

Abstract
Background:
Oxidative stress and increased DNA damage have been implicated in the etiopathogenesis of vitiligo. Oxidative DNA damage is mainly repaired by the base excision repair (BER) pathway.
Aim:
We sought to determine whether polymorphisms in DNA repair genes may have a role in the pathogenesis of vitiligo.
Materials and Methods:
We conducted a study including 100 patients with vitiligo and age- and sex-matched 193 control subjects to examine the role of single-nucleotide polymorphisms of BER genes, human 8-oxoG DNA N-glycosylase 1 (codon 326), apurinic/apyrimidinic endonuclease 1 (APE1) (codon 148), and X-ray repair cross-complementing group 1 (codon 399) as risk factors for vitiligo. These polymorphisms were determined by quantitative real-time polymerase chain reaction and melting curve analysis.
Results:
No significant association was observed between the variant alleles of studied genes and vitiligo.
Conclusion:
However, we showed that the presence of APE1 148Glu variant allele is associated with leukotrichia. This preliminary study suggests that APE1 (codon 148) polymorphism may play a role in vitiligo pathogenesis.
Key words: Apurinic/apyrimidinic endonuclease 1, DNA repair, gene polymorphism, human 8-oxoguanine DNA N-glycosylase 1, vitiligo, X-ray repair cross-complementing group 1
INTRODUCTION
Vitiligo is a progressive depigmentary disorder of skin, resulting in loss of functional melanocytes. Several theories are thought to play a role in this complex process, including autoimmune, cytotoxic metabolites, neural and the genetic theories.[1] Melanin content and synthesis are assumed to be effected by a defective free radical reservoir.[2] Recently, scientific evidences linking oxidative stress and immune system to vitiligo pathogenesis highlight a convergent terminal pathway of oxidative stress–autoimmunity-mediated melanocyte loss.[3] The oxidative stress theory has been studied and incriminated in the etiopathogenesis of vitiligo.[1,4,5] Skin, creating an interface between the body and the outside world, is exposed to chemical (heavy metals), physical (ultraviolet radiation), or biological (allergens, microbiological agents) stimuli causing a significant increase in reactive oxygen species (ROS).[6] ROS can also be produced endogenously, primarily through mitochondrial respiration. Furthermore, pigment-forming cells are a potential source of ROS too.[7] In the recent studies, production and accumulation of ROS (i.e., hydrogen peroxide and peroxynitrite) and reduced catalase activity have been shown in the epidermal layer of vitiliginous skin.[4,8,9,10] In addition, an imbalance was found in some important antioxidants, including catalase, glutathione peroxidase, superoxide dismutase, and Vitamin E in peripheral blood mononuclear cells of vitiligo patients, which is correlated with increased intracellular ROS. The latter study shows the presence of systemic oxidative stress in vitiligo.[5]
In cells, DNA is probably the most biologically significant target of oxidative attack by ROS. The lesions induced by ROS on DNA are typically single- and double-strand DNA breaks in apurinic/apyrimidinic (AP) sites or oxidative alteration in DNA bases.[6,11] Guanine is the most vulnerable DNA base to oxidative stress.[10] Therefore, 8-oxoG can be used as a stable biomarker to detect oxidative DNA damage.[11] Increased 8-oxoG levels have recently been reported in the skin,[10] serum,[10,12] and in peripheral blood mononuclear cells[5,13] of patients with vitiligo as compared to controls.
Oxidative DNA damages are mainly repaired by base excision repair (BER) which is a major DNA repair pathway in mammalian cells.[12,14,15] In the first step of this pathway, a lesion-specific DNA glycosylase removes the damaged base by hydrolyzing the N-glycosidic bond of the base. In humans, 8-oxoG DNA N-glycosylase 1 (hOGG1) is the the major glycosylase which recognize and excise 8-oxoG. After removing the damaged base, an abasic (AP) site occurs in DNA[14] and this site is incised by AP endonuclease (APE), leaving a single-nucleotide gap. A DNA polymerase fills this gap and a DNA ligase III completes the repair process by sealing the nick.[6,14] X-ray repair cross-complementing group 1 (XRCC1) is an important scaffold protein that interacts with all these proteins to facilitate efficient repair of DNA damage and coordinate the whole process.[16] Genetic variations in these genes can change BER function and impair DNA damage repair capacity.
