

ABSTRACT
MicroRNAs (miRNAs) exhibit aberrant expression in the initiation and progression of a variety of human cancers, including colorectal cancer (CRC). However, the exact mechanisms are not well defined. miRNA expression profiles were characterized by microarrays in CRC samples, and miRNA 18b (miR-18b) was increased significantly in tumor tissues. The expression of miR-18b was confirmed in the CRC cell lines SW480 and HCT116 and 44 clinical specimens by quantitative real-time PCR (qRT-PCR). Multiple linear regression analysis showed a strong correlation of miR-18b expression with lymph node and distant metastasis. Overexpression of miR-18b promoted cell proliferation by facilitating cell cycle progression, and knockdown of miR-18b significantly suppressed migration in CRC cells. CDKN2B was identified as a target of miR-18b by high-throughput RNA sequencing and bioinformatics. After transfection with a miR-18b mimic, expression of CDKN2B was reduced significantly in CRC cells, and the effect was restored when a miR-18b inhibitor was transfected. A luciferase assay indicated miR-18b directly binds to the 3′ untranslated region (UTR) of CDKN2B. Expression of CDKN2B was downregulated in patient cancer tissues and negatively correlated with miR-18b. In a model of ectopic expression of miR-18b and CDKN2B, CDKN2B overexpression antagonized the effects of miR-18b in vitro and in vivo. The data show that miR-18b is involved in CRC carcinogenesis through targeting CDKN2B.
KEYWORDS: microRNA 18b, microRNA array, colorectal cancer, CDKN2B, high-throughput RNA sequencing
INTRODUCTION
Colorectal cancer (CRC) is the fourth most common cancer and the second leading cause of cancer-related death worldwide (1). More than 1.2 million patients are diagnosed with CRC every year, and more than 600,000 people die from the disease (2). In China, the incidence of CRC has been increasing continually in the most recent years, and it ranks fourth in mortality of all malignant diseases. The long-term survival of patients with CRC remains poor despite recent therapeutic advances in treatment. Therefore, there is an urgent and growing need for better understanding of the molecular pathogenesis, for identifying novel biomarkers for diagnosis and prognosis, and for exploring new and more effective treatment strategies.
MicroRNAs (miRNAs) are a cluster of noncoding RNAs 18 to 25 nucleotides in length. The miRNA guide strand is associated with a protein complex termed the RNA-induced silencing complex and guides the complex to target mRNAs via base pairing between the miRNA and the cognate target mRNA sequence (3). Accumulating studies over the past decades suggest that miRNAs exhibit aberrant expression in the initiation and progression of a variety of human cancers. These miRNAs have been implicated in several cellular processes, including differentiation, proliferation, autophagy, and apoptosis. Some oncogenic miRNAs have been demonstrated to be involved in the development of CRC (4). However, the disease-specific mechanisms of most miRNAs involved in the progress of CRC remain unknown.
To investigate the CRC-specific miRNA profile in humans, we identified multiple miRNAs differentially expressed in CRC tissues by miRNA expression microarray experiments. Among them, miRNA 18b (miR-18b) represents one of the most significantly upregulated miRNAs in CRC tissues. miR-18b is located at chromosome X within the miR-106a-363 cluster, which is the paralog of the miR-17-92 cluster. Previous reports showed that miRNAs encoded by these two clusters could act as oncogenes (5). For example, it has been shown that miR-18b is differentially expressed in several human cancers, including hepatocellular carcinoma (6), gastric cancer (7), nasopharyngeal carcinoma (8), breast cancer (9), ovarian cancer (10), and lung adenocarcinomas (11). In addition, aberrant expression of miR-18b in the sera of some patients with breast cancer (12) and rectal cancer patients who were lymph node positive after preoperative chemoradiotherapy (13) was observed. Although accumulating evidence suggests that miR-18b plays a role in the development of human cancers, its pathophysiological functions, as well as cellular targets, in CRC remain unclear. In the present study, we analyzed the expression levels of miR-18b in CRC tissues and cell lines, identified its target genes, and investigated the mechanisms of miR-18b involved in CRC progression. Our data suggest that miR-18b has the potential to serve as a novel biomarker and therapeutic target in CRC.
RESULTS
Identification of miRNAs dysregulated in CRC tissues.
To identify miRNAs that potentially drive CRC carcinogenesis, microRNA expression profiles were examined by microarray analysis. Hierarchical clustering showed systematic variations in transcription levels between paired tumor and nontumor tissues from 3 CRC patients. Microarray data showed 17 miRNAs were upregulated and 7 were downregulated in tumor tissues compared with nontumor tissues (Fig. 1). To validate the microarray analysis findings, 8 miRNAs (miR-124-3p, miR-6750, miR-18b, miR-21-3p, miR-224, miR-146, miR-27a, and miR-550a) were analyzed by quantitative real-time PCR (qRT-PCR) in 10 pairs of randomly selected CRC patients (Fig. 2A). qRT-PCR analysis verified the microarray findings, showing that a set of miRNAs are dysregulated in CRC tissues. Among them, we focused on three miRNAs (miR-18b, miR-21-3p, and miR-244) for further study because they are more frequently dysregulated than others in patient tissues. The expression levels of miR-18b, miR-21-3p, and miR-224 were further measured by qRT-PCR in 44 pairs of human CRC tissues and paired nontumor samples. The mRNA expression of miR-18b, miR-21-3p, and miR-224 in CRC tissue was 3.16, 1.73, and 1.50 times as much as that in the control, respectively. We found that miR-18b was significantly upregulated compared to the control (P < 0.001) (Fig. 2B). Furthermore, we evaluated the association between clinical characteristics and miR-18b expression levels in CRC patients by multiple linear regression analysis (Table 1). Statistical analysis showed a strong correlation between miR-18b expression and the lymph node and distant metastasis (Table 1). However, the level of miR-18b was not associated with other clinical factors, including age, gender, location, vessel invasion, tumor differentiation grade, and tumor-node-metastasis (TNM) stage. Collectively, these findings strongly suggest that upregulation of miR-18b is correlated with CRC progression.
