
Figure 3.
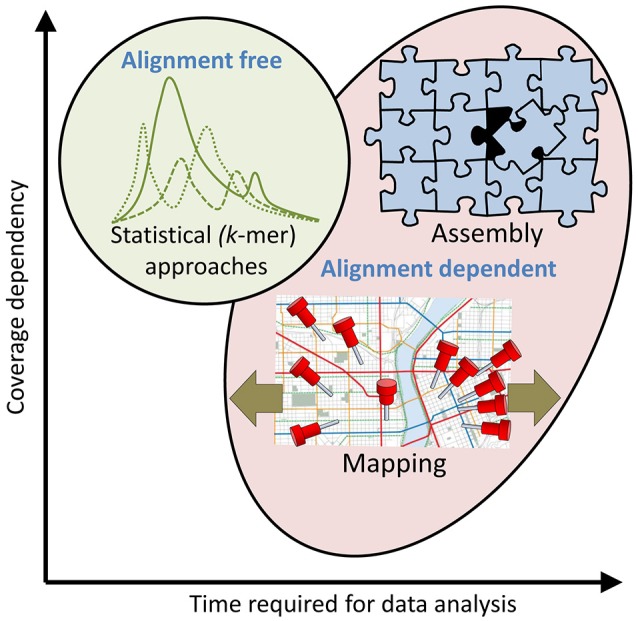
Approaches to HTS sequence read analysis and their dependence on alignment, time and coverage. At least three different approaches for analysis of HTS sequence reads can be selected, but combinations are often preferred. Top right: Assembly of sequences into contigs (see also Figure 2), scaffolds and complete genome assemblies is alignment dependent, time consuming and the success probability is usually correlated with the coverage. This approach is typically taken when time is not the limiting factor and a complete assembly is desired for successive analysis and reference applications. Bottom: Mapping (see also Figure 2) of reads to existing assembly/assemblies is also alignment dependent and time consuming but can also be performed successfully at low coverage (a single read can be mapped to a reference assembly). The size of the reference (e.g., database or genome) and the degree to which mismatches are accepted will have a significant impact on the time required for data analysis (olive arrows). This approach is typically taken to determine functional aspects of metagenome and transcriptome sequences and in metataxonomics. Top left: K-mer analysis (see also Figure 2) is a fast, alignment independent, statistical (probabilistic) approach to investigate properties of a sequenced genome such as its similarity and relationship to other (reference) genomes. It is typically used to screen sequenced genomes to identify genomes of particular interest for more comprehensive analysis.