

Abstract
Background
Farm workers in rural areas consume more alcohol than those who reside in urban areas. Occupational exposures such as agricultural work can pose hazards on the respiratory system. It is established that hog barn dust induces inflammation in the airway, including the release of cytokines such as tumor necrosis factor alpha (TNFα), interleukin-6 (IL-6) and interleukin-8 (IL-8). We have shown that alcohol alters airway epithelial innate defense through changes in both nitric oxide (NO) and cAMP-dependent protein kinase (PKA). Simultaneous exposure to hog barn dust and alcohol decreases inflammatory mediators, TNFα, IL-6 and IL-8, in mice. Previously, mice exposed to both alcohol and hog barn dust showed a depleted amount of lymphocytes compared to mice exposed only to hog barn dust. Weakening of the innate immune response could lead to enhanced susceptibility to disease. In addition, mice that were co-exposed to hog barn dust and alcohol also experienced increased mortality.
Methods
Because we recently demonstrated that PKA activation inhibits the TNFα sheddase, TNF-α converting enzyme (TACE), we hypothesized that an alcohol-mediated PKA pathway blocks TACE activity and prevents the normative inflammatory response to hog barn dust exposure. To delineate these effects, we used PKA pathway inhibitors (adenylyl cyclase, cAMP and PKA) to modulate the effects of alcohol on dust-stimulated TNFα release in the bronchial epithelial cell line, BEAS-2B. Alcohol pretreatment blocked TACE activity and TNFα release in hog barn dust-treated cells.
Results
Alcohol continued to block hog barn dust-mediated TNFα release in the presence of the particulate adenylyl cyclase inhibitor, SQ22,536. The soluble adenylyl cyclase (sAC) inhibitor, KH7, however, significantly increased the inflammatory response to hog barn dust. PDE4 inhibitors significantly elevated cAMP and enhanced alcohol-mediated inhibition of dust-stimulated TNFα release. In addition, the nitric oxide synthase inhibitor, L-NMMA, also reversed the alcohol-blocking effect on dust-stimulated TNFα.
Conclusions
These data suggest that alcohol requires a soluble cyclase-generated cAMP-PKA pathway that is dependent upon the action of NO to inhibit TACE and TNFα release. These findings support our observations that alcohol functions through a dual NO and PKA pathway in bronchial epithelial cells.
Introduction
The respiratory system encounters an onslaught of airborne matter. Agriculture workers, for instance, are exposed to various types of dusts on a daily basis. Animal husbandry dust, composed of feces, bacteria, and endotoxins, as well as many other components, is extremely complex and can include respirable particles that distribute deep in the airway (Gerald et al., 2014; May et al., 2012). In particular, concentrated animal feeding operations (CAFOs) can harbor higher levels of dust than ambient air due to the lack of normal airflow and poor ventilation (Cormier et al., 2000) . Swine CAFOs in particular are known to have higher levels of harmful dusts than those found in other agricultural settings (May et al., 2012). Inhalation of organic dust can cause airway inflammation (Poole and Romberger, 2012) and for farm workers, result in an increased prevalence of chronic bronchitis and other respiratory symptoms including runny nose, watery eyes and shortness of breath (Alterman et al., 2008). One explanation for the immunological responses to swine production dust is that it causes impaired human macrophage function (Poole et al., 2008) . Another explanation is found in the increased epithelial cell inflammatory cytokine production observed in mouse models exposed to organic dust (Wyatt et al., 2014). The release of pro-inflammatory cytokines TNFα, IL-6 and IL-8 are regulated by the intracellular activation of PKCα followed by the subsequent downstream activation of PKCε following organic dust exposure (Wyatt et al., 2014).
The effects of hog barn dust on the airways have been studied extensively; however, co-exposure to dust and alcohol is understudied despite mounting evidence that people exposed to CAFO dust also consume alcohol (Stallones and Xiang, 2003). Likewise, the demographics of farmers are changing rapidly and a new population of agricultural workers is emerging (Kandel, 2008). This emerging population often consumes higher levels of alcohol than their non-rural counterparts (Brumby et al., 2013; Stallones and Xiang, 2003).
Alcohol affects a variety of pulmonary functions. Chronic alcohol exposure induces cilia desensitization in vitro and in vivo (Wyatt and Sisson, 2001; Elliott et al., 2007). This leads to ineffective mucociliary clearance as alcohol impacts the airway through the nitric oxide (NO) and PKA pathway (Sisson et al., 1999). Alcohol alters airway epithelial innate defense through changes in both NO and the cAMP-dependent protein kinase (PKA) (Wyatt et al., 2003; Wyatt et al., 2013). In addition, alcohol can lead to leaky lung (Brown et al., 2004), a reduction in alveolar macrophages (Brown et al., 2007) and a decrease in glutathione levels (Holguin et al., 1998) . With a decrease in immune cells and antioxidants, the airway is primed for microbes to colonize. Without normal innate and adaptive immune processes infections are at risk for going unresolved.
Previous work in our laboratory has shown that TNFα, IL-6 and IL-8 cytokines are produced in the lungs following hog barn dust exposure in mice (Poole et al., 2009) . Alcohol exposure, however, negatively affects the normal inflammatory response to hog barn dusts in mice. Elevations in lung lavage inflammatory cytokines and mononuclear cell aggregates normally observed in histological evaluations as a result of repetitive hog barn dust extract exposure (HDE) are abrogated in mice that were previously fed alcohol (McCaskill et al., 2012). Mice treated with the concurrent exposure of alcohol and HDE had a decrease in weight and experienced 20% mortality. Previously, it was shown that alcohol effects on the inflammatory response can impair the host’s ability to clear debris, infectious agents and microbes (Happel and Nelson, 2005). Recently, we identified a cyclic AMP-mediated inhibition of the TNFα converting enzyme (TACE) that can block dust-stimulated TNFα release (Wyatt et al., 2014). Because alcohol elevates cAMP in airway epithelium (Sisson et al., 1999), we hypothesized that alcohol exposure blocks dust-mediated TNFα production in airway epithelium through a cAMP-dependent inhibition of TACE activation.
Materials and Methods
Cell culture
BEAS-2B, a human bronchial epithelial transformed cell line (ATCC, Manassas, VA), was treated with or without organic dust extract (5%), ethanol (EtOH; 100 mM) or the following inhibitors: SQ22, 536, KH7, H-89, L-NMMA and Rp-cAMPS (each obtained from Sigma, St. Louis, MO). Cells were exposed to EtOH (24 hr) before being exposed to inhibitors in the presence of EtOH for 1 hr. After treating with inhibitors for 1 hr, cells were treated with dust extract, EtOH and inhibitors for 6 hr. Human bronchial epithelial cells were cultured as described in Romberger et al. 2002 (Romberger et al., 2002) .