FIG 1.

Hierarchical clustering showing systematic variations in transcription levels between paired tumor and nontumor tissues from 3 CRC patients (>2- or <0.5-fold; P < 0.05).
FIG 2.

(A) Expression levels of 8 miRNAs were analyzed by qRT-PCR in 10 pairs of randomly selected human CRC samples. (B) The expression levels of miR-18b, miR-21-3p, miR-224 were analyzed by qRT-PCR in 44 pairs of CRC samples. The graphs show means ± SD. *, P < 0.05; **, P < 0.01.
TABLE 1.
Association between clinical characteristics of CRC patients and miR-18b expression levels revealed by multiple linear regression analysisa
| Patient characteristic | B | SE | R2 | F | P value |
|---|---|---|---|---|---|
| Age | 0.083 | 0.523 | 0.657 | 9.852 | 0.875 |
| Gender | −0.227 | 0.447 | 0.656 | 18.582 | 0.615 |
| Location | −0.091 | 0.463 | 0.657 | 11.801 | 0.846 |
| Tumor differentiation grade | −1.451 | 0.902 | 0.654 | 25.157 | 0.115 |
| Vessel invasion | 0.168 | 0.665 | 0.656 | 14.521 | 0.802 |
| Distant metastasis | 3.134 | 0.561 | 0.631 | 35.079 | <0.01 |
| Metastatic lymph node | 1.833 | 0.471 | 0.631 | 35.079 | <0.01 |
B, regression coefficient; F, variance test of the overall regression model.
miR-18b promotes CRC cell proliferation, migration, and cell cycle progression.
To investigate the biological significance of miR-18b upregulation in the development and progression of CRC, we performed gain-of-function and loss-of-function studies in CRC cell lines. After transfecting the mimic and inhibitor of miR-18b, we detected the level of miR-18b to confirm the efficiency of its up- or downregulation. The expression of miR-18b was upregulated in both HCT116 and SW480 cells (P < 0.05) (Fig. 3A). Both HCT116 and SW480 cells were suitable transfection host cells and have often been used in the study of miRNA function (14, 15). We found that inhibition of miR-18b expression in HCT116 and SW480 cells suppressed cell proliferation by CCK-8 compared with the nonspecific control (P < 0.05) (Fig. 3B). Conversely, overexpression of miR-18b dramatically promoted cell proliferation (Fig. 3B). To gain insights into the mechanism by which miR-18b enhances CRC cell proliferation, we analyzed cell cycle distributions in HCT116 and SW480 cells by flow cytometry after miR-18b overexpression or knockdown. We found that miR-18b overexpression decreased the percentage of cells in G1 phase and increased those in S phase, while miR-18b underexpression had the opposite effect (Fig. 3C). These data suggested that miR-18b could promote the transition from G1 to S phase.
FIG 3.

(A) miR-18b expression levels in CRC cells after transfection with miR-18b mimic, inhibitor, mimic negative control, and inhibitor negative control. (B) miR-18b significantly inhibited HCT116 and SW480 cell viability. Cell growth rates were detected by CCK-8 assay. OD, optical density. (C) miR-18b regulated cell cycle progression. miR-18b overexpression promoted the transition from G1 phase to S phase, which was detected by flow cytometry at 48 h posttransfection. (D) Reexpression of miR-18b in HCT116 and SW480 cells significantly inhibited cell migration ability as determined by a cell migration assay. Shown is quantitative analysis of migrated HCT116 and SW480 cells. The data are means from three independent experiments ± SD. *, P < 0.05. All experiments were performed in triplicate. nc, negative control.
Since miR-18b expression was correlated closely with the metastasis property of CRC, migration assays were performed by Transwell experiments, and we found that, compared with the controls, knockdown of miR-18b significantly suppressed migration of CRC cells. In contrast, overexpression of miR-18b promoted the effect (Fig. 3D). These results suggest that miR-18b expression promotes CRC cell proliferation and migration in vitro.
CDKN2B is a potential target of miR-18b.
To identify the target mRNAs of miR-18b, we performed high-throughput RNA sequencing (RNA-seq) experiments after knockdown of miR-18b in HCT116 cells. The miRNA-mRNA integration analysis workflow was as follows (Fig. 4A). We first used 3 different software programs for microRNA target prediction and identified 2,190 potential miR-18b targets based on their sequence complementarity (Fig. 4B). RNA-seq experiments were carried out in miR-18b-depleted HCT116 cells and normal HCT116 cells. Then, the gene expression profile in HCT116 cells regulated by miR-18b was analyzed by RNA-seq. Gene ontology (GO) analysis revealed that the differentially expressed genes were involved in molecule function, cellular components, and biological processes (data not shown). The resulting 678 differentially expressed genes (DEGs) were overlapped with the miR-18b targets predicted by bioinformatics analysis, and 19 candidate genes were identified: the CDKN2B, CDK5R2, CYLD, GORAB, TTBK2, TBATA, ONECUT1, GGN, IGSF9B, AQP1, KCNRG, CHD9, ZBTB24, CYP11A1, HARBI1, C2orf88, SARM1, ATG9A, and PPP2R1B genes (Fig. 4C and D). Among these candidates, the CDKN2B gene exhibited the most significant alteration. Our data showed that miR-18b promoted G1-S transition, and the CDKN2B gene is well known to inhibit the transition, so we verified whether the CDKN2B gene is a target of miR-18b. CDKN2B is a cyclin-dependent kinase inhibitor, forms a complex with cyclin-dependent kinase 4 (CDK4) or CDK6, and prevents the activation of the CDK kinases, thus functioning as a cell growth regulator that controls cell cycle G1 progression (16). In addition, an alignment of CDKN2B mRNA and miR-18b sequences using the RNA22 database predicted the binding site of miR-18b (Fig. 4E). Furthermore, in the current study, the RNA-seq result clearly showed that CDKN2B was enriched in miR-18b knockdown cells (Fig. 4F), which suggested that CDKN2B might be the potential target of miR-18b.
