
Figure 4. A model for Rac1-mediated, Nox2-dependent activation of p38MAPK in pancreatic β-cells under glucotoxic conditions.
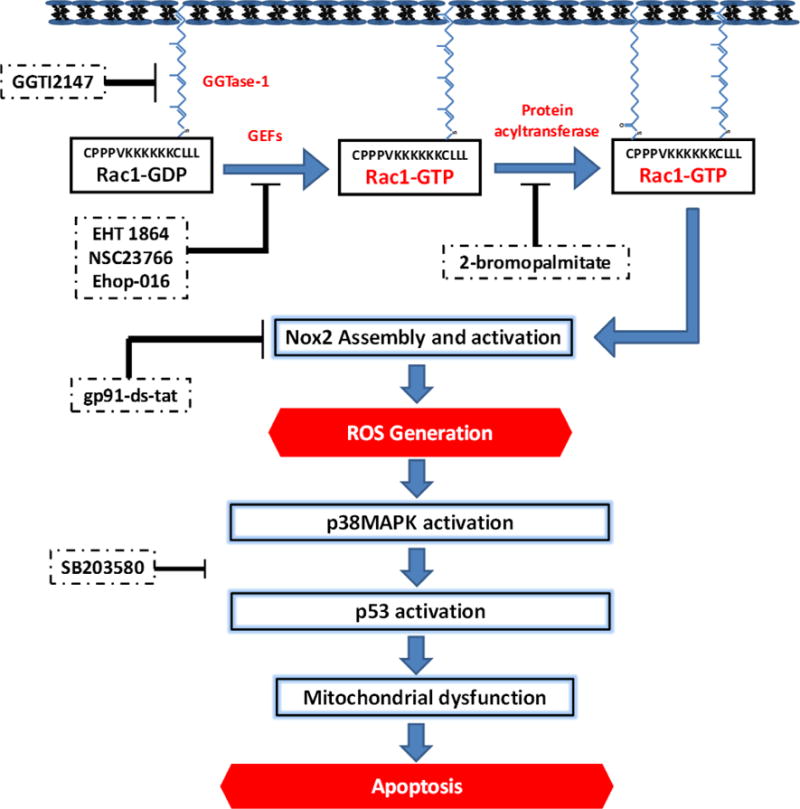
Based on the findings reviewed in this article, we propose a model to implicate Rac1 in Nox2-mediated activation of stress kinase [p38MAPK and p53] in pancreatic β-cells under the duress of glucotoxic conditions. Inhibition of Rac1 activation or function at the level of GEFs [EHT 1864, NSC23766 and Ehop-016] suppresses high glucose-induced activation of stress kinases. Furthermore, it appears that palmitoylation [sensitive to 2-brompalmitate; 2-BP], but not geranylgeranylation [inhibited by GGTI-2147] is necessary for high glucose-induced effects on stress kinase activation. Lastly, inhibition of Nox2 [with gp91-ds-tat peptide] markedly attenuated HG-induced Nox2 activation, ROS generation and p38MAPK activation. Together, our findings provide the first evidence to support our hypothesis that high glucose exposure conditions promote activation of Rac1, a key member of Nox2 holoenzyme, which, in turn, promotes the activation of Nox2 culminating in the generation of excessive ROS. An increase inROS leads to the onset of mitochondrial dysfunction as evidenced by caspase-3 activation, loss in metabolic function, including impaired GSIS. SB203580, a known inhibitor of p38MAPK, markedly attenuated high glucose-induced p53 activation, thus suggesting that p38MAPK activation is upstream to p53 activation. This figure is reproduced [with permission from Elsevier].45 Note that data on Rac1-mediated activation of p53 in pancreatic β-cells under the duress of glucotoxicity, as indicated in the figure, are published recently.46