Organic dust extract and collection
Dust was collected from raised surfaces of hog confinement facilities and an aqueous extract prepared as previously detailed (Dusad et al., 2013; Poole et al., 2014). The dust was made into an aqueous extract (HDE; hog barn dust extract) by vortexing 1g/ml of dust into Hank’s balanced salt solutions (HBSS) for 1 min and allowed to sit for 1 hr. The solution was then centrifuged for 20 min at 2000g, the supernatant fraction was removed and this step was repeated for an additional 30 min centrifugation. The supernatant fraction was then sterile filtered (0.22 microns). Aliquots were kept at −20°C until utilized. A 5% dust extract diluted in media was used for cell culture stimulations. Extensive characterization of the components in this extract has been previously reported with regard to endotoxin, muramic acid, ergosterol, fatty acids, and other bacterial components (Boissy et al., 2014; Poole et al., 2010).
Enzyme-linked immunosorbent assay (ELISA) for human IL-6, IL-8 and TNFα
BEAS-2B cells were grown to 95% confluency on collagen-coated 6-well tissue culture plates (Falcon, Pittsburg, PA) in LHC-9/RPMI media and incubated at 37°C, 5% CO2. TNFα, IL-6, and IL-8 concentrations were evaluated by antibody detection using a commercially prepared ELISA kit (R&D systems, Minneapolis, MN). Cytokines were normalized by the corresponding protein concentrations using a Nanodrop spectrophotometer (Thermo Fisher, Pittsburgh,PA).
Protein Kinase C activity assay
BEAS-2B cells were cultured on 60 mm tissue culture dishes (B-D Falcon, Franklin Lakes, NJ) to 95% confluence and pre-treated with 100 mM of EtOH 24 hr. The cells were then subjected to a 1 hr pretreatment of inhibitor and EtOH. Following this exposure, cultures were stimulated with dust extract (5%), EtOH and/or inhibitors. Phorbol-12-myristate-13-acetate (PMA; Sigma) was added to some dishes for 5–15 min as a positive control. Cell lysis buffer (35 mM Tris-HCl, 0.4 mM EGTA, and 10 mM MgCl) containing a 1:10 dilution of protease inhibitor cocktail (Sigma), 0.2 mM PMSF and 0.1% Triton X-100 was added and cell monolayers in culture dishes frozen in liquid nitrogen. Dishes were scraped, the supernatant fraction containing the cells was collected and sonicated for 5 s, crude cell protein was centrifuged at 10,000g for 30 min at 4°C. The cell fractions containing either PKC alpha or PKC epsilon were measured for activity as previously described (Wyatt et al., 2012) .
TACE activity assay
TNFα-Converting Enzyme (TACE) activity was measured using a commercially available fluorometric assay (SensoLyte 520 TACE activity assay kit, AnaSpec, Fremont, CA). On 6-well plates, BEAS-2B were grown confluent to ~90% and treated with above-mentioned conditions. Fractions from supernatant fractions were utilized using the supplied TACE substrate solution as per manufacturer’s instructions. To stop the horseradish peroxidase reaction, 2 M H2SO4 was added to each plate well and fluorescence recorded at 490/520 nm. Experimental sample readings were extrapolated to a standard curve of recombinant human TACE and results expressed as µg protein/mL of sample.
NF-κB Binding Assay
BEAS-2B cells were treated with media only, ethanol (100 mM), HDE (5%) or both ethanol and HDE. Beas-2B cells were exposed to ethanol 24 hr and then ethanol and HDE for 6 hours. NF-κB binding assay was completed as described (Wyatt et al., 2014).
Statistical Analyses
Each experiment contained three technical replicates and each experiment was conducted three separate times. GraphPad Prism software (San Diego, CA, version 6.0) was used to analyze values as well as statistical analyses. One-way analysis of variance (ANOVA) was used to determine statistical significance with a p<0.05 (95% confidence interval).
Results
Alcohol blocks HDE-mediated PKCε activation and IL-8 release in bronchial epithelial cell lines
BEAS-2B cells were exposed to 100 mM EtOH for 24 hr only, or EtOH followed by a 6 hr 5% HDE exposure (Figure 1A). Cells were also exposed to media alone. Supernatant fractions were collected following the 6 hr exposure and analyzed for IL-8 by ELISA. HDE exposure significantly increased IL-8 release as compared to the media control (p <0.001) while EtOH pretreatment significantly decreased HDE-stimulated IL-8 release (p <0.001). EtOH alone had no effect on IL-8 release. In Figure 1B, PKCε activity was measured in BEAS-2B cells that were exposed to media or HDE (5%) for 0, 1, 6 and 24 hr in the presence or absence of EtOH. HDE stimulated a significant increase in PKCε activity at 6 hr compared to the EtOH treated cells (p <0.001). EtOH-exposed cells did not show any increases in PKCε activity when compared to media-only treated cells. These data indicate that EtOH blocks HDE-stimulated IL-8 release and PKCε activity in BEAS-2B cells.
Figure 1. Alcohol blocks HDE-stimulated PKCε activation and IL-8 release in BEAS-2B.

A. BEAS-2B cells were exposed to media, 5% HDE, EtOH (100 mM) 24 hr and HDE and EtOH (24 hr prior to the 6 hour exposure) for 6 hr. Following treatment, IL-8 release was assayed. HDE stimulated IL-8 release compared to the controls, b denotes p<0.0001. Co-treatment of HDE-EtOH decreased IL-8 release significantly when compared to HDE-exposed cells, c denotes p<0.0001. B. BEAS-2B cells were exposed to media, 5% HDE and EtOH (100 mM) for 0,1, 6, and 24 hr. Post-treatment cells were assayed for the PKCε activity. HDE increased the PKCε activity when compared to the controls at 6 hr, b denotes p<0.001. EtOH failed to increase the PKCε activity at any of the time points, p<0.001. One-way ANOVA was conducted with a Tukey post-hoc test.
Alcohol blunts HDE-stimulated PKCε activation in primary airway epithelial cells
To ensure that alcohol is having the same effects in primary airway cells, human bronchial epithelial cells (HBECs) were exposed to EtOH (100 mM) for 24 hr, or 24 hr EtOH with a 6 hr post-treatment of 5% HDE. The supernatant fractions were collected and assayed by ELISA for IL-8 release (Figure 2A). HDE exposure increased IL-8 release and EtOH significantly blocked this HDE-mediated release (p <0.001). The media and EtOH-only controls showed nominal baseline levels of IL-8 release. In Figure 2B, HBECs were exposed to EtOH (100 mM), 5% HDE, EtOH and HDE or media only for 6 hr. HDE exposure increased PKCε activity while EtOH treatment decreased HDE-stimulated PKCε activity (p <0.001). These data confirm that EtOH and HDE affect IL-8 release and PKCε activity in airway primary cells and cell lines in a similar manner.
Figure 2. Alcohol blocks HDE-stimulated PKCε activation in primary HBEC.