FIG 4.

(A) Strategy for identification and validation of miR-18b targets. (B) miR-18b targets predicted by 3 different types of software. There were as many as 1,786 mRNAs in intersections of the 3 target-predicting software programs. (C) Nineteen mRNAs were in the intersection of RNA-seq and predicted mRNAs. (D) The concrete target mRNAs of miR-18b were based on the intersection between RNA-seq and its predicted targets. (E) Alignment of CDKN2B mRNA and the miR-18b sequence predicted by the RNA22 database (also termed miR-18-5p). (F) RNA-seq data indicated that there was enrichment of CDKN2B in miR-18b-depleted HCT116 cells compared with normal HCT116 cells.
miR-18b suppresses the cell cycle by targeting CDKN2B.
Since CDKN2B was predicted to be the target of miR-18b, we measured its mRNA and protein expression levels by qRT-PCR and Western blotting in CRC cell lines. The results showed that overexpression of a miR-18b mimic reduced the mRNA and protein levels of CDKN2B, whereas overexpression of a miR-18b inhibitor had the opposite effect in HCT116 and SW480 cells (Fig. 5A and B). Interestingly, a luciferase reporter assay indicated that miR-18b significantly inhibited luciferase activity in the reporter vector containing the wild-type 3′ untranslated region (UTR) of CDKN2B but not in the mutant 3′ UTR vector, demonstrating the specificity of miR-18b in CDKN2B 3′ UTR targeting (Fig. 5C). Consistently, protein expression and mRNA levels of CDKN2B were significantly downregulated in tumor tissues compared with nontumor controls (Fig. 6A and B). Moreover, we further analyzed the association between miR-18b and CDKN2B mRNA levels and found that miR-18b was negatively correlated with CDKN2B mRNA in CRC patients by Pearson correlation analysis, suggesting that miR-18b negatively regulates the expression of CDKN2B (Fig. 6C). Our data suggest that CDKN2B is a target of miR-18b in CRC development and progression.
FIG 5.
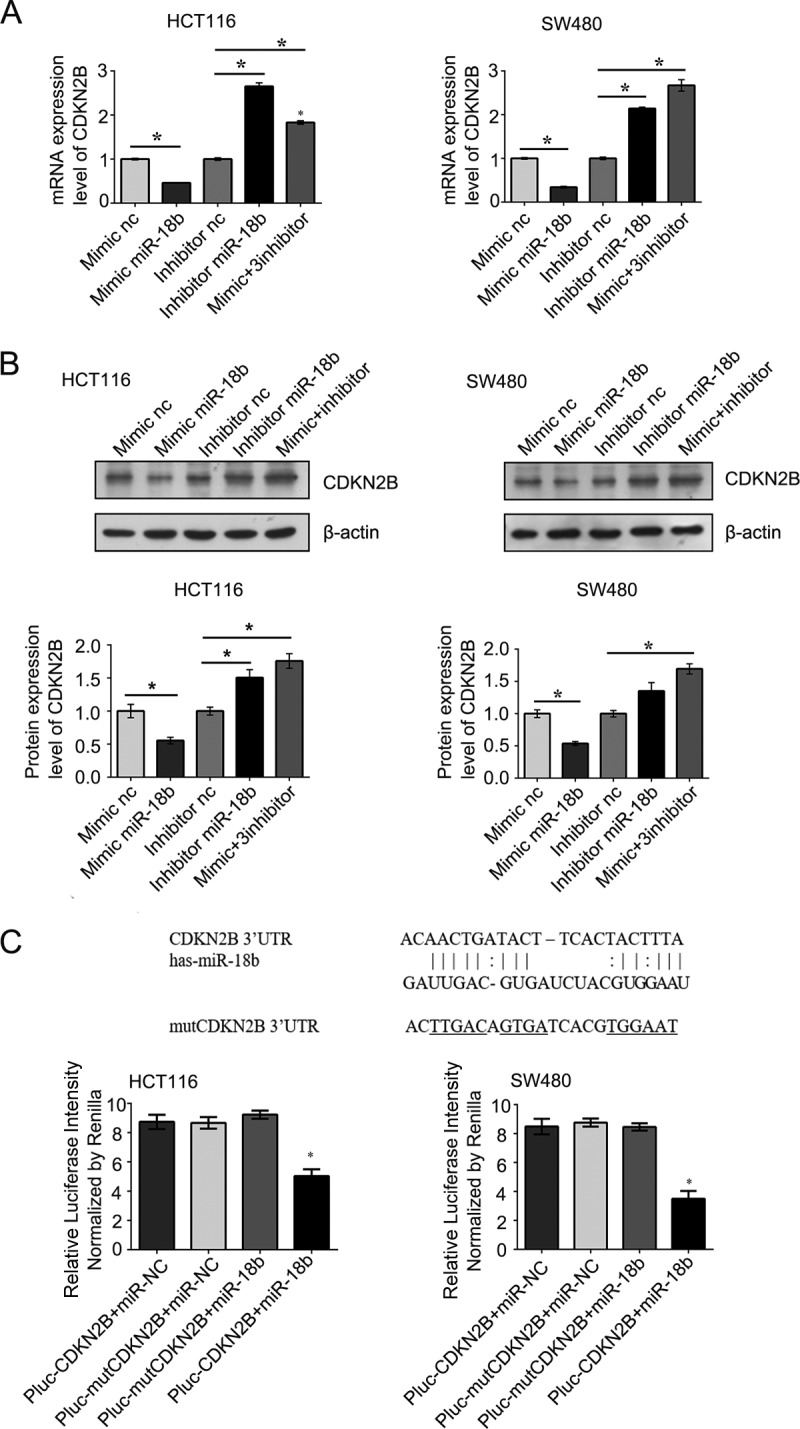
(A) CDKN2B mRNA expression was detected by qRT-PCR in CRC cells after transfection at 48 h. (B) The protein level of CDKN2B was verified by Western blotting in CRC cells; β-actin was used as a loading control. (C) Base pairing complement suggested that the putative miR-18b binding position was at the 3′ UTR of CDKN2B. Lines, strict base pairing between miRNA and mRNA; colons, nonstrict pairing; underlining, differences from CDKN2B. The luciferase activity was standardized to that of hRluc. The graphs show means ± SD. *, P < 0.05.