There are reports on some oxidative stress-related disorders incriminating DNA repair gene polymorphisms.[17,18,19,20] In the literature, there is only one study showing that APE1 Asp148Glu polymorphism aggravates oxidative stress in human melanocytes and contributes to genetic predisposition to vitiligo in Chinese people.[12] Accordingly, we aimed to investigate the BER gene polymorphisms of three genes; hOGG1 Ser326Cys, APE1 Asp148Glu, and XRCC1 Arg399Gln in the etiopathogenesis of vitiligo.
MATERIALS AND METHODS
Patients
A total of 100 vitiligo patients older than 18 years showing any clinical distribution except segmentary type and who were not on systemic or topical therapy were included in the study. Vitiligo was diagnosed on the basis of clinical findings, at the dermatology outpatient clinic. The control group consisted of age- and sex-matched 193 dermatology outpatients who accepted to join the study, with diagnoses of melanocytic nevus, callus, or fibroepithelial polyps that are not known to be associated with oxidative stress or DNA repair system. Additional criteria of no past history of any systemic, infectious, autoimmune, genetic, or atopic disease and a negative family history for vitiligo were also provided. Patients taking any tablet including vitamins were excluded from the study. The study was approved by the Local Ethics Committee and written informed consent was taken from all the participants.
DNA isolation
Genomic DNA was isolated from peripheral blood leukocytes using high pure polymerase chain reaction (PCR) template preparation kit (Roche Diagnostics GmbH, Mannheim, Germany).
Genotyping by fluorescence-based melting curve analysis
Detection of polymorphisms was done by rapid capillary PCR with melting curve analysis using fluorescence-labeled hybridization probes in a LightCycler (Roche Diagnostics GmbH, Mannheim, Germany). Primers and probes were prepared by Metabion International AG (Martinsried, Germany).
Analysis was done in 20 μl volumes using glass capillaries. The PCR mix contained ~50–100 ng of the genomic DNA, 1x LC™ FastStart DNA Master HybProbe kit (Roche Diagnostics), 0.5 μM of each primer, 0.15 μM of each probe, and 2.5 mM total MgCl2. Reaction conditions were as follows: initial denaturation at 95°C for 10 min then 40 cycles of denaturation at 95°C for 15 s, annealing at 58°C (hOGG1 codon 326) or 56°C (APE/Ref-1 codon 148) or 55°C (XRCC1 codon 399), and elongation at 72°C for 12 s. Melting curve analysis was done with an initial denaturation step at 95°C for 5 s and 20 s at 50°C (45°C for XRCC1 codon 399), slow heating to 75°C with a ramping rate of 0.15°C/s (0.1°C/s for XRCC1 codon 399), and continuous fluorescence detection. Melting curves were converted to melting peaks by plotting the negative derivatives of fluorescence against temperature (−dF/dT). A negative control containing all reagents but water instead of the DNA template was included to each amplification set. Melting curves were evaluated by two independent observers who were blinded to the analysis of the clinical data. In addition, 10% of randomly selected samples were repeated independently to verify genotyping results and 100% concordance was found.
Statistical analysis
Differences in genotype distributions and allele frequencies in the cases and the controls were compared for statistical significance using the Chi-square (χ2) test. The statistical significance for deviations from Hardy–Weinberg Equilibrium (HWE) was determined using the Pearson χ2-test. Odds ratios were calculated and given with 95% confidence intervals. The wild-type genotype/allele served as a reference category. Comparison of individual clinical variables between genotypes was assessed with χ2-test. Statistical analyses were performed with SPSS version 11.0 for Windows (SPSS Inc., Chicago, IL, USA). In addition, the NCSS 2000 statistical package was used to evaluate the power analysis.