A. HBECs were exposed to media, 5% HDE, EtOH (100µM) and HDE and EtOH for 6 hr. Ethanol treatment occurred 24 hr prior to the 6-hour exposure. Post-treatment IL-8 release was assayed. HDE stimulated IL-8 release when compared to the controls, b signifies p<0.0001. The co-treatment of HDE and EtOH decreased IL-8 release significantly when compared to the HDE-exposed cells, c signifies p<0.0001. B. HBECs were exposed to media, 5% HDE and EtOH (100µM) for 6 hr. Post-treatment cells were assayed for PKCε activity. HDE increased PKCε activity when compared to the controls, b signifies p<0.001. EtOH decreased PKCε activity significantly, a signifies p<0.001. A one-way ANOVA was conducted with a Tukey post-hoc test.
Alcohol does not alter HDE-stimulated PKCα activation in bronchial epithelial cells
Because HDE activates PKCα upstream of PKCε, we evaluated the effects of EtOH on PKCα. In Figure 3, PKCα and PKCε activities were observed in BEAS-2B cells that were exposed to 0%, 0.1%, 1% and 5% HDE over 1 hr with or without EtOH (100 mM). In Figure 3A, PKCα activity increased with 1% and 5% HDE exposure in comparison to cells that did not receive HDE (p< 0.05). EtOH, however, did not alter PKCα activity in cells exposed to HDE. Under the same conditions, cells were treated with various concentrations of HDE for 6 hr and assayed for PKCε activity (Figure 3B). HDE (1% and 5%) increased PKCε activity when compared to the media-only exposed cells (p <0.05). EtOH decreased the HDE-mediated PKCε activity (p<0.05 compared to 1% HDE; p<0.001 compared to the 5% HDE). PKCε activity is decreased by EtOH but PKCα activity is unaffected, which indicates that EtOH is affecting a target of HDE downstream of PKCα but upstream of PKCε.
Figure 3. Alcohol does not block HDE-stimulated PKCα activation in BEAS-2B.

A. BEAS-2B cells were exposed to media only, which served as the control, HDE (0.1, 1, 5%) in the presence or absence of EtOH (100 mM) for 6 hr. Post-treatment, cells were assayed for PKCα activity. HDE (1% and 5%) increased PKCα activity significantly compared to media only exposed cells, b denotes p<0.05. A co-exposure of HDE and EtOH did not alter PKCα activity, p >0.05. B. BEAS-2B cells were exposed to media, HDE (0.1, 1, 5%) in the presence or absence of EtOH (100 mM) for 6 hr. Post-treatment, cells were assayed for PKCε activity. HDE (1 and 5%) stimulated an increase of PKCε activity when compared to control cells, b denotes p≤ 0.05. When cells were exposed to HDE and EtOH PKCε activity was decreased, a signifies p<0.05 under a one-way ANOVA and Tukey post-test analysis.
PMA stimulation of PKCε is not affected by alcohol
To determine whether alcohol has a direct inhibitory effect on PKCε or an indirect effect on an upstream mediator of PKCε activation, we assayed the ability of a direct PKC activator, phorbol-12 myristate-13 acetate (PMA) to activate PKCε in the presence of alcohol (Figure 4). BEAS-2B were pretreated with media or 100 mM EtOH for 24 hr followed by a 6 hr treatment with either 5% HDE or 100 ng/ml PMA. Cells were then assayed for PKCε activity. Consistent with the data shown in Figure 3, HDE again stimulated PKCε activity (p<0.01) and EtOH significantly decreased HDE-stimulated activity. In contrast, PMA-stimulated PKCε activity was not decreased by EtOH pretreatment. These data show that alcohol-mediated inhibition of PKCε is specific to HDE stimulation and not due to a non-specific effect of alcohol on the enzyme.
Figure 4. PMA-stimulated PKCε activation is not blocked by alcohol.

BEAS-2B cells were exposed to media, 5% HDE, EtOH (100 mM), 5%HDE + EtOH (100 mM), PMA (100 ng/ml) and PMA + EtOH (100 mM) for 6 hr. PKCε activity increased with HDE exposure and co-exposure of HDE and EtOH decreased PKCε activity significantly, b denotes p<0.05. PMA stimulated PKCε activity and co-treatment of PMA and EtOH also stimulated PKCε activity, b also denotes p<0.05 under a one-way ANOVA and Tukey post-hoc test.
EtOH inhibits TACE activity
TACE is a sheddase that regulates sequestration of TNFα. Cleavage of TACE results in a biologically active form of TNFα. BEAS-2B were pretreated with or without 100 mM EtOH for 24 hr and then exposed to media or 5% HDE for 6 hr. TNFα release was then measured via ELISA (Figure 5A). HDE significantly increased TNFα release compared to control media (p<0.01) while EtOH significantly decreased HDE-stimulated TNFα release (p<0.05). Media and EtOH alone controls showed low baseline levels of TNFα release. TACE activity was then measured in BEAS-2B under the same conditions of exposure (Figure 5B). HDE stimulated TACE activity in comparison to the media-only exposed cells (p<0.05). EtOH pretreatment blunted HDE-mediated TACE activity when compared to the HDE alone (p <0.05). As a control, pretreating BEAS-2B with TNFα protease inhibitor 1 (Tapi-1; 20 µM) was highly effective at blunting HDE-stimulated TACE activity.
Figure 5. HDE-stimulated TACE activity and TNF release is blocked by alcohol.

A. BEAS-2B cells were exposed to media, 5% HDE, EtOH (100 mM), 5% HDE + EtOH (100 mM) for 6 hr. HDE stimulated an increase in TNFα release when compared to controls, b signifies p<0.01. The co-exposure of HDE and EtOH resulted in a significant decrease in TNFα release when compared to the HDE alone, c signifies p<0.05. Control and EtOH-treated cells were not significantly different.
B. BEAS-2B were subjected to the following treatments: media, 5% HDE, HDE + EtOH (100 mM), EtOH (100 mM) alone, HDE (5%)+Tapi-1 (10 µM) and Tapi-1 alone. TACE activity was increased with HDE exposure, but ameliorated when airway cells were exposed to HDE and alcohol. b signifies the increase of TACE activity with HDE exposure, while c denotes a decrease in TACE activity with a co-exposure of HDE and EtOH, p<0.05 under a one-way ANOVA with a Tukey post hoc analysis.
Inhibition of PKA results in restoration of HDE-mediated IL-6 release
Because our previous studies revealed that elevations in cAMP decreased HDE-stimulated TACE activation (Wyatt et al., 2014), we targeted the PKA pathway to deduce how alcohol blocks HDE-mediated pro-inflammatory cytokine release. BEAS-2B cells were exposed to media alone, 5% HDE alone, EtOH (100 mM) and HDE alone, and EtOH and HDE with or without the PKA inhibitor H-89 (10 µM) or the PKA antagonist Rp-cAMPS (5 µM). Similar to IL-8 release, HDE stimulated a significant release of IL-6 (p<0.05), while the combination of HDE and EtOH treatment blunted the release of IL-6 (p <0.05; Figure 6). Rp-cAMPS treatment fully restored the IL-6 response to HDE in the presence of EtOH (p <0.001). H-89 treatment was also able to fully restore HDE-stimulated IL-6 release in the presence of EtOH-exposed airway cells (p <0.0001). These data support a role for PKA in the alcohol-mediated inhibition of HDE-stimulated cytokine.