FIG 6.

(A) Western blotting was performed to show CDKN2B expression in human CRC. (B) Scattergram presenting mRNA expression levels of CDKN2B in 44 CRC patients. (C) Pearson correlation analysis revealed that miR-18b was negatively correlated with CDKN2B mRNA in CRC patients. Each data point represents an individual colon tissue sample. All experiments were performed in triplicate. The graphs show means ± SD. *, P < 0.05; **, P < 0.01.
CDKN2B overexpression antagonizes the effects of miR-18b in vitro and in vivo.
Although the downregulation of CDKN2B by miR-18b and the negative correlation of CDKN2B and miR-18b in CRC patients are convincing, the role of CDKN2B in antagonizing miR-18b function remains to be fully explored. In light of this, we examined the effect of ectopic expression of miR-18b and/or CDKN2B on cell growth and migration in SW480 cells. We noticed that the miR-18b levels in the Lenti-miR-18b and Lenti-miR-18b plus Lenti-CDKN2B groups were about 20.4-fold and 23.3-fold those in the Lenti-miR NC SW480 cells (Fig. 7A). Meanwhile, the expression of CDKN2B was decreased in Lenti-miR-18b plus Lenti-CDKN2B SW480 cells compared with Lenti-CDKN2B SW480 cells (Fig. 7B). We also conducted a CCK-8 assay and found that ectopic expression of CDKN2B significantly suppressed the effects of stimulation proliferation of miR-18b (P < 0.01) (Fig. 7C). The number of SW480 cells that migrated to the lower chamber (see Materials and Methods) in Lenti-miR-18b plus Lenti-CDKN2B SW480 cells was smaller than in Lenti-miR-18b SW480 cells (P < 0.05) (Fig. 7D).
FIG 7.

(A and B) miR-18b and CDKN2B mRNA expression was detected by qRT-PCR in stable overexpression of miR-18b and/or CDKN2B or the negative control (NC) in SW480 cells. U6 is a reference gene. (C) Ectopic expression of CDKN2B significantly suppressed the effects of stimulation of proliferation of miR-18b (P < 0.01). (D) Representative images of stable overexpression of miR-18b and CDKN2B cells that migrated into the lower chamber. The number of SW480 cells that migrated to the lower chamber in Lenti-miR-18b plus Lenti-CDKN2B SW480 cells was less the that in Lenti-miR-18b SW480 cells. (E) miR-18b expression was detected by qRT-PCR in colorectal cancer xenografts. (F) CDKN2B expression was detected in subcutaneous implantation of human colorectal cancer cells in 5 groups. (G) (Left) Tumor growth curves. miR-18b promoted colorectal cancer cell growth in vivo, and the growth effect could be antagonized by CDKN2B. The data are means ± SD (n = 4/group). (Right) Images of tumors induced by miR-18b and/or CDKN2B or negative-control SW480 cells.
Because miR-18b could promote CRC cell growth and was antagonized by CDKN2B in vitro, we next addressed the effects in vivo. In the model of tumor xenografts in nude mice, BALB/c nude mice were injected subcutaneously with SW480 cells stably expressing miR-18b and/or CDKN2B or the negative control. We detected the expression of miR-18b and CDKN2B in subcutaneous tumors and demonstrated that both of them were indeed elevated (Fig. 7E and F). Lenti-CDKN2B suppressed the tumor growth rate in nude mice compared with Lenti-CDKN2B-NC, and from day 13 on, Lenti-miR-18b exhibited a markedly increased growth rate compared with Lenti-miR-NC (P < 0.05). In contrast, from day16 on, the tumor volume of Lenti-miR-18b plus Lenti-CDKN2B mice was smaller than that of Lenti-miR-18b mice (P < 0.05) (Fig. 7G). These results indicated that miR-18b has the ability to promote CRC cell growth and that CDKN2B overexpression can antagonize the effects of miR-18b in vitro and in vivo.
DISCUSSION
In recent years, our understanding of the mechanisms underlying colorectal carcinogenesis has vastly expanded. The initiation and progression of CRC is a complex process that involves multiple factors and multistep alterations. A variety of factors—genetic, immunological, epigenetic, microbial, and environmental—all have different impacts on CRC carcinogenesis (17).
In the present study, we found that several miRNAs exhibited abnormal expression in human CRC by microarray profiling analysis, and miR-18b showed the most significant abnormality among these candidate miRNAs. Our data suggest that miR-18b is overexpressed in CRC patients, which is consistent with previous reports showing miR-18b overexpression in CRC, as well as in hepatic cell carcinoma and breast, gastric, and nasopharyngeal cancers (6–9, 18). These studies imply that miR-18b is indeed involved in the progression of various human cancers as an oncogene. However, there have been other reports revealing the opposite trend in several human cancers. For instance, miR-18b expression is downregulated in a radioresistant esophageal cell line (19). It has also been reported that stable overexpression of miR-18b produces potent tumor suppressor activity by suppressing melanoma cell viability, migration, and invasiveness; inducing apoptosis; and reducing tumor growth, which implies a novel role for miR-18b as a tumor suppressor in melanoma (20). In the study, we detected the expression of miR-18b in CRC cell and tumor xenografts in a nude mouse model and found miR-18b could promote cell growth in vitro and in vivo. The contradiction between different studies might derive from the different cancers with intrinsic biologic characteristics and microenvironments, consequently inducing different mechanisms to regulate miR-18b expression in different malignant tumors. For instance, miR-18b could be reduced by hypermethylation of CpG islands in the promoter and reinduced after 5-AZA-deoxycytidine treatments in melanoma cell lines (20, 21). miR-18b suppresses high-glucose-induced proliferation in human retinal endothelial cells (HRECs) by targeting IGF-1/IGF1R signaling pathways (22). Therefore, the function of miR-18b might be tissue specific and should be explored further.
