

Abstract
Generating aptamers that bind to specific metal ions is challenging because existing aptamer discovery methods typically require chemical labels or modifications that can alter the structure and properties of the ions. In this work, we report an aptamer discovery method that enables us to generate high-quality structure-switching aptamers (SSAs) that undergo a conformational change upon binding a metal ion target, without the requirement of labels or chemical modifications. Our method is more efficient than conventional selection methods because it enables direct measurement of target binding via fluorescence-activated cell sorting (FACS), isolating only the desired aptamers with the highest affinity. Using this strategy, we obtained a highly specific DNA SSA with ~30-fold higher affinity than the best aptamer for Hg2+ in the literature. We also discovered DNA aptamers that bind to Cu2+ with excellent affinity and specificity. Both aptamers were obtained within four rounds of screening, demonstrating the efficiency of our aptamer discovery method. Given the growing availability of FACS, we believe our method offers a general strategy for discovering high-quality aptamers for other ions and small-molecule targets in an efficient and reproducible manner.
Keywords: aptamers, molecular screening, metal ions, fluorescence-activated cell sorting
Graphical abstract
Metal ions play important roles in many biological and environmental systems. For example, metal ions are implicated in neurodegenerative diseases;1 specifically, Cu2+ is associated with the aggregation of amyloid-β (Aβ) peptides2 in Alzheimer’s disease.3 Furthermore, mercury and cadmium ions are environmental contaminants that are toxic to humans.4–8 Thus, the ability to detect and quantify metal ions without cumbersome laboratory instruments such as a mass spectrometer would be of considerable value in the field and help clarify the relationship between changes in metal ion concentrations and disease states.9,10 The development of such biosensors requires affinity reagents that bind specifically to the target metal ions. Unfortunately, monoclonal antibodies—the gold standard of affinity reagents—are notoriously difficult to generate for metal ions, because they typically do not elicit sufficient immune responses in animals.11,12 Thus, there is an urgent need for alternative affinity reagents that can bind to specific metal ions.
Aptamers offer an interesting alternative to antibodies, because they are discovered through in vitro screening methods13,14 rather than relying on in vivo processes,15 bypassing hurdles related to target immunogenicity. A number of groups have generated metal ion-binding aptamers for a variety of useful applications. For example, Chung and co-workers demonstrated the use of DNA aptamers to directly detect Pb2+ and Hg2+ in serum.16 Similarly, Li and Long17,18 used DNA aptamers that bind to Hg2+ to monitor water quality. The discovery of aptamers using conventional systematic evolution of ligands by exponential enrichment (SELEX) typically requires chemical labeling or modifications of the target. For example, chemical linkers are commonly used to immobilize the target molecule onto a solid support, and fluorescence reporters are often conjugated to the target molecule for detection.19–21 Unfortunately, these options are challenging to implement for metal ions and other small molecules, as they would significantly affect their structure22 and chemical properties.23 Therefore, it would be most desirable to have a screening method that works with the target in solution, without any need for target labeling or attachment to a solid support.
In this work, we describe an aptamer discovery method that can efficiently produce high-quality structure-switching DNA aptamers (SSA) that undergo large conformational changes upon binding to their metal ion target, without any labels or chemical modifications. Our aptamer discovery method (called structure-switching particle display, or SS-PD) transforms solution-phase aptamers into “aptamer particles” through emulsion polymerase chain reaction (PCR)21,24 and then uses fluorescence-activated cell sorting (FACS) to directly measure the binding interactions between each aptamer particle and the target metal ion, individually isolating only the desired aptamers with the highest affinity (Figure 1). SS-PD is highly efficient: within four rounds, we obtained DNA aptamers for Hg2+ with ~30-fold higher affinity than a previously described aptamer for this ion. We also discovered DNA aptamers that bind to Cu2+ with excellent affinity and specificity within three rounds of SS-PD.
Figure 1.
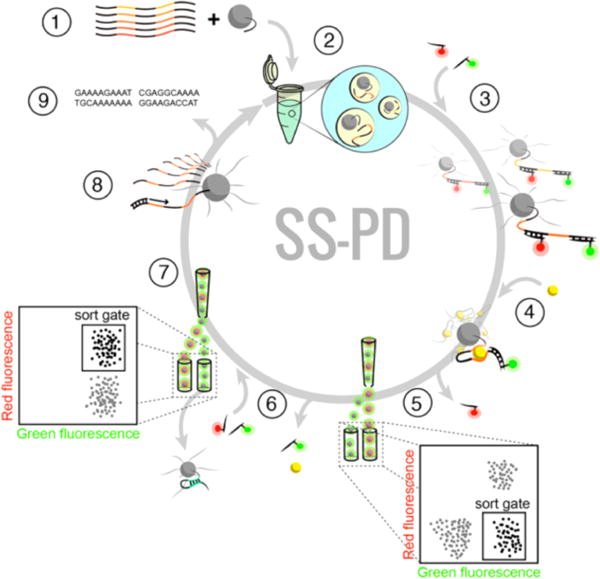
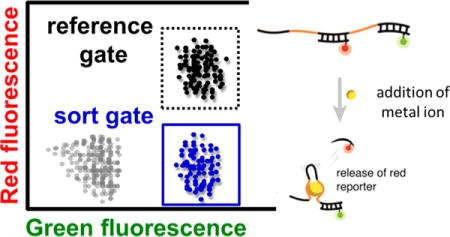
Overview of the SS-PD screening method: (1) The solution-phase aptamer library is combined with forward primer-conjugated magnetic particles, (2) then subjected to emulsion PCR to produce monoclonal aptamer particles, which are (3) hybridized with red and green reporters. After (4) incubation with a target metal ion, (5) we perform a “binding screen” using FACS to isolate aptamer particles with high green and low red fluorescence. (6) We then elute the green reporters and metal ion targets from the selected aptamer particles and rehybridize them with red and green reporters. (7) Self-hybridizing, false-positive aptamer particles are then eliminated with a “folding screen”, which discards aptamer particles that cannot rehybridize to the red reporter. (8) Aptamer particles that survive both FACS screens are either PCR amplified and subjected to additional screening or (9) cloned and sequenced.