RESULTS
A total of 293 subjects (100 vitiligo and 193 controls) were included in this case–control study. Table 1 depicts the clinical characteristics of the vitiligo patients including clinical type of the disease, duration, family history, leukotrichia, stability within 1 year, and associated diseases. The mean age was 35.6 ± 9.6 years (100 female and 93 male, range 18–72 years) for controls. There was no significant difference among the study and control groups in terms of mean age and sex distribution (P = 0.061 and P = 0.435, respectively). We had an 87.5% power to detect an effect size (W) of 0.20 using a 2 degrees of freedom (α = 0.05).
Table 1.
Characteristics of the patients with vitiligo

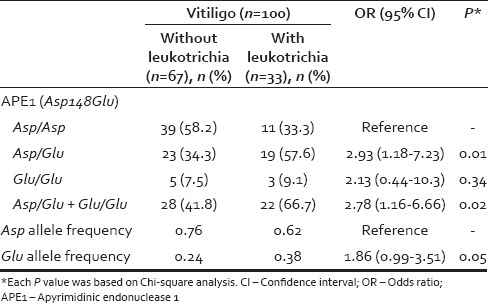
The genotypic and allelic distributions of the polymorphisms in hOGG1, APE1, and XRCC1 genes in the patients and controls are shown in Table 2. The distributions of the hOGG1 codon 326, APE/Ref-1 codon 148, and XRCC1 codon 399 genotypes were in accordance with the HWE among the controls (P = 0.77, P = 0.61 and P = 0.76, respectively) and the patients with vitiligo (P = 0.56, P = 0.95 and P = 0.85, respectively) The variant allele frequencies found in the control group were consistent with our previous studies.[17,21,22,23] A relation between these polymorphisms and the clinical features of vitiligo patients (type of the disease, duration/age of onset, family history, leukotrichia, stability within 1 year and associated diseases) was also analyzed. Among these parameters, only leukotrichia was found to be related with the Asp148Glu variant allele of APE1 polymorphism [Table 3].
Table 2.
Distribution of genotypes and allele frequencies in patients with vitiligo (n=100) and control group (n=193)
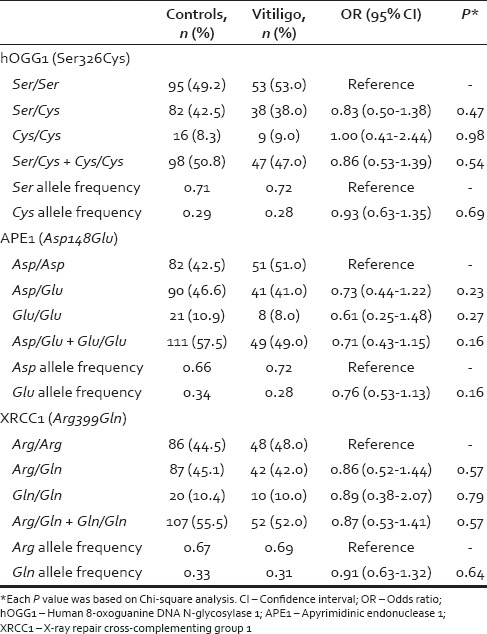
Table 3.
Distribution of apyrimidinic endonuclease 1 (Asp148Glu) genotypes and allele frequencies in patients with vitiligo with and without leukotrichia
DISCUSSION
The BER pathway is the most important cellular protection mechanism responding to oxidative DNA damage, and hOGG1 is a key enzyme removing damaged bases by cleavage of the N-glycosylic bonds between the bases and the deoxyribose moieties. Previous studies have revealed the presence of several polymorphisms in the gene encoding hOGG1 protein. Of these, Ser326Cys is the most common polymorphism and has attracted widespread attention.[17] In this polymorphism, the C to G substitution occurs at position 1245 in exon 7 of hOGG1 gene, resulting in an amino acid substitution of serine with cysteine in codon 326. It has been found that hOGG1 Ser326Cys polymorphism is associated with a reduced capacity to repair oxidative DNA damage.[21,22] Although some studies have shown conflicting results.[23] Moreover, there are some epidemiological studies showing increased risk of disease in the presence of this polymorphism.[16,17,19] Contrary to the findings of increased 8-oxoG levels in vitiligo and the role of hOGG1 in removal of 8-oxoG, we did not find any association between allele or genotype frequencies of the hOGG1 Ser326Cys variant and the risk for vitiligo.