Figure 6. Rp-cAMPS or H89 allow HDE-stimulated IL-6 in presence of EtOH.

A. BEAS-2B cells were treated with the following treatments for 6 hr: media, HDE, HDE + EtOH and HDE + ETOH + inhibitors (Rp-cAMPS (5 µmol) or H-89 (10 µM)). Supernatant fractions were extracted and IL-6 was measured via ELISA. Both the antagonist analog (Rp-cAMPS) and PKA inhibitor (H-89) were capable of fully restoring the HDE-mediated cytokine release that was blunted by alcohol when compared to the co-exposed cultures, d denotes p<0.0001 and e signifies p<0.001 under a one-way ANOVA and Tukey post-hoc test.
Soluble adenylyl cyclase is responsible for partially restoring TNFα
Because PKA is the primary target for cAMP, we sought to determine if inhibiting adenylyl cyclase (AC) would reverse the alcohol-mediated inhibition of HDE-stimulated TNFα release. BEAS-2B cells were exposed to media, 5% HDE, EtOH (100 mM) and EtOH and HDE with or without the AC inhibitors SQ22,536 and KH7. In Figure 7a, HDE-exposed cells manifested an increase in TNFα release when compared to media-only exposed cells (p<0.05) while EtOH pretreatment significantly decreased HDE-stimulated TNFα release (p<0.05). Preincubation with 10 µM KH7 (soluble AC inhibitor) significantly reversed alcohol-mediated inhibition of HDE-stimulated TNFα (p<0.05). However, the inhibitor of transmembrane AC, SQ22,536 did not rescue HDE-mediated TNFα release from alcohol inhibition. In addition to blocking the formation of cAMP through cyclase inhibition, we investigated the effects of protecting the cAMP signal through phosphodiesterase (PDE) inhibition. BEAS-2B cells were pretreated with or without PDE inhibitors IBMX (200µM), Rolipram (8 µM), or Ro 20–1724 (20 µM) followed by exposure to media, 5% HDE, EtOH (100 mM), or EtOH and HDE. At 6 hrs, TNFα release was measured. TNFα release increased with HDE exposure while TNFα release was decreased in HDE-exposed cells pretreated with EtOH. Importantly, both PDE4-selective inhibitors (Rolipram and Ro 20–1724) and the nonselective PDE inhibitor (IBMX) enhanced the alcohol-mediated decrease in HDE-stimulated TNFα release (Figure 7B). Collectively, these data suggest cAMP signaling is responsible for the alcohol-mediated decrease in pro-inflammatory cytokine release.
Figure 7. KH7 inhibitor, not SQ22,536 allowed HDE-stimulated TNFα/IL-8 in presence of EtOH.

A. BEAS-2B were subjected to the following treatments for 6 hr: media, 5% HDE, HDE + EtOH (100 mM) and HDE + EtOH + inhibitor (SQ22,536,50 µM; or KH7, 10µM)). Supernatant fractions were extracted and TNFα, IL-8 and IL-6 were measured via ELISA. The KH7 sAC inhibitor was able to partially restore cytokine production (d signifies p <0.05 compared to b (HDE)) whereas the SQ22, 536 pAC inhibitor was not different from the co-exposed (HDE + EtOH) group cultures. B. BEAS-2B cells were exposed to media, HDE, EtOH and HDE with or without IBMX (0.2 mmol/L), Rolipram (8 µM), Ro 20–1724 (20µM). HDE increased TNFα release while EtOH decreased HDE-induced TNFα release b signifies p<0.05 compared to media (a). Treatment with PDE inhibitors resulted in decreased TNFα release, c denotes p<0.05 compared to b. Significance was determined under a one-way ANOVA and Tukey post-hoc test.
Inhibition of NO fully restores pro-inflammatory cytokine production
We have previously established that alcohol requires the generation of nitric oxide (NO) to activate the cAMP/PKA pathway (Sisson et al., 1999). We therefore examined the role of NO in the alcohol-mediated inhibition of HDE-stimulated cytokine release. BEAS-2B cells were pre-treated with or without L-NMMA (10 µM), an NO inhibitor, for 1 hr followed by a 6 hr exposure to either media, HDE (5%), EtOH (100 mM), or HDE and EtOH (Figure 8). Compared to controls, HDE significantly stimulated TNFα release (p<0.05) while EtOH pretreatment decreased this HDE-stimulated TNFα (p<0.05). L-NMMA reversed alcohol-mediated inhibition of HDE-stimulated TNFα release.
Figure 8. L-NMMA allows HDE-stimulated TNFα/IL-8 in presence of EtOH.

BEAS-2B were subjected to the following treatments for 6 hr: media, 5% HDE, HDE + EtOH (100 mM) and HDE + ETOH + L-NMMA (100 mM). Supernatant fractions were extracted and pro-inflammatory cytokines TNFα, IL-8 and IL-6 were measured via ELISA. L-NMMA restored HDE-mediated cytokine production compared to HDE-exposed cell cultures, b denotes p>0.05 under a one-way ANOVA and Tukey post-hoc analysis.
NF-κB binding remains unaffected despite ethanol treatment
To ensure that ethanol is not inhibiting or reducing pro-inflammatory transcription factor, NF-κB, its expression was analyzed post-ethanol and HDE treatment. In Figure 9, we treated BEAS-2B cells with 100 mM ethanol and 5% HDE. The media only and ethanol treatments did not significantly alter the NF-κB binding (p >0.05). However, HDE induced NF-κB expression (p <0.05) and ethanol did not reduce this phenomenon in the combined treatment (p <0.05). From this data, we conclude that ethanol does not inhibit NF-κB expression as the mechanism for the reduction of TACE and TNF-α. These data suggest that alcohol requires the dual signaling pathways of both NO and cAMP to activate PKA for the inhibition of TACE-mediated TNFα release in response to HDE (Figure 10).
Figure 9. Ethanol does not decrease TNF-α levels via NF-κB expression.
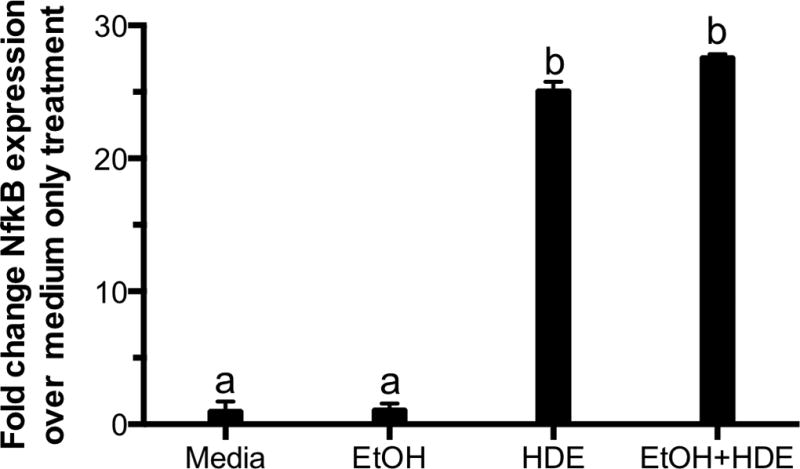
BEAS-2B cells were incubated with 100 mM ethanol 24 hr followed by a 6-hour treatment of 5% HDE and ethanol. NF-κB activation was assayed as described and expressed as a fold change activation over medium-only treated cells. Media and ethanol were not significantly different. a denotes p>0.05, whereas HDE and HDE and ethanol significantly increased NF-κB expression (b signifies p-value<0.05) under a one-way ANOVA and Tukey post-hoc test.
