, Tao Z, Dong JF, Moake J. Human endothelial cells synthesize and release ADAMTS-13. J Thromb Haemost. 2006;4:1396–1404. doi: 10.1111/j.1538-7836.2006.01959.x. [DOI] [PubMed] [Google Scholar]
- 21.Uemura M, Tatsumi K, Matsumoto M, Fujimoto M, Matsuyama T, Ishikawa M, Iwamoto TA, Mori T, Wanaka A, Fukui H, et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005;106:922–924. doi: 10.1182/blood-2005-01-0152. [DOI] [PubMed] [Google Scholar]
- 22.Zhou W, Inada M, Lee TP, Benten D, Lyubsky S, Bouhassira EE, Gupta S, Tsai HM. ADAMTS13 is expressed in hepatic stellate cells. Lab Invest. 2005;85:780–788. doi: 10.1038/labinvest.3700275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki M, Murata M, Matsubara Y, Uchida T, Ishihara H, Shibano T, Ashida S, Soejima K, Okada Y, Ikeda Y. Detection of von Willebrand factor-cleaving protease (ADAMTS-13) in human platelets. Biochem Biophys Res Commun. 2004;313:212–216. doi: 10.1016/j.bbrc.2003.11.111. [DOI] [PubMed] [Google Scholar]
- 24.Furlan M, Robles R, Morselli B, Sandoz P, Lammle B. Recovery and half-life of von Willebrand factor-cleaving protease after plasma therapy in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 1999;81:8–13. [PubMed] [Google Scholar]
- 25.Majerus EM, Zheng X, Tuley EA, Sadler JE. Cleavage of the ADAMTS13 propeptide is not required for protease activity. J Biol Chem. 2003;278:46643–46648. doi: 10.1074/jbc.M309872200. [DOI] [PMC free article] [PubMed] [Google Scholar]