APE1 is another essential enzyme in BER pathway which incises AP site, in the DNA by hydrolyzing the phosphodiester backbone.[16] The Asp148Glu polymorphism of APE1 gene predicts an amino acid change from aspartic acid to glutamic acid change at amino acid 148 of APE1 protein. Wei et al. investigated APE1 Asp148Glu polymorphism as a candidate risk factor for vitiligo in a Han Chinese population.[12] They found that 148Glu variant genotypes (Asp/Glu heterozygotes and Glu/Glu variant genotype) were associated with a slight increased risk (1.24- and 1.48-fold, respectively) of vitiligo compared with the wild type genotype (Asp/Asp). This association was more pronounced in the subgroup of nonsegmental, male, active vitiligo patients with an age of onset after 20 years.[12] Furthermore, in cultured human melanocytes, 8-oxoG levels were found to be increased in patients carrying APE1-148Glu variant. Consequently, the authors suggested that APE1 polymorphisms may increase oxidative DNA damage of human melanocytes and affect the risk of vitiligo in the described subgroup of their study population.[12] However, our study did not show a direct significant association of this polymorphism and vitiligo. We carried out a detailed investigation of the associations, if any, between APE1 polymorphisms and subgroups of vitiligo patients with different clinical characteristics. Within this frame type of the vitiligo, age of onset, duration of the disease, family history, stability within 1 year, presence of leukotrichia, and presence of associated diseases were evaluated. We found out that the presence of APE1 148Glu variant allele was related only with leukotrichia.
This is the first study showing APE1 148Glu variant allele is associated with leukotrichia. Leukotrichia is defined as the presence of a localized patch of white hair and has been reported in 9%–45% of vitiligo patients.[24] Leucotrichia can follow or precede depigmentation of surrounding epidermis. It is caused by loss of hair melanocytes found deep in the proximal anagen hair bulb. Vitiliginous areas with overlying leukotrichia usually fail to achieve repigmentation by conventional medical treatments. Thus, leukotrichia is known to be a poor prognostic sign. Actually, hair bulb is an immune priviledged site and its involvement may be a sign of severe damage. Therapy resistance may be the result of deficient melanocyte reservoir or repair difficulties in this location. Follicular pigmentation is controlled by different intrinsic and extrinsic factors. Since oxidative stress is a well-known factor in the destruction of follicular melanocytes, it is assumed that BER mechanisms should be working in this process. The functional significance of 148 Glu variant of APE1 protein has not been elucidated. Previously, it was suggested that genetic variants of APE1 may cause reduced or missed enzymatic activity leading to inadequate repair. Recently, altered endoribonuclease activity of APE1 variants has been identified in the human population.[25] With reference to what is known, we think APE1 polymorphism may contribute severe oxidative damage of melanocytes by lack of repair.
XRCC1 is an important platform protein participating in the BER pathway, which is recruited to the site of repair till the last stage of ligation. It regulates and coordinates the whole process by interacting with DNA ligase and certain polymerases.[26,27] XRCC1 also facilitates exchange of DNA glycosylase with APE1 at the damaged substrate. Although over 300 polymorphisms have been described in XRCC1 gene, the Arg399Gln has been the most extensively studied. The G → A substitution in codon 399 of exon 10 in XRCC1 gene alter amino acid sequence of XRCC1 protein from arginine to glutamine. This alteration may affect the capacity to undergo DNA repair and produces significant conformational changes in the XRCC1 protein, including the loss of secondary structural features that may be critical for protein–protein interactions in BER.[28] XRCC1-codon 399, the variant allele has been suggested to be associated with increased DNA damage in lymphocytes,[29] mononuclear cells,[30] and leukocytes.[31] It has been found to be likely that an impaired DNA damage may induce an autoimmune response in susceptible individuals. In our previous research, we have studied XRCC1 gene polymorphisms as a candidate risk factor for various disease.[17,18,32,33,34] However, in this study, there was no significant association between XRCC1 Arg399Gln polymorphism and the risk of vitiligo. Similarly, Wei et al. did not detect any significant association of this polymorphism in their study group of Chinese vitiligo patients.[12]
This study has some limitations. Although we are aware of the preference of larger sample size, we were obliged to work on a relatively small number of subjects due to budget restrictions.