In addition, we found that miR-18b expression was much higher in CRC patients with lymph node and distant metastasis, suggesting that miR-18b is related to survival. Previous reports also suggested that the higher expression of miR-18b was associated with poor survival in human cancers, such as rectal cancer, hepatocellular carcinoma (HCC), and breast cancer (6, 13, 23). miR-18b also exhibited dysregulation in some benign diseases, including hypertrophic cardiomyopathy and multiple sclerosis (24, 25). Yin et al. reported that miR-18b was involved in the process of benign colorectal adenomas developing into malignancy (18). Taken together, the data show that miR-18b is responsible for various human diseases, particularly in malignant tumors, including CRC. Since it also contributes to colorectal adenomas during the transition process from benign lesions into malignant disease, miR-18b may serve as a diagnostic biomarker and a novel therapeutic target in CRC.
Previous studies and our data all showed that miR-18b plays a critical role in the pathogenesis of human cancer. However, to date, studies have shown only miR-18b abnormal expression; its function and underlying mechanism remain to be explored. Further, we aimed to identify the potential miRNA targets for CRC. High-throughput RNA-seq suggested that miR-18b might play an effective role by targeting some genes involving in growth and proliferation, invasion and metastasis, glucose metabolism, cell cycle regulation, epithelial-mesenchymal transition, and apoptosis of CRC. Compared with traditional systematic analysis of miRNAs in colorectal tumorigenesis (18), which searches for miRNA-regulated genes by computer-aided algorithms, such as TargetScan, miRanda, and miRwalk, our study proposes a novel and efficient strategy to improve the accuracy of miRNA targets through combined use of RNA-seq and predicted target gene bioinformatics software. In the present study, we identified CDKN2B mRNA as the most likely target of miR-18b in CRC. CDKN2B is a cyclin-dependent kinase inhibitor and functions as a cell growth regulator that controls cell cycle G1 progression (16). Previous studies have confirmed CDKN2B as a critical tumor suppressor, and deletion of its enhancer element is associated with some human neoplasms (26, 27). Silencing of CDKN2B gene expression by epigenetic modification, such as promoter hypermethylation, plays a key role in gastric adenocarcinoma and multiple myelomas (28, 29). Reexpression of CDKN2B in tumor-derived cells significantly attenuates the tumorigenic potential of the cells and delays tumor progression (30). miRNAs can regulate gene expression by translation suppression, mRNA cleavage, or both. We showed that miR-18b significantly inhibited CDKN2B at both the mRNA and protein levels, whereas its knockdown had the opposite effect. Our luciferase assay indicated miR-18b directly binds to the 3′ UTR of CDKN2B. Correlation analysis indicated that the mRNA level of miR-18b is negatively related to the level of CDKN2B in CRC tissues, suggesting that miR-18b negatively regulates CDKN2B expression in CRC development. Furthermore, we investigated the effects of miR-18b and CDKN2B on CRC cells in vitro and in vivo and verified that CDKN2B overexpression could antagonize the effect of miR-18b on CRC cell proliferation, migration, and tumor growth rate, which demonstrated that miR-18b indeed plays a key role in the pathogenesis of CRC through targeting CDKN2B.
Conclusions.
Our study demonstrates that miR-18b contributes to CRC carcinogenesis by targeting CDKN2B and regulating cell cycle progression. Our findings also suggest that miR-18b might be a potential biomarker and therapeutic target for CRC.
MATERIALS AND METHODS
Sample preparation.
All CRC patients were recruited at the Department of Oncology, Zhongnan Hospital of Wuhan University (Wuhan, China), from July 2014 to September 2014. CRC tissues and paired tumor-adjacent nontumor samples were obtained from 44 patients with confirmed histology during surgery. The nontumor samples were more than 5 cm away from tumor tissues. The tissues were immersed in RNAlater (Sigma-Aldrich, St. Louis, MO) quickly in case of RNA degradation. After overnight incubation, the samples were transferred into a −80°C refrigerator and stored for further use. None of the patients had received preoperative adjuvant chemotherapy or radiotherapy. The study was approved by the Institutional Review Board for Clinical Research and the Ethics Committee of the Zhongnan Hospital. Written informed consent was also obtained from all the subjects before initiating the study protocol.
Cell culture and transfection.
HCT116 and SW480 cells were purchased from the China Center for Type Culture Collection (Wuhan, China). All the cells were grown in Dulbecco's modified Eagle's medium (DMEM) (HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS) (HyClone), 100 U/ml penicillin, and 100 mg/ml streptomycin. The cells were maintained in a 5% CO2 humidified atmosphere at 37°C. Oligonucleotides containing a miR-18b mimic, inhibitor, mimic negative control, and inhibitor negative control were synthesized by Suzhou Gene Pharma Co., Ltd. (Suzhou, China). The sequences were as follows: mimic sense, 5′-UAAGGUGCAUCUAGUGCAGUUAG-3′, and antisense, 5′-AACUGCACUAGAUGCACCUUAUU-3′; mimic negative control sense, 5′-UUCUCCGAACGUGUCACGUTT-3′, and antisense, 5′-ACGUGACACGUUCGGAGAATT-3′; inhibitor, 5′-CUAACUGCACUAGAUGCACCUUA-3′; inhibitor negative control, 5′-CAGUACUUUUGUGUAGUACAA-3′. The human miR-18b mimic, inhibitor, mimic negative control, and inhibitor negative control (Genepharma, Suzhou, China) were transfected into CRC cells with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to procedures recommended by the manufacturer. The cells were harvested 48 to 72 h posttransfection.
Microarrays and computational analysis.