RESULTS AND DISCUSSION
Overview of SS-PD Screening
To directly measure the binding of metal ions to aptamers, we converted the solution-phase aptamer library into monoclonal aptamer particles using emulsion PCR21 (Figure 1, step 1). Briefly, we made water-in-oil droplets that contain PCR reagents and paramagnetic particles with covalently conjugated DNA forward primers. The droplets contained, on average, no more than one aptamer template, following a Poisson distribution (see Methods). We then performed PCR within the droplet to create a library of aptamer particles. Each aptamer particle displays approximately 2 × 105 copies of a single aptamer sequence on its surface (Figure 1, step 2).
Our aptamer library design enables us to directly measure the binding of individual aptamer particles to metal ions without any labels, solid support, or modifications. This design, first described by Nutiu and co-workers,25 features a constant domain in the center of the aptamer flanked by two random domains that are screened for metal ion binding (Figure 2A). The constant domain is initially hybridized to a complementary, fluorescently labeled (Alexa Fluor 647) “red reporter” DNA strand. If the binding of the metal ion to the random domains is sufficiently strong, it can cause the red reporter to dehybridize from the constant domain (Figure 2B). Release of the red reporter causes a decrease in the red fluorescence of the aptamer particle that can be readily detected by FACS. To normalize the density of the aptamers on the surface of each aptamer particle, we also incorporated a “green reporter” complementary DNA strand, labeled with carboxyfluorescein, which hybridizes to the reverse primer-binding domain of the aptamer (see Methods for sequence and fluorophore information).
Figure 2.
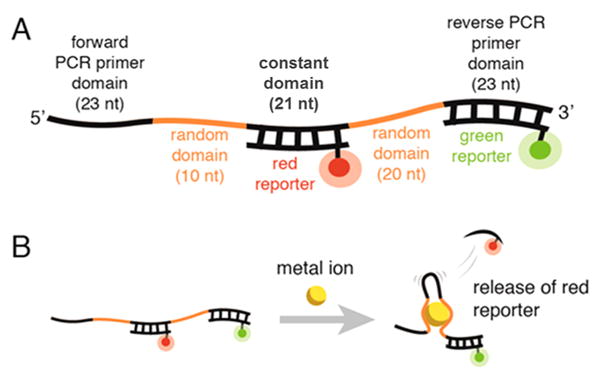
(A) Design of the SS-PD aptamer library. (B) Binding between the metal ion and the aptamer causes the release of the red reporter. The green reporter indicates the density of aptamers displayed on the aptamer particle surface.
Once the red and green reporters are hybridized to the aptamer particles (Figure 1, step 3), we perform the “binding screen” by incubating the library of aptamer particles with the metal ions (step 4). As described above, aptamer particles with high affinity for the metal ions display a reduced red fluorescence (as the red reporter is released), while retaining green fluorescence. These aptamer particles with low red fluorescence are sorted and collected via FACS (step 5). After the binding screen, we use heat to release the metal ions and green reporters from the collected aptamer particles. We then rehybridize these particles with both red and green reporters and perform a “folding screen” in the absence of the metal ion target (step 6), isolating only those particles that exhibit both strong red and green fluorescence (step 7, inset). In this way, the folding screen eliminates false positive aptamer particles that release the red reporter without the target (presumably through self-hybridization). Although the folding screen may decrease the diversity of the aptamer pool, this step allows us to identify aptamers with the desired structure switching function. Aptamer particles that survive both the binding and folding screens are amplified through PCR and used for the next round of screening (step 8) or for sequencing (step 9).
SS-PD Screen for Hg2+ Aptamers
As an initial demonstration, we performed four rounds of SS-PD to obtain aptamers that bind specifically to Hg2+ ions. In each round, we systematically increased the screening stringency by decreasing the target concentration (see Table S1). We started with 1 mM Hg2+ in rounds 1 and 2 and then dropped the concentration to 500 μM (round 3) and 50 μM (round 4). The binding screen results are shown in Figure 3A, and folding screen results are shown in Supplemental Figure S1. We observed clear enrichment of Hg2+-binding aptamers in four rounds (Figure 3A). In round 1, only 0.4% of aptamers exhibited signal above background at 1 mM Hg2+. This is calculated by taking the difference between the aptamer population in the sort gate at 1 mM Hg2+ (0.6%) versus the population in the sort gate with no Hg2+ (0.2%). By round 4, 1.6% of the aptamers showed binding even at a 20-fold lower concentration of Hg2+ (50 μM). We stopped after four rounds because the output of the fourth round of screening exhibited a change in red fluorescence similar to that of the pool generated in round 3 at the same Hg2+ concentration (see Supplemental Figure S2).
Figure 3.