Figure 10. Summary model diagram.
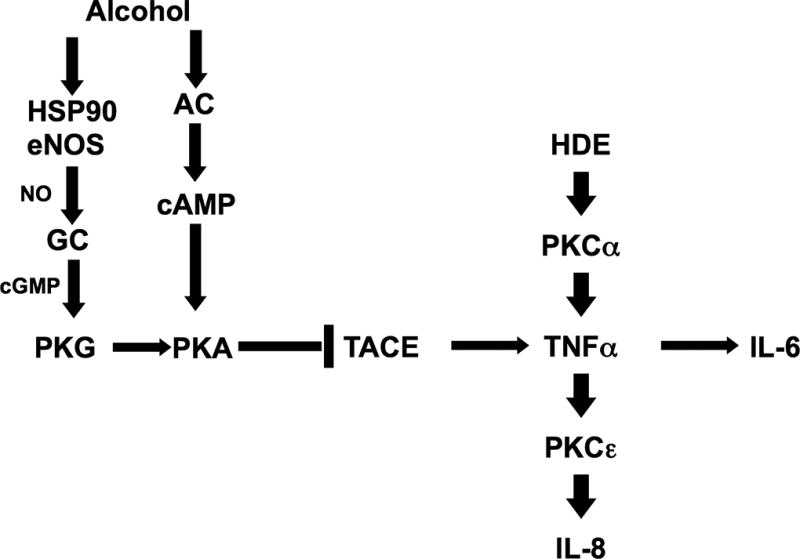
Proposed model of alcohol’s effect on HDE-stimulated pro-inflammatory cytokine release. Alcohol stimulates both nitric oxide (NO) and adenynyl cyclase, which leads to the activation cAMP and subsequent PKA activation, which then inhibits TACE activity. By blunting TACE activity, the HDE-stimulated release of pro-inflammatory cytokines TNFα, IL-8 and IL-6 are decreased. PKCα is activated by dust upstream of TACE and independent of TACE action, while PKCε activity is regulated by dust downstream of TACE and TNFα.
Discussion
Alcohol has the ability to greatly modulate host innate immunity. The tracheal cilia can become desensitized when exposed to chronic alcohol, which leads to defective mucociliary clearance (Sisson, 2007). Alcohol feeding of animals (acutely) reduces TNFα production by alveolar macrophages (Kolls et al., 1995). Alveolar macrophage phagocytosis can also be modified under acute and chronic alcohol treatments (Zhang et al., 1998; Spitzer and Zhang, 1996; Rimland and Hand, 1980; Brown et al., 2007). Mice exposed to a bacterial infection or LPS experienced blunted chemokine production and neutrophil recruitment (Boe et al., 2003; Zhang et al., 1998). In our current study, we investigated similar effects of alcohol, but in epithelial cells and using organic dust. Hog barn dust exposure in agricultural workers is of concern because dusts can pose respiratory problems (Poole and Romberger, 2012). Alcohol use can result in a loss of normal immunological and inflammatory responses. In the airways of heavy alcohol consumers, a condensation “rain effect” occurs, allowing alcohol to accumulate in localized high (mM) concentrations, leading to the impairment of normal airway defenses (Sisson, 2007). Using a dose of 100 mM ethanol is not cytotoxic to BEAS-2B cells (Simet et al., 2012). In fact, the highest known non-lethal blood alcohol concentration in a human at the time of hospital admission was 1510 mg/dL or 327 mM (Johnson et al., 1982). Blood alcohol concentrations can be lower than lung alcohol concentrations due to the rapid warming and cooling of ethanol in the bronchial airways leading to condensation. This is known as the rain effect. Airway epithelial cells also metabolize alcohol via CYP2E1. There is evidence that ethanol can impact such pathways. In McCaskill et al., mice fed 20% ethanol via diet for 6 weeks experienced increased gene expression of lung CYP2E1 (McCaskill et al., 2015) . With this knowledge of alcohol’s ability to interfere with respiratory innate defenses, we tested our hypothesis that alcohol blocks dust-mediated pro-inflammatory cytokine production through a cAMP-dependent inhibition of TACE.
Our current investigations indicate that alcohol reduces HDE-stimulated pro-inflammatory cytokine release by bronchial epithelial cells via inhibiting NO, adenylyl cyclase and PKA. Our lab has established that alcohol activates the NO pathway, resulting in PKG activation, which is required for the downstream activation of PKA by EtOH (Sisson et al., 1999). Our data show that the NO pathway is also involved with alcohol’s role in diminishing HDE-mediated inflammation. Alcohol stimulates the production of cAMP through an alcohol-sensitive AC (Tabakoff et al., 2001) , and our results demonstrate that cAMP production is being activated by alcohol, leading to blunted pro-inflammatory TNFα and downstream IL-6 and IL-8 release. By inhibiting sAC, the cytokine release that normally occurs in response to HDE was partially restored in the presence of alcohol. Interestingly, the transmembrane AC inhibitor had little effect on cytokine released in response to HDE+EtOH. This observation is consistent with our previous findings that the ciliated airway epithelium contains a soluble adenylyl cyclase that is stimulated by alcohol (Sisson et al., 2009). We also demonstrate the effects of inhibiting phosphodiesterases (PDE) on alcohol-mediated inhibition of dust-stimulated cytokine release using both a non-selective inhibitor, IBMX, and selective PDE4 inhibitors, rolipram and Ro 20–1724 (Figure 7B). We have previously established that in bovine epithelial cells EtOH modulates PDE4 activity (Forget et al., 2003). Previously, we showed that by inhibiting PDE4, eventual cAMP degradation in response to alcohol-stimulated cAMP production is blocked, thus allowing for an enhanced accumulation of cAMP after alcohol exposure (Forget et al., 2003). Both selective and non-selective PDE4 inhibitors enhanced the EtOH-mediated inhibition of HDE-stimulated TNFα release. These data provide several lines of evidence that EtOH is inhibiting TNFα release through a cAMP-dependent manner.