CONCLUSION
APE1 polymorphism has been found to be associated with leukotrichia in our study group of vitiligo patients. The preliminary results of this study may prove to be valuable in understanding possible underlying mechanisms and the therapy resistance in this subset of patients. Furthermore, it may help develop treatments targeting DNA repair pathways besides surgical procedures. Finally, we admit the need for controlling these findings with larger and population-based studies on various geographic areas and with different genetic backgrounds.
Financial support and sponsorship
This study was supported by the Research Fund of the Turkish Dermatological Society.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Schallreuter KU, Bahadoran P, Picardo M, Slominski A, Elassiuty YE, Kemp EH, et al. Vitiligo pathogenesis: Autoimmune disease, genetic defect, excessive reactive oxygen species, calcium imbalance, or what else? Exp Dermatol. 2008;17:139–40. doi: 10.1111/j.1600-0625.2007.00666_1.x. [DOI] [PubMed] [Google Scholar]
- 2.Laddha NC, Dwivedi M, Mansuri MS, Gani AR, Ansarullah M, Ramachandran AV, et al. Vitiligo: Interplay between oxidative stress and immune system. Exp Dermatol. 2013;22:245–50. doi: 10.1111/exd.12103. [DOI] [PubMed] [Google Scholar]
- 3.Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90–100. doi: 10.1034/j.1600-0749.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 4.Schallreuter KU, Moore J, Wood JM, Beazley WD, Gaze DC, Tobin DJ, et al. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J Investig Dermatol Symp Proc. 1999;4:91–6. doi: 10.1038/sj.jidsp.5640189. [DOI] [PubMed] [Google Scholar]
- 5.Dell'Anna ML, Maresca V, Briganti S, Camera E, Falchi M, Picardo M. Mitochondrial impairment in peripheral blood mononuclear cells during the active phase of vitiligo. J Invest Dermatol. 2001;117:908–13. doi: 10.1046/j.0022-202x.2001.01459.x. [DOI] [PubMed] [Google Scholar]
- 6.Iyama T, Wilson DM., 3rd DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 2013;12:620–36. doi: 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smit NP, van Nieuwpoort FA, Marrot L, Out C, Poorthuis B, van Pelt H, et al. Increased melanogenesis is a risk factor for oxidative DNA damage – Study on cultured melanocytes and atypical nevus cells. Photochem Photobiol. 2008;84:550–5. doi: 10.1111/j.1751-1097.2007.00242.x. [DOI] [PubMed] [Google Scholar]
- 8.Ohno M, Miura T, Furuichi M, Tominaga Y, Tsuchimoto D, Sakumi K, et al. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 2006;16:567–75. doi: 10.1101/gr.4769606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schallreuter KU, Wood JM, Berger J. Low catalase levels in the epidermis of patients with vitiligo. J Invest Dermatol. 1991;97:1081–5. doi: 10.1111/1523-1747.ep12492612. [DOI] [PubMed] [Google Scholar]
- 10.Schallreuter KU, Moore J, Wood JM, Beazley WD, Peters EM, Marles LK et al. Epidermal H2O2 accumulation alters tetrahydrobiopterin (6BH4) recycling in vitiligo: Identification of a general mechanism in regulation of all 6BH4-dependent processes? J Invest Dermatol. 2001;116:167–74. doi: 10.1046/j.1523-1747.2001.00220.x. [DOI] [PubMed] [Google Scholar]