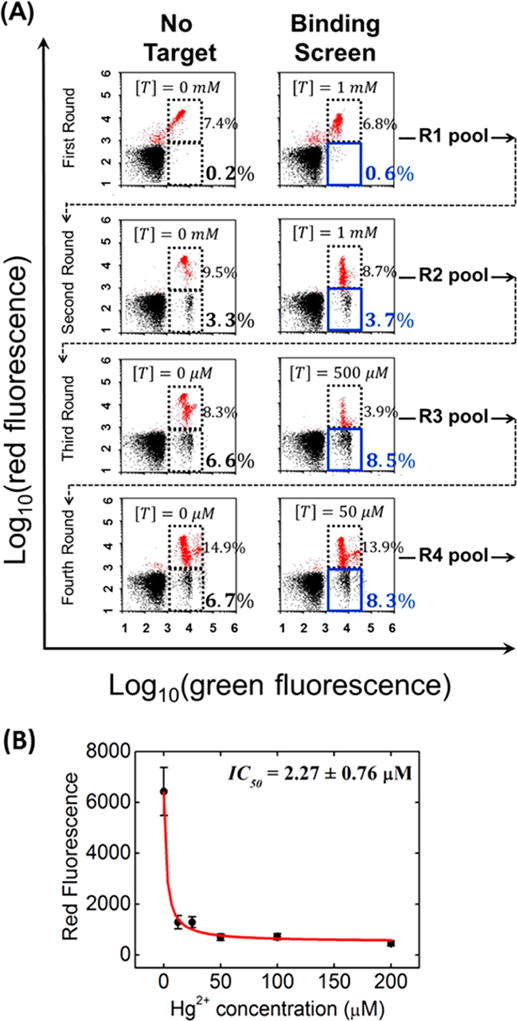
SS-PD screening for Hg2+-specific SSAs. (A) FACS plots show four rounds of the binding screen at different target concentrations ([T]) of Hg2+. In each plot, the blue solid box denotes the sort gate and the dashed box denotes the reference gate. The red dots in the plot indicate those aptamer particles that are hybridized to both the red and green reporters. The output of each round n (labeled as Rn on the right side of each plot) is tested and sorted in the next round (per the dashed arrow). (B) IC50 measurement for SSA-HgII. Error bars represent the standard deviation from 10 000 particle measurements. These data fit well our theoretical model of IC50 (see Supporting Information).
To identify the sequences of individual aptamers in this final R4 pool, we cloned it into the pCR4-TOPO vector and transformed it into competent E. coli cells. Interestingly, we found that many of the aptamers contained point mutations and truncations within the constant domain and primer-binding sites. Similar results have been previously reported for small-molecule structure-switching aptamer selection.26 We believe these are partly the result of selection pressure that favors constant domains with lower hybridization energy to the red reporter, with the mutation process exacerbated by the presence of Hg2+ during PCR.27 We chose seven clones with unaltered primer-binding domains and fewer than five point mutations or truncations in the constant domain for further analysis (sequences shown in Table S2).
To rapidly identify the aptamers that bind most strongly to Hg2+, we synthesized seven sets of aptamer particles, each displaying multiple copies of a single aptamer sequence and hybridized the red reporters. We then measured red reporter release at various concentrations of Hg2+ to obtain the IC50: the concentration at which the red fluorescence drops to half its maximum value. All sequences and their IC50 values are shown in Table S2. The IC50 values ranged from 2.27 to 26.35 μM. The SSA-HgII aptamer showed the best IC50 (2.27 ± 0.76 μM), as shown in Figure 3B.
We note that the IC50 is different from the true equilibrium dissociation constant (KD). This is because the IC50 is measured in the presence of the red reporter, which acts as a binding competitor. The theoretical relationship between IC50 and KD for our aptamer design is provided in the Supporting Information. To experimentally obtain the KD of SSA-HgII, we used a microscale thermophoretic technique28 without hybridizing the red reporter to the aptamer. This technique utilizes the difference in the mobility of target-bound and unbound aptamers in response to a temperature gradient to obtain the KD (see Methods). The KD of SSA-HgII was 1.49 ± 0.28 μM (Supplemental Figure S3A), which is approximately 30-fold better than a previously published Hg2+ aptamer29 measured with the same method (see Supplemental Figure S3B). SSA-HgII does not share sequence homology with this previously reported Hg2+ aptamer,29 suggesting that the mechanisms of binding are different between the two aptamers. Interestingly the affinity of a truncated derivative of SSA-HgII, containing 30 contiguous selected bases, without the constant domain, was approximately 4-fold higher than the full-length SSA-HgII aptamer (Supplemental Figure S3C), suggesting that the constant domain does not participate in binding of Hg2+ to the aptamer. Nuclear magnetic resonance (NMR)-based structural studies are under way to gain a deeper understanding of this binding mechanism.
SS-PD Screen for Cu2+ Aptamers
To show that the SS-PD platform is generalizable to other metal ion targets, we also identified aptamers for Cu2+, a target for which no aptamers have been described in the literature. We achieved a high degree of enrichment in just three rounds of SS-PD. Binding screen results are shown in Figure 4A, and folding screen results are shown in Supplemental Figure S4. In the first round, only 0.6% of aptamers exhibited signal above background at 400 μM Cu2+. This is calculated by taking the difference between the aptamer population in the sort gate at 400 μM Cu2+ (0.8%) versus the population in the sort gate without Cu2+ (0.2%). By round 3, we observed a similar difference in the aptamer population, but at a ~16-fold lower Cu2+ concentration (25 μM). This indicates significant enrichment of Cu2+-binding aptamers.
Figure 4.
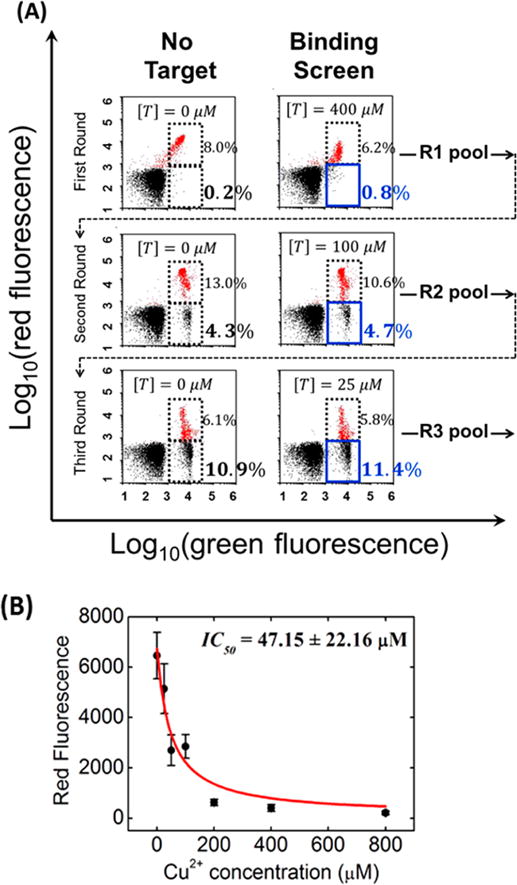
SS-PD screening of Cu2+ aptamers. (A) FACS plots show three binding screen rounds at different target concentrations ([T]) of Cu2+. Data are presented in the same format as in Figure 3. (B) IC50 measurement for SSA-CuII. Error bars represent the standard deviation from 10 000 particle measurements. The data fit well to our theoretical model of IC50 (see Supporting Information).