The observation that alcohol is working through a cAMP-dependent manner is an important key to the regulation of HDE-stimulated TACE. With the addition of cAMP, PKCε activity is decreased whereas PKCα activity remains unaltered. This data led us to believe that the effects of alcohol on airway epithelial cells occurs downstream of PKCα, but upstream of PKCε. Our previous study describes the sequential nature of PKCα and PKCε (Wyatt et al., 2010). PKCα activation occurs around 1 hr while PKCε activation occurs later, at 6 hr. PKCε cannot be activated by HDE without the earlier activation of PKCα. This is the reasoning behind Figure 3A, which shows no increase of PKCα activity at 1 hr but PKCε activity is increased after a 6 hr HDE exposure. In the study of Song et al. 2005, alcohol was shown to disrupt TACE activity (Song et al., 2005). TACE is a sheddase, which cleaves TNFα into its active signaling form (Rose-John, 2013). We have previously shown that TNFα is released following PKCα activation and is required for PKCε activation and IL-8 release (Wyatt et al., 2010; Wyatt et al., 2014). We have recently shown that HDE-mediated increases in TACE activity could be inhibited using cell-permeable cAMP analogs to directly activate PKA (Wyatt et al., 2014) . In our study, alcohol inhibited TACE activity, a concept also observed by others (Song et al., 2005). Our data confirms TNFα release after HDE exposure corresponds temporally with TACE activity. We found that alcohol, through a cAMP-PKA dependent manner, reduces HDE-stimulated TACE activity, which corresponds with a reduction in inflammatory cytokine release from bronchial epithelial cells stimulated with HDE. In addition, as shown as in Figure 9, NF-κB transcription remains unaffected after alcohol treatment, which leads us to conclude that alcohol is decreasing the HDE-mediated cytokine through the PKA pathway. The specific mechanism by which PKA inhibits TACE requires further investigation. Events upstream of TACE could be the reason for this difference. One of the proteins that could be affected is furin. Furin is a proprotein convertase that cleaves TACE into its soluble form. In the studies published by Howarth and colleagues in 2012 (Howarth et al., 2012), hepatocytes that were exposed to EtOH showed endoplasmic reticulum (ER) fragmentation. This dysfunction is of importance in the case of furin because furin is manufactured in the ER and undergoes folding as well proteolytic cleavage (Creemers et al., 1995). Future studies showing furin presence and activity in relation to TACE and TNFα in the context of alcohol and dust exposure are warranted.
HDE stimulates inflammatory cytokine release in vitro and this response is reduced by alcohol treatment. Although this reduction in inflammation associated with EtOH may be construed as a protective effect, clinical findings suggest this inhibition of inflammation in the lungs has negative consequences with regards to lung immunity. There are studies demonstrating that hog barn dust induces oxidants. In Pender et al., THP-1 cells that were exposed to hog barn dust (5 and 10 %) show increased levels of ROS production (Pender et al., 2014). Wyatt et al. show increased NO in bovine airway cells exposed to hog barn dust (Wyatt et al., 2008). Simet and colleagues also demonstrate the efficacy of commercially available antioxidants (NAC) to alleviate alcohol-mediated ciliary desensitization (Simet et al., 2013). Diallyl disulfide also reduces DNA damage and CYP2E1 levels when exposed to 80 mM alcohol in human bronchial epithelium (Sapkota et al., 2014) . Chronic alcohol exposure is known to induce alcohol induced ciliary dysfunction (AICD) in vivo and in vitro (Wyatt et al., 2004; Sisson et al., 1999). This results in the mucociliary apparatus becoming defective due to cilia desensitization. Similarly, pathogens trapped in the mucus blanket will not be cleared from the airway as efficiently, potentially leading to infection. Furthermore, alcohol use can lead to sustained infections due to impaired leukocyte functions (Happel et al., 2006). We have previously shown that alcohol exposure in BEAS-2B cells increases permeability and alters tight junction proteins (Simet et al., 2012). Concurrent HDE and EtOH treatment results in a decrease of the natural airway response (IL-8 and TNFα release) to dust. The normative inflammatory response incurred as a result of HDE exposure is thus altered by alcohol treatment.
These cellular observations support existing data using an in vivo animal model of dust exposure in the context of alcohol consumption. In studies conducted with a mouse model, mice that consumed ad libitum EtOH for 6 wk and inhaled HDE for 3 wk had fewer neutrophils and lymphocytes present in bronchoalveolar lavage fluid (BALF) compared to mice that were only exposed to HDE (McCaskill et al., 2012). In our study, we observed pro-inflammatory cytokines that are responsible for neutrophil and lymphocyte recruitment increase with a 5% HDE exposure in the BEAS-2B cell line. We also observed a blunting effect in the airway cells that were exposed to HDE and EtOH, which would be in accordance with our in vivo model histological findings (McCaskill et al., 2012). A decrease in pro-inflammatory cytokines (TNFα, IL-6, IL-8) as seen in Figures 1, 5 and 6 corroborates the published observations of decreased lung lavage lymphocytes and mononuclear cell aggregates in alcohol-fed mice challenged with HDE. The macrophages collected from the BALF were significantly lower in the HDE and HDE and ethanol treated groups when compared to control and ethanol exposed groups. In that study, mice fed alcohol and exposed to HDE experienced a significant reduction in body weight and 20% mortality, an observation not seen in alcohol-fed mice not exposed to dust (McCaskill et al., 2012) . In the Mason et al. 2004 study, mice that were fed alcohol chronically and exposed to tuberculosis experienced worse infections than those who were not drinking alcohol (Mason et al., 2004). Thus, alcohol has the ability to dampen the immune response and therefore leave the individual immunocompromised and prone to infection. Our findings here suggest that alcohol reduces the inflammatory response of bronchial epithelial cells to HDE via a mechanism involving the inhibition of TACE by NO and PKA pathways.
In summary, alcohol exposure reduces HDE-mediated inflammatory cytokine release from bronchial epithelial cells in a cAMP-dependent manner. Our findings suggest alcohol modulates TACE activity to prevent inflammatory responses in the bronchial epithelium. Future studies are warranted to further investigate the precise mechanism of PKA-mediated TACE inhibition. We have shown that chronic alcohol use can have negative effects on the airway epithelium in response to HDE. Such observations may have important ramifications on the health of alcohol-consuming workers exposed to organic agricultural dusts.