FlashTag biotin HSR was added to microRNA before microRNA array analysis. MicroRNA array results were scanned with a GeneChip Scanner 7G (Affymetrix, Santa Clara, CA). The quality of RNA was examined with an Agilent 2100 bioanalyzer and RNA LabChip kits. All the data were normalized, and further analyses were carried out with Expression Console (Affymetrix, Santa Clara, CA). The miRNAs were considered to be differentially expressed only when they exhibited a mean change of ≥2.0-fold or <0.5-fold. Hierarchical clustering was performed based on the significantly differential expression of miRNAs, using Cluster and Treeview software from Stanford University (Stanford, CA).
RNA extraction and qRT-PCR.
Total RNA was extracted from tissues and cells with a YPH EASYspin tissue/cell RNA quick extraction kit (YPH, Beijing, China). miRNA reverse transcription (RT) was performed using the All-in-One miRNA qRT-PCR detection kit (GeneCopoeia, Rockville, MD) according to the manufacturer's instructions. Reference gene RUN-6 and miRNA primers were purchased from GeneCopoeia Inc. (catalog no. HmiRQP9001 and HmiRQP0256, respectively). In addition, mRNA RT was performed with a ReverTra Ace kit (Toyobo, Osaka, Japan). The cDNA then served as the template for SYBR real-time PCR. Primer sequences were as follow: CDKN2B, 5′-TGGCTACGAATCTTCCG-3′ (forward) and 5′-CCTCCTCCACTTTGTCCT-3′ (reverse); GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 5′-GAAGGTGAAGGTCGGAGTC-3′ (forward) and 5′-GAAGATGGTGATGGGATTTC-3′ (reverse). All reactions were run in triplicate on a real-time PCR detection system (Bio-Rad, Hercules, CA).
Cell proliferation assays.
HCT116 (2 × 103) or SW480 (1.5 × 103) cells were transfected with miR-18b mimic or inhibitor and then seeded in 96-well plates. The cells were then counted with a Cell Counting Kit 8 (Dojindo Molecular Technologies, Kumamoto, Japan) at 4 to 48 h posttransfection. The plates were incubated for an additional 2 h at 37°C. The absorbance at 450 nm was measured with a microplate reader (ELX-800; BioTek, Winooski, VT).
Cell migration assays.
Cell migration assays were performed using Transwell chambers (8-μm pore size; BandD, NJ) according to the manufacturer's instructions. The cells (2 × 105) were plated in the insert of the well. After incubation for 48 h, cells that had migrated to the lower surface were stained with crystal violet (Sigma-Aldrich, St. Louis, MO) and photographed. The numbers of cells that migrated were quantified by counting six random fields. Representative photos were taken at ×200 magnifications.
Flow cytometry assay.
At 48 h after transfection with miR-18b mimic or inhibitor, CRC cells were collected with trypsin. The cells were fixed with 70% ethanol and stored overnight at −20°C. The fixed cells were washed with PBS, treated with 50 μg/ml RNase A (Sangon Biotech Co., Ltd., Shanghai, China), and stained with 50 μg/ml propidium iodide for 30 min in the dark. The stained cells were analyzed using flow cytometry (FACSCalibur; Becton Dickinson, Franklin Lakes, NJ).
Bioinformatics analysis and RNA-seq library construction.
An RNA-seq library was constructed with the Illumina TruSeq library construction kit (Illumina, CA). The RNA-seq libraries were sequenced using HiSeq2000 for 100-bp paired-end sequencing. Quality control of the RNA-seq data was performed using Fatsqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and low-quality bases were trimmed. The data were mapped to the hg19 genome reference with Tophat2, allowing a maximum of 2 mismatches (31–33). Cufflinks was used to find the gene which was differentially expressed (31). Gene ontology analysis was performed using DAVID (http://david.abcc.ncifcrf.gov). The PITA, miRanda, and RNA22 databases were used to predict the potential target genes of miR-18b.
Luciferase assay.
The luciferase assay was performed as described previously. Briefly, a 322-bp fragment of the wild-type CDKN2B UTR containing the putative miR-18b binding site or a mutant CDKN2B sequence was cloned into the pMIR-REPORT luciferase plasmid (ViewSolid Biotech, Beijing, China). HCT116 or SW480 cells were transfected with different reporter vectors (p-Luc-3′UTR-CDKN2B or p-Luc-3′UTR-mut-CDKN2B) and cotransfected with the negative control or miR-18b mimic. Reporter vectors containing hRluc, a humanized Renilla luciferase, were used for normalizing the transfection rates. The cells were assayed for luciferase activity 48 h after transfection with a Dual-Light reporter assay system (Promega, Madison, WI). Measurement periods were 2 s for luc2 activity and 2 s for hRluc activity. The background activity was subtracted prior to evaluation. Firefly luciferase activity was normalized for Renilla luciferase activity as recommended by the manufacturer (Promega). The assessment was performed in duplicate for each construct and control. The oligonucleotides used for cloning the CDKN2B 3′ UTR into pMIR-REPORT were as follows: CDKN2B wild type, CGGCTAGCGATAGATGCACTTATGCAGTA and GCGTCGACGGGAACCCCTGTGAACCTTTA; CDKN2B mutant, ATTCCACGTGATCACTGTCAAGTTCCAATGATATAATGATCAA and ACTTGACAGTGATCACGTGGAATATAAAGAATTAACAGAGATT.
Western blot analysis.
Total protein was extracted from CRC cells and patient tissues. Equal amounts of proteins were separated by SDS-15% PAGE and transferred to a nitrocellulose membrane (Bio-Rad). After being blocked with 5% nonfat milk, the membrane was incubated with anti-CDKN2B (ABcolonal, Cambridge, MA) antibody or anti-β-actin (ABcolonal, Cambridge, MA). All of the Western blots were repeated three times independently, and only representative images are shown in the figures.
Stable overexpression of miR-18b and CDKN2B in SW480 cells.