We stopped after three rounds of SS-PD because the aptamers resulting from round 3 exhibited a similar change in red fluorescence in comparison to those resulting from round 2 at the same Cu2+ concentration (see Supplemental Figure S5). We cloned the aptamers resulting from round 3 and chose two sequences with no mutations in the primer-binding domains and fewer than five point mutations/truncations in the constant domain (sequences shown in Table S2). We note that we observed a similar mutation rate for this Cu2+ screening process to that for the previously described Hg2+ screening process. The SSA-CuII aptamer exhibited the strongest binding, with an IC50 of 47.15 μM (Figure 4B). Unfortunately, we were unable to determine the KD of the SSA-CuII aptamer using microscale thermophoresis because Cu2+ causes quenching of the fluorescence signal.30 However, given that the IC50 and KD values were very close each other for the SSA-HgII aptamer (within a factor of 2), we also expect the KD of SSA-CuII to be similar to its IC50.
Hg2+ and Cu2+ Aptamers Exhibit High Specificity
We quantified the specificity of our SSA-HgII and SSA-CuII aptamers and found that they show minimal binding to other divalent metal ions. We synthesized aptamer particles displaying SSA-HgII and SSA-CuII and hybridized them with the red reporter. We then measured the change in red fluorescence after incubating with Ca2+, Mg2+, Pb2+, and Zn2+; we also challenged SSA-HgII and SSA-CuII with Cu2+ or Hg2+, respectively (see Supporting Information for methods). Both aptamers were highly specific and showed minimal binding to these nontarget ions (Figure 5). As shown in Figure 5A, the Hg2+ aptamer SSA-HgII shows minimal binding to the nontarget divalent metal ions tested. Similarly, the Cu2+ aptamer SSA-CuII also shows minimal cross reactivity (Figure 5B).
Figure 5.
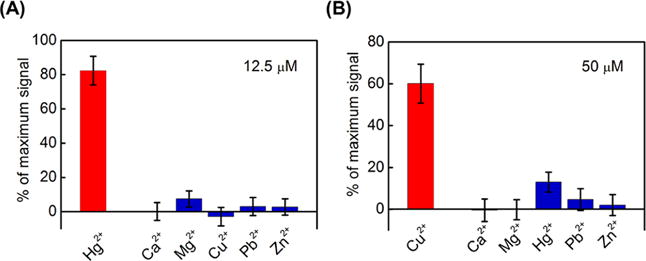
Specificity measurements of our metal ion-binding aptamers. The red bars indicate the signal from an aptamer binding to its target, while the blue bars indicate the signal from nontarget ions. We determined the relative binding of aptamer SSA-HgII at metal ion concentrations of 12.5 μM (A) and aptamer SSA-CuII at metal ion concentrations of 50 μM (B). Error bars represent standard deviations from 10 000 particle measurements.
CONCLUSION
We report a screening strategy that employs direct binding measurement to efficiently isolate high-quality structure-switching aptamers for metal ion targets without labeling, solid support, or chemical modifications. Using our SS-PD screening method, we obtained a DNA SSA for Hg2+ with ~30-fold higher affinity than existing aptamers and also discovered SSA for Cu2+ with excellent specificity. The label-free nature of our approach offers a critical advantage, ensuring minimal interference in aptamer–target interaction, and our two-color reporter system generates the self-calibrated fluorescent binding signals needed for FACS-based aptamer isolation. Although we used metal ions as the target in this work, we believe it should be feasible to use SS-PD to generate DNA SSAs for a broad spectrum of small-molecule targets with minimal alteration to the method. In this way, we believe SS-PD offers a highly accessible method for the research community to generate high-quality aptamer reagents against challenging molecular targets in an efficient and reproducible manner.
METHODS
DNA and Library Preparation
The single-stranded DNA (ssDNA) library, primers, and selected aptamer sequences were purchased from Integrated DNA Technologies (IDT). The nucleic acid library was synthesized with hand mixing and normal desalting purification. Each 97-nucleotide (nt) library member featured a 21-nt constant domain (with TTT sequences at either side) between a pair of randomized domains (10- and 20-nt), which were in turn flanked by two 23-nt PCR primer sites. The sequence of each library member was as follows: 5′-cctctctatgggcagtcggtgat-[10N]-TTT-CTGCAGCGATTCTTG-TTT-[20N]-ggagaatgaggaacccagtgcag-3′ (lowercase letters represent primer sites). The library had an equal probability (25%) of each base (A, T, G, or C) at each randomized site. The central constant domain was designed to hybridize to the AlexaFluor 647-modified 15-nt red reporter oligonucleotide (5′-CAAGAATCGCTGCAG-AlexaFluor 647-3′). During screening, the aptamers were also hybridized to the carboxyfluorescein- (FAM)-modified 23-nt green reporter oligonucleotide (5′-FAM-CTGCACTGGGTTCCTCATTCTCC-3′) and a forward primer (FP) complementary oligonucleo-tide (5′-ATCACCGACTGCCCATAGAGAGG-3′) to block the other primer site. Unlabeled, 5′-amino-modified, and FAM-modified (green reporter) PCR primers and red reporter DNA were obtained from IDT with HPLC purification.
