
FIGURE 1.
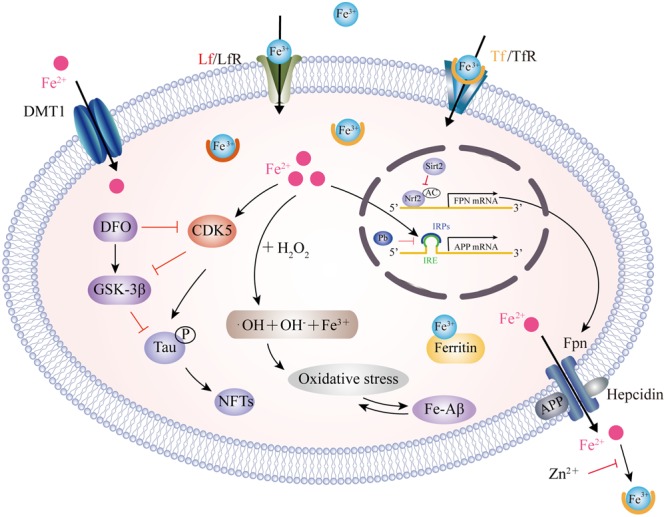
Model for iron transport in neurons and iron imbalance associated with AD. Two pathways have been well-documented for cellular iron uptake: TfR endocytosis-mediated Tf-Fe3+ entry and the direct import of Fe2+ by DMT1. The only way for iron to be transported out of the cell is by Fpn with the ferroxidase activity of CP or HP. However, hepcidin binding to Fpn causes its internalization to prevent Fe2+ export. Intracellular Fe2+ binds to the IRE in the 5′ UTR of APP mRNA, which reverses the repression of IRP1 and promotes APP translation. Pb enhances the IRP1/IRE-mediated repression of APP. APP also has ferroxidase activity and interacts with Fpn to oxidize Fe2+ into Fe3+ for Tf binding. APP ferroxidase is inhibited by extracellular Zn2+. In addition, sirt2 regulates Fpn expression via the deacetylation of Nrf2, which in turn controls cellular iron homeostasis. In the AD brain, increased levels of Fe2+ induces the Fenton reaction to produce •OH, resulting in oxidative damage and Aβ production. In addition, Fe2+ can bind to Aβ and enhance its aggregation, while Fe2+ increases tau phosphorylation via activation of CDK5 and GSK3β. This process can be inhibited by DFO administration. CP, ceruloplasmin; HP, hephaestin; IRE, iron responsive element; IRP1, iron regulatory protein; Pb, lead; Sirt2, sirtuin 2; Nrf2, nuclear factor erythroid-derived 2-related factor 2; CDK5, cyclin-dependent kinase 5; GSK3β, glycogen synthase kinase 3β; DFO, desferrioxamine.