SW480 cells were stably transduced with commercially available lentiviral microRNA particles expressing miR-18b, termed Lenti-miR-18b; control lentivirus particles, termed Lenti-miR-NC; CDKN2B, termed Lenti-CDKN2B; and control lentivirus particles, termed Lenti-CDKN2B-NC (GeneCopoeia, Rockville, MD). In another group, SW480 cells were stably transduced simultaneously with lentiviral microRNA particles expressing miR-18b and lentiviral particles expressing CDKN2B, termed Lenti-miR-18b plus Lenti-CDKN2B. The plasmid is based on the pSMART vector with a puromycin selection, Cherry, or green fluorescent protein (GFP) cassette. SW480 cells were transduced with lentiviral microRNA particles at a multiplicity of infection (MOI) of 50 and 6 μg/ml Polybrene for 24 h. The SW480 cells were then maintained in 6-well plates and purified with 5 μg/ml puromycin for 1 week before further experiments.
Model of tumor xenografts in nude mice.
SW480 cells (2 × 106) stably expressing miR-18b and/or CDKN2B or negative-control cells were injected subcutaneously into the left flank of male BALB/c nude mice (4 weeks old; n = 4 mice per group). The tumor size was measured every 3 days using calipers, and the tumor volume (V) was calculated as follows: (l × w × w)/2, with l indicating length and w indicating width. The mice were killed by cervical dislocation on day 28, and the tumors were excised and snap-frozen for RNA extraction. All experimental procedures were approved by the Animal Ethics Committee of Wuhan University.
Statistical analysis.
Data are presented as means and standard deviations (SD) of the results from at least three independent experiments performed in triplicate. SPSS 17.0 software (IBM, Chicago, IL) and Prism 5.0 (GraphPad, San Diego, CA) were used for statistical analysis. Multiple linear regression analysis was used to evaluate the association between clinical characteristics and miR-18b expression levels of CRC patients. Two groups were compared by Student's t test, and the correlation was determined by Pearson correlation analysis. A P value of 0.05 or less was considered to indicate statistical significance.
Accession number(s).
We have mapped our RNA-Seq data to human genome hg19 and uploaded the bedgraph format files to the UCSC Ggenome Browser ( https://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=leipinji&hgS_otherUserSessionName=hg19%2DHCT116%2Dliyiming).
ACKNOWLEDGMENTS
This project was supported by a grant from the National Natural Science Foundation of China (no. 81270468).
M.Y. and M.W. designed the project and wrote the manuscript. Y.L., X.Y., and J.Z. contributed to all experiments. Y.L. was mainly responsible for acquisition and analysis of data and writing the manuscript. M.C. carried out animal experiments. M.W. and L.L. conducted the experiments and data analysis. X.Y. and J.L. collected human tissue samples. We all read and gave our approval to the final version of the manuscript.
There were no conflicts of interest involved in this study.
REFERENCES
- 1.Amin M, Lockhart AC. 2015. The potential role of immunotherapy to treat colorectal cancer. Expert Opin Investig Drugs 24:329–344. doi: 10.1517/13543784.2015.985376. [DOI] [PubMed] [Google Scholar]
- 2.Brenner H, Kloor M, Pox CP. 2014. Colorectal cancer. Lancet 383:1490–1502. doi: 10.1016/S0140-6736(13)61649-9. [DOI] [PubMed] [Google Scholar]
- 3.Macfarlane LA, Murphy PR. 2010. MicroRNA: biogenesis, function and role in cancer. Curr Genomics 11:537–561. doi: 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okugawa Y, Toiyama Y, Goel A. 2014. An update on microRNAs as colorectal cancer biomarkers: where are we and what's next? Expert Rev Mol Diagn 14:999–1021. doi: 10.1586/14737159.2014.946907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendell JT. 2008. miRiad roles for the miR-17-92 cluster in development and disease. Cell 133:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murakami Y, Tamori A, Itami S, Tanahashi T, Toyoda H, Tanaka M, Wu W, Brojigin N, Kaneoka Y, Maeda A, Kumada T, Kawada N, Kubo S, Kuroda M. 2013. The expression level of miR-18b in hepatocellular carcinoma is associated with the grade of malignancy and prognosis. BMC Cancer 13:99. doi: 10.1186/1471-2407-13-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo J, Miao Y, Xiao B, Huan R, Jiang Z, Meng D, Wang Y. 2009. Differential expression of microRNA species in human gastric cancer versus non-tumorous tissues. J Gastroenterol Hepatol 24:652–657. doi: 10.1111/j.1440-1746.2008.05666.x. [DOI] [PubMed] [Google Scholar]
- 8.Yu X, Zhen Y, Yang H, Wang H, Zhou Y, Wang E, Marincola FM, Mai C, Chen Y, Wei H, Song Y, Lyu X, Ye Y, Cai L, Wu Q, Zhao M, Hua S, Fu Q, Zhang Y, Yao K, Liu Z, Li X, Fang W. 2013. Loss of connective tissue growth factor as an unfavorable prognosis factor activates miR-18b by PI3K/AKT/C-Jun and C-Myc and promotes cell growth in nasopharyngeal carcinoma. Cell Death Dis 4:e634. doi: 10.1038/cddis.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fonseca-Sanchez MA, Perez-Plasencia C, Fernandez-Retana J, Arechaga-Ocampo E, Marchat LA, Rodriguez-Cuevas S, Bautista-Pina V, Arellano-Anaya ZE, Flores-Perez A, Diaz-Chavez J, Lopez-Camarillo C. 2013. microRNA-18b is upregulated in breast cancer and modulates genes involved in cell migration. Oncol Rep 30:2399–2410. doi: 10.3892/or.2013.2691. [DOI] [PubMed] [Google Scholar]
- 10.Kim TH, Kim YK, Kwon Y, Heo JH, Kang H, Kim G, An HJ. 2010. Deregulation of miR-519a, 153, and 485-5p and its clinicopathological relevance in ovarian epithelial tumours. Histopathology 57:734–743. doi: 10.1111/j.1365-2559.2010.03686.x. [DOI] [PubMed] [Google Scholar]