Coupling FPs to Particles
A 500 μL amount of 1 μm MyOne carboxylic acid magnetic particles (107/μL, Life Technologies) was washed once with 500 μL of 0.01 N NaOH and five times with 500 μL of nuclease-free water, then resuspended in a 150 μL reaction mixture containing 200 mM NaCl, 0.2 mM 5′-amino-modified FP (5′-amino-PEG18-CCTCTCTATGGGCAGTCGGTGAT-3′), 1 mM imidazole chloride, 50% v/v dimethyl sulfoxide (DMSO), and 250 mM 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC). Amino group modification enables covalent coupling, keeping FPs attached to the particles during thermal cycling. 5′-PEG18 was incorporated as a spacer to minimize undesired interactions among the aptamers on the particle surface. Magnetic particles were mixed well with the above reagents and then vortexed, sonicated, and incubated overnight at room temperature on a rotator. After coupling was complete, we passivated the particle surfaces with PEG12. This was done by converting the remaining carboxyl groups on the particles to amino-reactive NHS-ester in the presence of 250 mM EDC and 100 mM N-hydroxysuccinimide (NHS) in 2-(N-morpholino)ethanesulfonic acid (MES) buffer (100 mM, pH 4.7) for 30 min at room temperature, followed by conjugation with 20 mM amino-PEG12 in MES buffer for 1 h. The particles were washed four times with 500 μL of 1× TE buffer (10 mM Tris, pH 8.0, 0.1 mM EDTA), then resuspended in 500 μL of 1× TE buffer and stored at 4 °C. Unless otherwise specified, all chemical reagents were purchased from Pierce Biotechnology.
Aptamer Particle Generation
We generated our aptamer particles via emulsion PCR. The oil phase (prepared fresh each day) was composed of 4.5% Span 80, 0.40% Tween 80, and 0.05% Triton X-100 in mineral oil, all purchased from Sigma-Aldrich. The aqueous phase consisted of 1× GoTaq PCR Master Mix (Promega), 25 mM MgCl2, 3.5 mM of each dNTP (Promega), 40 nM FP, 3 μM reverse primer (RP), 0.25 U/μL of GoTaq Hot Start Polymerase (Promega), 2 pM template DNA, and 3 × 108 FP-coated particles in a total volume of 1 mL. Water-in-oil emulsions were prepared by adding 1 mL of the aqueous phase to 7 mL of oil phase in a DT-20 tube (IKA) locked into the Ultra-Turrax device (IKA). This addition was performed dropwise over 30 s while the mixture was being stirred at 620 rpm in the Ultra-Turrax. After adding the aqueous phase, we continued stirring the mixture for 5 min. The emulsions were distributed in 100 μL aliquots into ~80 wells of one 96-well PCR plate.
We performed PCR under the following cycling conditions: 95 °C for 3 min, followed by 50 cycles of 93 °C for 15 s, 59 °C for 30 s, and 72 °C for 75 s. After PCR, the emulsions were collected into an emulsion collection tray (Life Technologies) by centrifuging at 500g for 2 min. We broke the emulsions by adding 10 mL of 2-butanol to the tray and transferred the collected sample to a 50 mL tube. After vortexing for 30 s, the particles were pelleted by centrifugation at 3000g for 5 min. After carefully removing the oil phase, we resuspended the particles in 1 mL of emulsion breaking buffer (100 mM NaCl, 1% Triton X-100, 10 mM Tris-HCl, pH 7.5, and 1 mM EDTA) and transferred them to a new 1.5 mL tube. After resuspending the particles via vortexing and centrifugation for 1 min at 21000g, we carefully removed the supernatant and transferred the particles to a new 1.5 mL tube. We repeated this washing step one more time and then placed the tube on a magnetic separator (MPC-S, Life Technologies) and pipeted off the remaining supernatant. Particles were washed once with 1× TE buffer using magnetic separation, then resuspended in 100 μL of 1× TE buffer.
To generate ssDNA, we magnetically concentrated the particles for 1 min and removed the supernatant with a pipet tip. We then resuspended the particles in 200 μL of 100 mM NaOH and incubated them for 20 min. We placed the tube in the magnetic separator for 1 min and carefully removed the supernatant. Particles were washed twice with 1× TE buffer using magnetic separation and then resuspended in 100 μL of 1× TE buffer.
SS-PD Screening
Prior to the screen, aptamer particles were hybridized with 2 μM each of red reporter and green reporter, as well as an FP-complementary oligonucleotide in PBSMCT buffer (DPBS with 2.5 mM MgCl2, 1 mM CaCl2, 0.025% Tween 20) for Hg2+. For Cu2+ selection, we used MESSM buffer (MES with 100 mM NaCl, 2 mM MgCl2, and 0.025% Tween 20) instead of PBSMCT, because Cu2+ has a strong interaction with phosphate groups in the PBSMCT buffer. Approximately 80% of the aptamer particles after emulsion PCR are predicted to display only FP on their surface based on Poisson statistics and thus should not exhibit significant binding to the small-molecule targets. These FP-only particles serve as an excellent negative control for establishing the “reference gate” (Figure 3 and Figure 4, dashed boxes). During each binding screen, we incubated ~108 aptamer particles in 2 mL of PBSMCT with Hg2+ or in 2 mL of MESSM with Cu2+ for 20 min at room temperature. Target concentrations used for each round are listed in Table S1. Two-color labeling allowed us to monitor the population of aptamer particles displaying structure-switching, low-affinity, and self-hybridizing aptamers via flow cytometry (BD FACS Aria II). The FACS Aria II instrument provides quantitative information as it counts the number of particles that pass through the interrogation region. When gates are drawn around a population of interest, the Aria II instrument displays the percentage of particles in a given gate. The Aria II also counts the number of particles that are sorted from a desired gate. We monitored the fluorescence distribution of aptamer particles at a range of different target concentrations (50 μM to 1 mM of Hg2+ or 25 to 400 μM of Cu2+) and chose the target concentration at which 0.3% to 2% of the aptamer particles displayed a reduction in red fluorescence in response to the target. After collecting the aptamer particles with low red and high green fluorescence, we concentrated the collected aptamer particles and rehybridized them with 2 μM of red reporter, green reporter, and FP complement in the appropriate buffer. These aptamer particles were then washed three times with buffer and sorted via FACS in the absence of target for the folding screen. This time, we isolated aptamer particles that showed high levels of both red and green fluorescence. After collecting these aptamer particles, we PCR amplified the isolated aptamers to generate an enriched pool for a subsequent round of aptamer particle synthesis.