- 11.Salem MM, Shalbaf M, Gibbons NC, Chavan B, Thornton JM, Schallreuter KU. Enhanced DNA binding capacity on up-regulated epidermal wild-type p53 in vitiligo by H2O2-mediated oxidation: A possible repair mechanism for DNA damage. FASEB J. 2009;23:3790–807. doi: 10.1096/fj.09-132621. [DOI] [PubMed] [Google Scholar]
- 12.Wei C, Jian Z, Wang L, Qiang H, Shi Q, Guo S, et al. Genetic variants of the APE1 gene and the risk of vitiligo in a Chinese population: A genotype-phenotype correlation study. Free Radic Biol Med. 2013;58:64–72. doi: 10.1016/j.freeradbiomed.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Giovannelli L, Bellandi S, Pitozzi V, Fabbri P, Dolara P, Moretti S. Increased oxidative DNA damage in mononuclear leukocytes in vitiligo. Mutat Res. 2004;556:101–6. doi: 10.1016/j.mrfmmm.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Parsons JL, Dianov GL. Co-ordination of base excision repair and genome stability. DNA Repair (Amst) 2013;12:326–33. doi: 10.1016/j.dnarep.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Seeberg E, Eide L, Bjørås M. The base excision repair pathway. Trends Biochem Sci. 1995;20:391–7. doi: 10.1016/s0968-0004(00)89086-6. [DOI] [PubMed] [Google Scholar]
- 16.Karahalil B, Bohr VA, Wilson DM., 3rd Impact of DNA polymorphisms in key DNA base excision repair proteins on cancer risk. Hum Exp Toxicol. 2012;31:981–1005. doi: 10.1177/0960327112444476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanrikulu S, Dogru-Abbasoglu S, Ozderya A, Ademoglu E, Karadag B, Erbil Y, et al. The 8-oxoguanine DNA N-glycosylase 1 (hOGG1) Ser326Cys variant affects the susceptibility to Graves' disease. Cell Biochem Funct. 2011;29:244–8. doi: 10.1002/cbf.1742. [DOI] [PubMed] [Google Scholar]
- 18.Dogru-Abbasoglu S, Aykaç-Toker G, Hanagasi HA, Gürvit H, Emre M, Uysal M. The Arg194Trp polymorphism in DNA repair gene XRCC1 and the risk for sporadic late-onset Alzheimer's disease. Neurol Sci. 2007;28:31–4. doi: 10.1007/s10072-007-0744-x. [DOI] [PubMed] [Google Scholar]
- 19.Sun C, Liu X, Zhang H, Guo W, Cai Z, Chen H, et al. Functional polymorphism of hOGG1 gene is associated with type 2 diabetes mellitus in Chinese population. Mol Cell Endocrinol. 2010;325:128–34. doi: 10.1016/j.mce.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Kang SY, Lee KG, Lee W, Shim JY, Ji SI, Chung KW, et al. Polymorphisms in the DNA repair gene XRCC1 associated with basal cell carcinoma and squamous cell carcinoma of the skin in a Korean population. Cancer Sci. 2007;98:716–20. doi: 10.1111/j.1349-7006.2007.00436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohno T, Shinmura K, Tosaka M, Tani M, Kim SR, Sugimura H, et al. Genetic polymorphisms and alternative splicing of the hOGG1 gene, that is involved in the repair of 8-hydroxyguanine in damaged DNA. Oncogene. 1998;16:3219–25. doi: 10.1038/sj.onc.1201872. [DOI] [PubMed] [Google Scholar]
- 22.Vodicka P, Stetina R, Polakova V, Tulupova E, Naccarati A, Vodickova L, et al. Association of DNA repair polymorphisms with DNA repair functional outcomes in healthy human subjects. Carcinogenesis. 2007;28:657–64. doi: 10.1093/carcin/bgl187. [DOI] [PubMed] [Google Scholar]