- 11.Dacic S, Kelly L, Shuai Y, Nikiforova MN. 2010. miRNA expression profiling of lung adenocarcinomas: correlation with mutational status. Mod Pathol 23:1577–1582. doi: 10.1038/modpathol.2010.152. [DOI] [PubMed] [Google Scholar]
- 12.Cookson VJ, Bentley MA, Hogan BV, Horgan K, Hayward BE, Hazelwood LD, Hughes TA. 2012. Circulating microRNA profiles reflect the presence of breast tumours but not the profiles of microRNAs within the tumours. Cell Oncol 35:301–308. doi: 10.1007/s13402-012-0089-1. [DOI] [PubMed] [Google Scholar]
- 13.Azizian A, Kramer F, Jo P, Wolff HA, Beissbarth T, Skarupke R, Bernhardt M, Grade M, Ghadimi BM, Gaedcke J. 2015. Preoperative prediction of lymph node status by circulating mir-18b and mir-20a during chemoradiotherapy in patients with rectal cancer. World J Surg 39:2329–2335. doi: 10.1007/s00268-015-3083-8. [DOI] [PubMed] [Google Scholar]
- 14.Almeida MI, Nicoloso MS, Zeng L, Ivan C, Spizzo R, Gafa R, Xiao L, Zhang X, Vannini I, Fanini F, Fabbri M, Lanza G, Reis RM, Zweidler-McKay PA, Calin GA. 2012. Strand-specific miR-28-5p and miR-28-3p have distinct effects in colorectal cancer cells. Gastroenterology 142:886–896. doi: 10.1053/j.gastro.2011.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qu LL, He L, Zhao X, Xu W. 2015. Downregulation of miR-518a-3p activates the NIK-dependent NF-kappaB pathway in colorectal cancer. Int J Mol Med 35:1266–1272. doi: 10.3892/ijmm.2015.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ortega S, Malumbres M, Barbacid M. 2002. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta 1602:73–87. [DOI] [PubMed] [Google Scholar]
- 17.Birt DF, Phillips GJ. 2014. Diet, genes, and microbes: complexities of colon cancer prevention. Toxicol Pathol 42:182–188. doi: 10.1177/0192623313506791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin Y, Song M, Gu B, Qi X, Hu Y, Feng Y, Liu H, Zhou L, Bian Z, Zhang J, Zuo X, Huang Z. 2016. Systematic analysis of key miRNAs and related signaling pathways in colorectal tumorigenesis. Gene 578:177–184. doi: 10.1016/j.gene.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 19.Basu RK, Wong HR, Krawczeski CD, Wheeler DS, Manning PB, Chawla LS, Devarajan P, Goldstein SL. 2014. Combining functional and tubular damage biomarkers improves diagnostic precision for acute kidney injury after cardiac surgery. J Am Coll Cardiol 64:2753–2762. doi: 10.1016/j.jacc.2014.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dar AA, Majid S, Rittsteuer C, de Semir D, Bezrookove V, Tong S, Nosrati M, Sagebiel R, Miller JR III, Kashani-Sabet M. 2013. The role of miR-18b in MDM2-p53 pathway signaling and melanoma progression. J Natl Cancer Inst 105:433–442. doi: 10.1093/jnci/djt003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Formosa A, Lena AM, Markert EK, Cortelli S, Miano R, Mauriello A, Croce N, Vandesompele J, Mestdagh P, Finazzi-Agro E, Levine AJ, Melino G, Bernardini S, Candi E. 2013. DNA methylation silences miR-132 in prostate cancer. Oncogene 32:127–134. doi: 10.1038/onc.2012.14. [DOI] [PubMed] [Google Scholar]
- 22.Wu JH, Wang YH, Wang W, Shen W, Sang YZ, Liu L, Chen CM. 2016. miR-18b suppresses high-glucose-induced proliferation in HRECs by targeting IGF-1/IGF1R signaling pathways. Int J Biochem Cell Biol 73:41–52. doi: 10.1016/j.biocel.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Yoshimoto N, Toyama T, Takahashi S, Sugiura H, Endo Y, Iwasa M, Fujii Y, Yamashita H. 2011. Distinct expressions of microRNAs that directly target estrogen receptor alpha in human breast cancer. Breast Cancer Res Treat 130:331–339. doi: 10.1007/s10549-011-1672-2. [DOI] [PubMed] [Google Scholar]
- 24.Otaegui D, Baranzini SE, Armananzas R, Calvo B, Munoz-Culla M, Khankhanian P, Inza I, Lozano JA, Castillo-Trivino T, Asensio A, Olaskoaga J, de Munain AL. 2009. Differential micro RNA expression in PBMC from multiple sclerosis patients. PLoS One 4:e6309. doi: 10.1371/journal.pone.0006309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. 2007. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol 42:1137–1141. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, Berns A. 2007. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 448:943–946. doi: 10.1038/nature06084. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Knobloch TJ, Poi MJ, Zhang Z, Davis AT, Muscarella P, Weghorst CM. 2014. Genetic alterations of RD(INK4/ARF) enhancer in human cancer cells. Mol Carcinog 53:211–218. doi: 10.1002/mc.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.do Nascimento Borges B, Burbano RM, Harada ML. 2013. Analysis of the methylation patterns of the p16 INK4A, p15 INK4B, and APC genes in gastric adenocarcinoma patients from a Brazilian population. Tumour Biol 34:2127–2133. doi: 10.1007/s13277-013-0742-y. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Bi L, Lin Y, Lu Z, Hou G. 2014. Clinicopathological significance and potential drug target of p15INK4B in multiple myeloma. Drug Des Devel Ther 8:2129–2136. doi: 10.2147/DDDT.S71088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camacho CV, Mukherjee B, McEllin B, Ding LH, Hu B, Habib AA, Xie XJ, Nirodi CS, Saha D, Story MD, Balajee AS, Bachoo RM, Boothman DA, Burma S. 2010. Loss of p15/Ink4b accompanies tumorigenesis triggered by complex DNA double-strand breaks. Carcinogenesis 31:1889–1896. doi: 10.1093/carcin/bgq153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]