Cloning and Sequencing
The SS-PD pool from the final round for HgCl2 (R4) and CuCl2 (R3) was cloned using a TOPO TA cloning kit (Life Technologies). Following PCR of the pool from the final round, the DNA was directly inserted into the pCR4-TOPO plasmid vector. The vectors were then transformed into chemically competent TOP10 E. coli cells. The cells were grown overnight on kanamycin-treated plates at 37 °C. We picked clones from 40 individual bacterial colonies from each pool and sent them off for Sanger sequencing (Genewiz).
Monoclonal Aptamer Particle Synthesis
After cloning and sequencing, we identified the subset of sequences with the fewest truncations or point-mutations in the constant domains and performed particle PCR to synthesize aptamer particles displaying each of these unique aptamer sequences. We first performed asymmetric PCR directly from the colonies. Each 50 μL reaction contained 1× GoTaq PCR Master Mix (Promega), 100 nM FP, and 1 μM unlabeled RP. We added template by touching a pipet tip into the desired colony and then using this pipet tip to mix the 50 μL of PCR mixture. We used the following PCR conditions: 95 °C for 2 min followed by 20 cycles of 95 °C for 15 s, 59 °C for 30 s, and 72 °C for 45 s, followed by a 7 min final extension at 72 °C. We then synthesized monoclonal aptamer particles by using the asymmetric PCR product for particle PCR. Each 60 μL particle PCR contained 1× GoTaq PCR Master Mix (Promega), 20 mM MgCl2, 1 μM green reporter (FAM-modified RP), 3 μL of asymmetric PCR product. and 3 × 107 FP-coated particles. PCR was carried out at 95 °C for 2 min followed by 24 to 32 cycles of 95 °C for 15 s, 59 °C for 30 s, and 72 °C for 75 s, followed by a 7 min final extension step at 72 °C. To avoid aggregation of the particles, we mixed the reactions by vortexing and sonicating the reagent tubes every four cycles in the middle of the 72 °C elongation step. Particle PCR progress was monitored and adjusted according to the FAM fluorescence intensity of the aptamer particles, as measured by flow cytometry. Following amplicon generation on the surface of the particles, we removed the reverse strand by treatment with 200 μL of 100 mM NaOH for 20 min.
Flow Cytometry-Based IC50 Assays
Following cloning, sequencing, and particle PCR, we determined the IC50 value of all sequences by a flow cytometry binding assay. The aptamer particles were hybridized with 2 μM red reporter, green reporter, and FP complementary strands in PBSMCT buffer for Hg2+ or MESSM buffer for Cu2+ aptamer particles and then washed three times with buffer to remove excess reporters and FP complement. We incubated 1.5 μL aliquots (~4.5 × 105 beads) of hybridized aptamer particles in 50 μL of buffer with titrations of Hg2+ ranging from 12.5 to 200 μM or of Cu2+ ranging from 25 to 800 μM at room temperature for 20 min on a rotator. AlexaFluor 647 fluorescence was then quantitated using an Accuri C6 flow cytometer (BD Biosciences). All IC50 and KD measurements were obtained using buffers that contained metal ion cofactors, such as Mg2+, and these cofactors could help to stabilize aptamer structures in solution. Furthermore, these cofactors may compete with the binding of target metal ions to the aptamer. As a result, it should be noted that the measured IC50 and KD values may be influenced by the actions of these metal ion cofactors.
Derivation of the Relationship between IC50 and True KD
We expect the IC50 value obtained via fluorescence measurement to be different from the KD. This is because there are two binding reactions in competition with each other: the hybridization of the red reporter to the constant domain of the aptamer and the binding of the aptamer to the target. In conditions where the concentration of red reporter ([Reporter]) is much larger than its dissociation constant (valid for most circumstances), the chemical equilibrium of the hybridization of red reporter can be described by
where [Reporter] is the concentration of unhybridized reporter (single-stranded form) and [Duplex] is the concentration of aptamer–reporter duplexes (double-stranded form). After incubating with target at a concentration of [Target], the aptamer–target chemical equilibrium can be described as
where [Complex] is the concentration of aptamer–target complexes.
We subsequently derived the aptamer particle fluorescence F as follows:
| (1) |
where Fm is the maximum fluorescence of one particle, Fb is the background fluorescence, and is the “effective” dissociation constant of the target in the presence of the red reporter. This approximation holds when , which is valid under our experimental conditions.
Note that the true dissociation constant 31,32 is expected to be smaller than IC50 by a factor of . for our experimental conditions, due to the intensive washing of the aptamer particles after hybridizing with the red reporter, the population of free red reporter in solution ([Reporter]) roughly equals that of the aptamers immobilized on the aptamer particles. We estimated this to be 4 × 10−10 M, equal to the concentration of particles (103/μL) times the coverage of aptamers for each aptamer particle (2.4 × 105). Given the free energy of hybridization for the red reporter (ΔG = −17.1 kcal/mol),33 we estimated the dissociation constant to be , or ~10−12−10−11 M, which is consistent with the literature.34 Given this, we estimate the true dissociation constant KD to be smaller than the measured IC50 value by approximately an order of magnitude.