- 23.Dherin C, Radicella JP, Dizdaroglu M, Boiteux S. Excision of oxidatively damaged DNA bases by the human alpha-hOgg1 protein and the polymorphic alpha-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res. 1999;27:4001–7. doi: 10.1093/nar/27.20.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ortonne JP, Bahadoran P, Fitzpatrick TB, Mosher DB, Hori Y. Hypomelanosis and hypermelanosis. In: Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. Fitzpatrick's Dermatology in General Medicine. New York: McGraw-Hill; 2003. p. 841. [Google Scholar]
- 25.Kim WC, Ma C, Li WM, Chohan M, Wilson DM, rd, Lee CH. Altered endoribonuclease activity of apurinic/apyrimidinic endonuclease 1 variants identified in the human population. PLoS One. 2014;9:e90837. doi: 10.1371/journal.pone.0090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tudek B. Base excision repair modulation as a risk factor for human cancers. Mol Aspects Med. 2007;28:258–75. doi: 10.1016/j.mam.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Campalans A, Marsin S, Nakabeppu Y, O'connor TR, Boiteux S, Radicella JP. XRCC1 interactions with multiple DNA glycosylases: A model for its recruitment to base excision repair. DNA Repair (Amst) 2005;4:826–35. doi: 10.1016/j.dnarep.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 28.Monaco R, Rosal R, Dolan MA, Pincus MR, Brandt-Rauf PW. Conformational effects of a common codon 399 polymorphism on the BRCT1 domain of the XRCC1 protein. Protein J. 2007;26:541–6. doi: 10.1007/s10930-007-9095-y. [DOI] [PubMed] [Google Scholar]
- 29.Lei YC, Hwang SJ, Chang CC, Kuo HW, Luo JC, Chang MJ, et al. Effects on sister chromatid exchange frequency of polymorphisms in DNA repair gene XRCC1 in smokers. Mutat Res. 2002;519:93–101. doi: 10.1016/s1383-5718(02)00127-4. [DOI] [PubMed] [Google Scholar]
- 30.Duell EJ, Wiencke JK, Cheng TJ, Varkonyi A, Zuo ZF, Ashok TD, et al. Polymorphisms in the DNA repair genes XRCC1 and ERCC2 and biomarkers of DNA damage in human blood mononuclear cells. Carcinogenesis. 2000;21:965–71. doi: 10.1093/carcin/21.5.965. [DOI] [PubMed] [Google Scholar]
- 31.Matullo G, Palli D, Peluso M, Guarrera S, Carturan S, Celentano E, et al. XRCC1, XRCC3, XPD gene polymorphisms, smoking and (32) P-DNA adducts in a sample of healthy subjects. Carcinogenesis. 2001;22:1437–45. doi: 10.1093/carcin/22.9.1437. [DOI] [PubMed] [Google Scholar]
- 32.Vural P, Degirmencioglu S, Dogru-Abbasoglu S, Saral NY, Akgül C, Uysal M. Genetic polymorphisms in DNA repair gene APE1, XRCC1 and XPD and the risk of pre-eclampsia. Eur J Obstet Gynecol Reprod Biol. 2009;146:160–4. doi: 10.1016/j.ejogrb.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Parildar-Karpuzoglu H, Dogru-Abbasoglu S, Hanagasi HA, Karadag B, Gürvit H, Emre M, et al. Single nucleotide polymorphisms in base-excision repair genes hOGG1, APE1 and XRCC1 do not alter risk of Alzheimer's disease. Neurosci Lett. 2008;442:287–91. doi: 10.1016/j.neulet.2008.07.047. [DOI] [PubMed] [Google Scholar]
- 34.Dogru-Abbasoglu S, Tanrikulu S, Ademoglu E, Erbil Y, Ozderya A, Karadag B, et al. Polymorphisms of DNA base-excision repair genes APE/Ref-1 and XRCC1 are not associated with the risk for Graves' disease. Cell Biochem Funct. 2009;27:462–7. doi: 10.1002/cbf.1595. [DOI] [PubMed] [Google Scholar]