References
- 1.Alterman T, Steege AL, Li J, Petersen MR, Muntaner C. Ethnic, racial, and gender variations in health among farm operators in the United States. Ann Epidemiol. 2008;18:179–186. doi: 10.1016/j.annepidem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Boe DM, Nelson S, Zhang P, Quinton L, Bagby GJ. Alcohol-induced suppression of lung chemokine production and the host defense response to Streptococcus pneumoniae. Alcohol Clin Exp Res. 2003;27:1838–1845. doi: 10.1097/01.ALC.0000095634.82310.53. [DOI] [PubMed] [Google Scholar]
- 3.Boissy RJ, Romberger DJ, Roughead WA, Weissenburger-Moser L, Poole JA, LeVan TD. Shotgun pyrosequencing metagenomic analyses of dusts from swine confinement and grain facilities. PloS one. 2014;9:e95578. doi: 10.1371/journal.pone.0095578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown LAS, Harris FL, Ping X, Gauthier TW. Chronic ethanol ingestion and the risk of acute lung injury: a role for glutathione availability? Alcohol. 2004;33:191–197. doi: 10.1016/j.alcohol.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Brown LA, Ping XD, Harris FL, Gauthier TW. Glutathione availability modulates alveolar macrophage function in the chronic ethanol-fed rat. Am J Physiol Lung Cell Mol Physiol. 2007;292:L824–32. doi: 10.1152/ajplung.00346.2006. [DOI] [PubMed] [Google Scholar]
- 6.Brumby S, Kennedy A, Chandrasekara A. Alcohol consumption, obesity, and psychological distress in farming communities—An Australian study. The Journal of Rural Health. 2013;29:311–319. doi: 10.1111/jrh.12001. [DOI] [PubMed] [Google Scholar]
- 7.Cormier Y, Israel-Assayag E, Racine G, Duchaine C. Farming practices and the respiratory health risks of swine confinement buildings. Eur Respir J. 2000;15:560–565. doi: 10.1034/j.1399-3003.2000.15.22.x. [DOI] [PubMed] [Google Scholar]
- 8.Creemers JW, Vey M, Schafer W, Ayoubi TA, Roebroek AJ, Klenk HD, Garten W, Van de Ven WJ. Endoproteolytic cleavage of its propeptide is a prerequisite for efficient transport of furin out of the endoplasmic reticulum. J Biol Chem. 1995;270:2695–2702. doi: 10.1074/jbc.270.6.2695. [DOI] [PubMed] [Google Scholar]
- 9.Dusad A, Thiele GM, Klassen LW, Gleason AM, Bauer C, Mikuls TR, Duryee MJ, West WW, Romberger DJ, Poole JA. Organic dust, lipopolysaccharide, and peptidoglycan inhalant exposures result in bone loss/disease. American journal of respiratory cell and molecular biology. 2013;49:829–836. doi: 10.1165/rcmb.2013-0178OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elliott MK, Sisson JH, Wyatt TA. Effects of Cigarette Smoke and Alcohol on Ciliated Tracheal Epithelium and Inflammatory Cell Recruitment. Am J Respir Cell Mol Biol. 2007;36:452–459. doi: 10.1165/rcmb.2005-0440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forget MA, Sisson JH, Spurzem JR, Wyatt TA. Ethanol increases phosphodiesterase 4 activity in bovine bronchial epithelial cells. Alcohol. 2003;31:31–38. doi: 10.1016/j.alcohol.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Gerald C, McPherson C, McDaniel TM, Xu Z, Holmes B, Williams L, Whitley N, Waterman JT. A BIOPHYSIOCHEMICAL ANALYSIS OF SETTLED LIVESTOCK AND POULTRY HOUSING DUSTS. American Journal of Agricultural and Biological Sciences. 2014;9:153. [Google Scholar]
- 13.Happel KI, Odden AR, Zhang P, Shellito JE, Bagby GJ, Nelson S. Acute alcohol intoxication suppresses the interleukin 23 response to Klebsiella pneumoniae infection. Alcoholism: Clinical and Experimental Research. 2006;30:1200–1207. doi: 10.1111/j.1530-0277.2006.00144.x. [DOI] [PubMed] [Google Scholar]
- 14.Happel KI, Nelson S. Alcohol, immunosuppression, and the lung. Proc Am Thorac Soc. 2005;2:428–432. doi: 10.1513/pats.200507-065JS. [DOI] [PubMed] [Google Scholar]
- 15.Holguin F, Moss I, Brown LA, Guidot DM. Chronic ethanol ingestion impairs alveolar type II cell glutathione homeostasis and function and predisposes to endotoxin-mediated acute edematous lung injury in rats. J Clin Invest. 1998;101:761–768. doi: 10.1172/JCI1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howarth DL, Vacaru AM, Tsedensodnom O, Mormone E, Nieto N, Costantini LM, Snapp EL, Sadler KC. Alcohol disrupts endoplasmic reticulum function and protein secretion in hepatocytes. Alcohol Clin Exp Res. 2012;36:14–23. doi: 10.1111/j.1530-0277.2011.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson RA, Noll EC, Rodney WM. Survival after a serum ethanol concentration of 1 1/2% Lancet. 1982;2:1394. doi: 10.1016/s0140-6736(82)91285-5. [DOI] [PubMed] [Google Scholar]
- 18.Kandel W. Profile of hired farmworkers, a 2008 update. Economic Research Report Number 60 Economic Research Report Number 60 2008 [Google Scholar]
- 19.Kolls JK, Xie J, Lei D, Greenberg S, Summer WR, Nelson S. Differential effects of in vivo ethanol on LPS-induced TNF and nitric oxide production in the lung. Am J Physiol. 1995;268:L991–8. doi: 10.1152/ajplung.1995.268.6.L991. [DOI] [PubMed] [Google Scholar]
- 20.Mason CM, Dobard E, Zhang P, Nelson S. Alcohol exacerbates murine pulmonary tuberculosis. Infect Immun. 2004;72:2556–2563. doi: 10.1128/IAI.72.5.2556-2563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.May S, Romberger DJ, Poole JA. Respiratory health effects of large animal farming environments. J toxicol Environ Health, Pt B. 2012;15:524–541. doi: 10.1080/10937404.2012.744288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCaskill ML, Hottor HT, Sapkota M, Wyatt TA. Dietary diallyl disulfide supplementation attenuates ethanol-mediated pulmonary vitamin D speciate depletion in C57Bl/6 mice. BMC Nutrition. 2015;1:18. doi: 10.1186/s40795-015-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCaskill ML, Romberger DJ, DeVasure J, Boten J, Sisson JH, Bailey KL, Poole JA, Wyatt TA. Alcohol exposure alters mouse lung inflammation in response to inhaled dust. Nutrients. 2012;4:695–710. doi: 10.3390/nu4070695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pender RJ, Minor RC, Hurley SL, Conklin DR, Waterman JT. Exposure to swine housing dust modulates macrophage morphology and function. American Journal of Immunology. 2014;10:35–45. [Google Scholar]