Microscale Thermophoresis-Based KD Assays
The true KD’s of SSA-HgII, the truncated SSA-HgII, and a previously reported Hg2+ aptamer, HgA, were experimentally measured by 2Bind (Regensburg, Germany) using microscale thermophoresis assays for molecular interactions. All aptamers were synthesized by IDT with Cy5 fluorophore modifications at the 5′ end. Cy5-labeled aptamers (5 nM) were incubated in PBSMCT buffer with a 2-fold titration series of HgCl2 ranging from low nanomolar to midmicromolar concentrations. SSA-HgII samples were then analyzed on a Monolith Pico device (NanoTemper Technologies) at 25 °C. The KD of the Hg2+ aptamers was determined based on the fluorescence shift due to normal thermophoresis. Error bars in the plots represent the standard deviation of two measurement trials, and the red solid lines are the fit using the regular binding equation, where binding fraction η = 1/(1 + KD/[Ligand]).
Specificity Assay
The specificity of SSA-HgII and SSA-CuII was tested by challenging monoclonal SSA-HgII and SSA-CuII aptamer particles with various nontarget divalent ions. First, monoclonal SSA-HgII and SSA-CuII aptamer particles were hybridized to 2 μM of red reporter, green reporter, and FP complement in PBSMCT buffer for SSA-HgII aptamer particles or MESSM buffer for SSA-CuII aptamer particles and then washed three times with buffer. We aliquoted 1.5 μL (~4.5 × 105 beads) of SSA-HgII or SSA-CuII aptamer particles into 50 μL of buffer with nontarget divalent ions at two different concentrations and incubated them for 20 min at room temperature on a rotator. The concentrations and ions used were 12.5 μM Ca2+, Mg2+, Pb2+, Zn2+, and Cu2+ for SSA-HgII and 50 μM Ca2+, Mg2+, Pb2+, Zn2+, and Hg2+ for SSA-CuII. The AlexaFluor 647 fluorescence for SSA-HgII or SSA-CuII in each reaction tube was quantitated using an Accuri C6 flow cytometer.
We determined the relative binding to nontarget ions as a percentage of each aptamer’s maximal binding to its target ion. We first calculated the fluorescence change for each ion by subtracting the fluorescence of SSA-HgII or SSA-CuII aptamer particles incubated with a given ion (Fi) from that of the aptamer particle alone (Fmax). Next, we computed the maximum fluorescence change by subtracting the fluorescence measurement obtained from SSA-HgII incubated with 200 μM Hg2+ or SSA-CuII incubated with 800 μM Cu2+ (Fmin) from the Fmax measurement obtained in the absence of any target. Since the fluorescence change for each aptamer particle is directly proportional to the quantity of either the bound aptamers on the surface of the particle or the released red reporter strands, the magnitude of binding for each ion can be given as a percentage of the maximum signal, expressed as (Fmax − Fi)/(Fmax − Fmin).
Supplementary Material
Acknowledgments
We are grateful for the financial support of the Garland Initiative, ARO Institute for Collaborative Biotechnologies (W911F-09-D-0001, W81XWH-09-0698), DARPA (N66001-14-2-4055), and National Institutes of Health (U01 HL099773). We thank Prof. James A. Thomson, Prof. Peter Coffey, Dr. Susanne Meyer, and Dr. Monte Radeke for assistance with the cell sorter instrument. We thank Margaret A. Nakamoto for assistance in editing the manuscript.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.6b02558.
Additional information (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Gaeta A, Hider RC. The Crucial Role of Metal Ions in Neurodegeneration: The Basis for a Promising Therapeutic Strategy. Br J Pharmacol. 2005;146:1041–1059. doi: 10.1038/sj.bjp.0706416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drew SC, Barnham KJ. The Heterogeneous Nature of Cu 2+ Interactions with Alzheimer’s Amyloid-β Peptide. Acc Chem Res. 2011;44:1146–1155. doi: 10.1021/ar200014u. [DOI] [PubMed] [Google Scholar]
- 3.Finder VH, Glockshuber R. Amyloid-beta Aggregation. Neurodegener Dis. 2007;4:13–27. doi: 10.1159/000100355. [DOI] [PubMed] [Google Scholar]
- 4.Clarkson TW, Magos L. The Toxicology of Mercury and Its Chemical Compounds. Crit Rev Toxicol. 2006;36:609–662. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- 5.Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and Cellular Mechanisms of Cadmium Carcinogenesis. Toxicology. 2003;192:95–117. doi: 10.1016/s0300-483x(03)00305-6. [DOI] [PubMed] [Google Scholar]
- 6.Duruibe JO, Ogwuegbu MOC, Egwurugwu JN. Heavy Metal Pollution and Human Biotoxic Effects. Int J Phys Sci. 2007;2:112–118. [Google Scholar]
- 7.He ZL, Yang XE, Stoffella PJ. Trace Elements in Agroecosystems and Impacts on the Environment. J Trace Elem Med Biol. 2005;19:125–140. doi: 10.1016/j.jtemb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 8.Järup L. Hazards of Heavy Metal Contamination. Br Med Bull. 2003;68:167–182. doi: 10.1093/bmb/ldg032. [DOI] [PubMed] [Google Scholar]
- 9.Domaille DW, Que EL, Chang CJ. Synthetic Fluorescent Sensors for Studying the Cell Biology of Metals. Nat Chem Biol. 2008;4:168–175. doi: 10.1038/nchembio.69. [DOI] [PubMed] [Google Scholar]
- 10.Dean KM, Qin Y, Palmer AE. Visualizing Metal Ions in Cells: An Overview of Analytical Techniques, Approaches, and Probes. Biochim Biophys Acta, Mol Cell Res. 2012;1823:1406–1415. doi: 10.1016/j.bbamcr.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayat A, Marty JL. Aptamer Based Electrochemical Sensors for Emerging Environmental Pollutants. Front Chem. 2014;2:41. doi: 10.3389/fchem.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiang Y, Lu Y. DNA as Sensors and Imaging Agents for Metal Ions. Inorg Chem. 2012;29:997–1003. doi: 10.1021/ic4019103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuerk C, Gold L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 14.Ellington AD, Szostak JW. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Lett To Nat. 1990;346:183–187. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 15.Jayasena SD. Aptamers: An Emerging Class of Molecules That Rival Antibodies in Diagnostics. Clin Chem. 1999;45:1628–1650. [PubMed] [Google Scholar]
- 16.Chung CH, Kim JH, Jung J, Chung BH. Nuclease-Resistant DNA Aptamer on Gold Nanoparticles for the Simultaneous Detection of Pb2+ and Hg2+ in Human Serum. Biosens Bioelectron. 2013;41:827–832. doi: 10.1016/j.bios.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 17.Li T, Dong S, Wang E. Label-Free Colorimetric Detection of Aqueous Mercury Ion Label-Free Colorimetric Detection of Aqueous Mercury Ion (Hg2+) Using Hg2+-Modulated G-Quadruplex-Based DNAzymes. Anal Chem. 2009;81:2144–2149. doi: 10.1021/ac900188y. [DOI] [PubMed] [Google Scholar]
- 18.Long F, Zhu A, Shi H, Wang H, Liu J. Rapid On-Site/In-situ Detection of Heavy Metal Ions in Environmental Water Using a Structure-Switching DNA Optical Biosensor. Sci Rep. 2013;3:1–7. doi: 10.1038/srep02308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoltenburg R, Reinemann C, Strehlitz B. FluMag-SELEX as an Advantageous Method for DNA Aptamer Selection. Anal Bioanal Chem. 2005;383:83–91. doi: 10.1007/s00216-005-3388-9. [DOI] [PubMed] [Google Scholar]
- 20.Ahmad KM, Oh SS, Kim S, McClellen FM, Xiao Y, Soh HT. Probing the Limits of Aptamer Affinity with a Microfluidic SELEX Platform. PLoS One. 2011;6:e27051. doi: 10.1371/journal.-pone.0027051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Gong Q, Maheshwari N, Eisenstein M, Arcila ML, Kosik KS, Soh HT. Particle Display: A Quantitative Screening Method for Generating High-Affinity Aptamers. Angew Chem, Int Ed. 2014;53:4796–4801. doi: 10.1002/anie.201309334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKeague M, Derosa MC. Challenges and Opportunities for Small Molecule Aptamer Development. J Nucleic Acids. 2012;2012:1–20. doi: 10.1155/2012/748913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lomenick B, Olsen RW, Huang J. Identification of Direct Protein Targets of Small Molecules. ACS Chem Biol. 2011;6:34–46. doi: 10.1021/cb100294v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trevino SG, Levy M. High-Throughput Bead-Based Identification of Structure-Switching Aptamer Beacons. Chem Bio Chem. 2014;15:1877–1881. doi: 10.1002/cbic.201402037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nutiu R, Li Y. In Vitro Selection of Structure-Switching Signaling Aptamers. Angew Chem, Int Ed. 2005;44:1061–1065. doi: 10.1002/anie.200461848. [DOI] [PubMed] [Google Scholar]
- 26.XXXStoltenburg R, Nikolaus N, Strehlitz B. Capture-SELEX: Selection of DNA Aptamers for Aminoglycoside Antibiotics. J Anal Methods Chem. 2012;1 doi: 10.1155/2012/415697. 110.1155/2012/415697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biles BD, Connolly BA. Low-Fidelity Pyrococcus furiosus DNA Polymerase Mutants Useful in Error-Prone PCR. Nucleic Acids Res. 2004;32:e176. doi: 10.1093/nar/gnh174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seidel SAI, Dijkman PM, Lea WA, van den Bogaart G, Jerabek-Willemsen M, Lazic A, Joseph JS, Srinivasan P, Baaske P, Simeonov A, Katritch I, Melo FA, Ladbury JE, Schreiber G, Watts A, Braun D, Duhr S. Microscale Thermophoresis Quantifies Biomolecular Interactions under Previously Challenging Conditions. Methods. 2013;59:301–315. doi: 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ono A, Togashi H. Highly Selective Oligonucleotide-Based Sensor for Mercury(II) in Aqueous Solutions. Angew Chem, Int Ed. 2004;43:4300–4302. doi: 10.1002/anie.200454172. [DOI] [PubMed] [Google Scholar]
- 30.Maji D, Zhou M, Sarder P, Achilefu S. Near infrared fluorescence quenching properties of copper (II) ions for potential applications in biological imaging. Proc SPIE. 2014;8956:1–6. [Google Scholar]
- 31.Schneider EV, Böttcher J, Huber R, Maskos K, Neumann L. Structure-Kinetic Relationship Study of CDK8/CycC Specific Compounds. Proc Natl Acad Sci U S A. 2013;110:8081–8086. doi: 10.1073/pnas.1305378110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng Y, Prusoff WH. Relationship between the Inhibition Constant (K1) and the Concentration of Inhibitor which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 33.Markham NR, Zuker M. DINAMelt Web Server for Nucleic Acid Melting Prediction. Nucleic Acids Res. 2005;33:577–581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stevens PW, Henry MR, Kelso DM. DNA Hybridization on Microparticles: Determining Capture-Probe Density and Equilibrium Dissociation Constants. Nucleic Acids Res. 1999;27:1719–1727. doi: 10.1093/nar/27.7.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.