- 25.Poole JA, Alexis NE, Parks C, MacInnes AK, Gentry-Nielsen MJ, Fey PD, Larsson L, Allen-Gipson D, Von Essen SG, Romberger DJ. Repetitive organic dust exposure in vitro impairs macrophage differentiation and function. J Allergy Clin Immunol. 2008;122:375–382. doi: 10.1016/j.jaci.2008.05.023. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poole JA, Anderson L, Gleason AM, West WW, Romberger DJ, Wyatt TA. Pattern recognition scavenger receptor A/CD204 regulates airway inflammatory homeostasis following organic dust extract exposures. Journal of immunotoxicology. 2014:1–10. doi: 10.3109/1547691X.2014.882449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poole JA, Dooley GP, Saito R, Burrell AM, Bailey KL, Romberger DJ, Mehaffy J, Reynolds SJ. Muramic acid, endotoxin, 3-hydroxy fatty acids, and ergosterol content explain monocyte and epithelial cell inflammatory responses to agricultural dusts. Journal of Toxicology and Environmental Health, Part A. 2010;73:684–700. doi: 10.1080/15287390903578539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poole JA, Romberger DJ. Immunological and inflammatory responses to organic dust in agriculture. Curr Opin Allergy Clin Immunol. 2012;12:126–132. doi: 10.1097/ACI.0b013e3283511d0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poole JA, Wyatt TA, Oldenburg PJ, Elliott MK, West WW, Sisson JH, Von Essen SG, Romberger DJ. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296:L1085–95. doi: 10.1152/ajplung.90622.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rimland D, Hand WL. The effect of ethanol on adherence and phagocytosis by rabbit alveolar macrophages. J Lab Clin Med. 1980;95:918–926. [PubMed] [Google Scholar]
- 31.Romberger DJ, Bodlak V, Von Essen SG, Mathisen T, Wyatt TA. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J Appl Physiol. 2002;93:289–296. doi: 10.1152/japplphysiol.00815.2001. 1985. [DOI] [PubMed] [Google Scholar]
- 32.Rose-John S. ADAM17, shedding, TACE as therapeutic targets. Pharmacological Research. 2013;71:19–22. doi: 10.1016/j.phrs.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 33.Sapkota M, Hottor TK, DeVasure JM, Wyatt TA, McCaskill ML. Protective role of CYP2E1 inhibitor diallyl disulfide (DADS) on alcohol-induced malondialdehyde-deoxyguanosine (M1dG) adduct formation. Alcohol Clin Exp Res. 2014;38:1550–1558. doi: 10.1111/acer.12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simet S, Wyatt TA, DeVasure J, Yanov D, Allen-Gipson D, Sisson JH. Alcohol Increases the Permeability of Airway Epithelial Tight Junctions in Beas-2B and NHBE Cells. Alcoholism: Clinical and Experimental Research. 2012;36:432–442. doi: 10.1111/j.1530-0277.2011.01640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simet SM, Pavlik JA, Sisson JH. Dietary antioxidants prevent alcohol-induced ciliary dysfunction. Alcohol. 2013;47:629–635. doi: 10.1016/j.alcohol.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sisson JH. Alcohol and airways function in health and disease. Alcohol. 2007;41:293–307. doi: 10.1016/j.alcohol.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sisson JH, May K, Wyatt TA. Nitric Oxide Dependent Ethanol Stimulation of Ciliary Motility Is Linked to cAMP-Dependent Protein Kinase (PKA) Activation in Bovine Bronchial Epithelium. Alcoholism: Clinical and Experimental Research. 1999;23:1528–1533. [PubMed] [Google Scholar]
- 38.Sisson JH, Pavlik JA, Wyatt TA. Alcohol stimulates ciliary motility of isolated airway axonemes through a nitric oxide, cyclase, and cyclic nucleotide-dependent kinase mechanism. Alcohol Clin Exp Res. 2009;33:610–616. doi: 10.1111/j.1530-0277.2008.00875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song K, Zhao XJ, Marrero L, Oliver P, Nelson S, Kolls JK. Alcohol reversibly disrupts TNF-alpha/TACE interactions in the cell membrane. Respir Res. 2005;6:123. doi: 10.1186/1465-9921-6-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spitzer JA, Zhang P. Gender differences in phagocytic responses in the blood and liver, and the generation of cytokine-induced neutrophil chemoattractant in the liver of acutely ethanol-intoxicated rats. Alcohol Clin Exp Res. 1996;20:914–920. doi: 10.1111/j.1530-0277.1996.tb05271.x. [DOI] [PubMed] [Google Scholar]
- 41.Stallones L, Xiang H. Alcohol consumption patterns and work-related injuries among Colorado farm residents. Am J Prev Med. 2003;25:25–30. doi: 10.1016/s0749-3797(03)00096-5. [DOI] [PubMed] [Google Scholar]
- 42.Tabakoff B, Nelson E, Yoshimura M, Hellevuo K, Hoffman PL. Phosphorylation cascades control the actions of ethanol on cell cAMP signalling. J Biomed Sci. 2001;8:44–51. doi: 10.1007/BF02255970. [DOI] [PubMed] [Google Scholar]
- 43.Wyatt T, Wells S, Alsaidi Z, DeVasure J, Klein E, Bailey K, Sisson J. Asymmetric dimethylarginine blocks nitric oxide-mediated alcohol-stimulated cilia beating. Mediators Inflamm. 2013 doi: 10.1155/2013/592892. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wyatt TA, Forget MA, Sisson JH. Ethanol stimulates ciliary beating by dual cyclic nucleotide kinase activation in bovine bronchial epithelial cells. The American journal of pathology. 2003;163:1157–1166. doi: 10.1016/S0002-9440(10)63475-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wyatt TA, Slager RE, Heires AJ, DeVasure JM, VonEssen SG, Poole JA, Romberger DJ. Sequential activation of protein kinase C isoforms by organic dust is mediated by tumor necrosis factor. American journal of respiratory cell and molecular biology. 2010;42:706–715. doi: 10.1165/rcmb.2009-0065OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wyatt TA, Gentry-Nielsen MJ, Pavlik JA, Sisson JH. Desensitization of PKA-stimulated ciliary beat frequency in an ethanol-fed rat model of cigarette smoke exposure. Alcohol Clin Exp Res. 2004;28:998–1004. doi: 10.1097/01.ALC.0000130805.75641.F4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wyatt TA, Poole JA, Nordgren TM, DeVasure JM, Heires AJ, Bailey KL, Romberger DJ. cAMP-dependent protein kinase activation decreases cytokine release in bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2014;307:L643–51. doi: 10.1152/ajplung.00373.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyatt TA, Sisson JH. Chronic ethanol downregulates PKA activation and ciliary beating in bovine bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L575–81. doi: 10.1152/ajplung.2001.281.3.L575. [DOI] [PubMed] [Google Scholar]
- 49.Wyatt TA, Sisson JH, Allen-Gipson DS, McCaskill ML, Boten JA, DeVasure JM, Bailey KL, Poole JA. Co-exposure to cigarette smoke and alcohol decreases airway epithelial cell cilia beating in a protein kinase Cepsilon-dependent manner. Am J Pathol. 2012;181:431–440. doi: 10.1016/j.ajpath.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wyatt TA, Sisson JH, Von Essen SG, Poole JA, Romberger DJ. Exposure to hog barn dust alters airway epithelial ciliary beating. Eur Respir J. 2008;31:1249–1255. doi: 10.1183/09031936.00015007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang P, Bagby GJ, Xie M, Stoltz DA, Summer WR, Nelson S. Acute ethanol intoxication inhibits neutrophil beta2-integrin expression in rats during endotoxemia. Alcohol Clin Exp Res. 1998;22:135–141. [PubMed] [Google Scholar]