

Abstract
Background
The ratio of sugars to organic acids, two of the major metabolites in fleshy fruits, has been considered the most important contributor to fruit sweetness. Although accumulation of sugars and acids have been extensively studied, whether plants evolve a mechanism to maintain, sense or respond to the fruit sugar/acid ratio remains a mystery. In a prior study, we used an integrated systems biology tool to identify a group of 39 acid-associated genes from the fruit transcriptomes in four sweet orange varieties (Citrus sinensis L. Osbeck) with varying fruit acidity, Succari (acidless), Bingtang (low acid), and Newhall and Xinhui (normal acid).
Results
We reanalyzed the prior sweet orange fruit transcriptome data, leading to the identification of 72 genes highly correlated with the fruit sugar/acid ratio. The majority of these sugar/acid ratio-related genes are predicted to be involved in regulatory functions such as transport, signaling and transcription or encode enzymes involved in metabolism. Surprisingly, only three of these sugar/acid ratio-correlated genes are weakly correlated with sugar level and none of them overlaps with the acid-associated genes. Weighted Gene Coexpression Network Analysis (WGCNA) has revealed that these genes belong to four modules, Blue, Grey, Brown and Turquoise, with the former two modules being unique to the sugar/acid ratio control.
Conclusion
Our results indicate that orange fruits contain a possible mechanistically distinct class of genes that may potentially be involved in maintaining fruit sugar/acid ratios and/or responding to the cellular sugar/acid ratio status. Therefore, our analysis of orange transcriptomes provides an intriguing insight into the potentially novel genetic or molecular mechanisms controlling the sugar/acid ratio in fruits.
Electronic supplementary material
The online version of this article (10.1186/s12870-017-1138-8) contains supplementary material, which is available to authorized users.
Keywords: Citrus, Orange, Fruit, Acids, Sugars, Sugar/acid ratio, Transcriptome, Gene networks
Background
Fleshy fruits contain various sugars and organic acids that are among the most important considerations for improving sensory traits such as taste and flavor. Fruit sweetness is determined by the levels of sugars and acids and their ratio [1–3]. The major sugars in fruits include sucrose, glucose and fructose, and malate and citrate are two major types of organic acids in fruits. Although the genes encoding enzymes involved in sugar or acid metabolism have long been proposed to control fruit sugar or acid levels, the intensive molecular biology studies so far have only led to the demonstration of two acid metabolism-related genes (a specific isoform of aconitase and a malate dehydrogenase) in the control of fruit acidity [4, 5].
To identify additional genes responsible for controlling fruit sweetness, many genetic, molecular and transcriptomic studies have been focused on the transport, storage and degradation of sugars and acids in both climacteric fruits such as tomato [4–9], and non-climacteric fruits such as citrus [10–15]. Recent genetic evidence has revealed important roles of the malate transporter MdMa1 [16], biochemically unknown CmPH gene [17] and the MdMYB transcription factor [18] in controlling apple or melon fruit acidity. Other genetic loci are yet to be cloned, and for many other genes correlated with sugar and/or acid levels, their functions in the control of fruit sweetness remain to be demonstrated by convincing genetic evidence.
Surprisingly, very few studies have examined the sugar/acid ratios. Mathematically, the sugar/acid ratio can be determined by changing the sugar or acid level alone or altering both sugar and acid levels in opposite directions. A recent genetic study showed that antisense suppression of a malate dehydrogenase gene MDH2 in tomato fruits increased the malate level but also led to a reduction in soluble sugar content [4], thus greatly increasing the sugar/acid ratio. Therefore, even a mild change in sugar and acid levels in opposite directions may lead to a significant alteration of the sugar/acid ratio. Such a tight link between sugar and acid metabolism has led to the argument that the sugar/acid ratio can only be regarded as a subjective parameter to evaluate the consumer’s perception of fruit sweetness rather than a horticultural trait. However, results from a genetic analysis involving a regular melon cultivar and its acidic melon bred line suggest distinct genetic variants controlling sugar content, acid content and the sugar/acid ratios [19]. Therefore, examining the genes or genetic variants correlated with the sugar/acid ratios has a potential to facilitate the identification of additional regulatory genes involved in the control of fruit sweetness.
In this study, we use citrus as a model to reanalyze the fruit transcriptome data recently published from our group [14]. Citrus, representing one of the largest fresh fruit and juice industry in the world, has the potential for use as an economically important tree fruit model organism to study non-climacteric fruit development. Citrus has a relatively small genome size (approximately 367 Mb), and efficient genetic transformation techniques have been developed for various citrus cultivars or varieties [20, 21]. In our prior study, we have identified a total of 39 genes strongly correlated with fruit acidity using the four varieties of sweet orange, ranging from acidless (Succari) to low acid (Bingtang) and normal acid (Newhall and Xinhui), which contain similar acid levels at Stage I (cell division; 45 days post anthesis/DPA) but exhibit differing acidity at Stage II (cell expansion; 142 DPA). Although these varieties were originally designed for the acid-related transcriptomic studies, we have found that they exhibit different sugar/acid ratios. Because there has been no transcriptomic study aiming at the genes specifically involved in the control of fruit sugar/acid ratio, we believe that reanalysis of this data will provide important insights. Towards this, we have identified a total of 72 genes strongly correlated with the sugar/acid ratios, and yet most of them do not correlate with either sugar or acid contents alone. Furthermore, since our first report of using systems biology approach in unraveling the citrus host response to the attack by one of the most constructive disease Huanglongbing [22], the gene coexpression-based approach has been increasingly used by us and other groups to further understand the gene networks involved in Huanglongbing pathogenesis and fruit development or predict gene annotations [14, 23–25]. Therefore, in this study we also use the network approach to analyze the fruit transcriptome data, leading us to conclude that these 72 sugar/acid ratio-related genes belong to four modules of the fruit development gene coexpression network, two of which have no overlap with the sugar- and acid-related network modules. Taken together, our results have uncovered the genes and coexpression modules uniquely associated with the control of sugar/acid ratio in expanding fruits.
Results
Analysis of the fruit sugar/acid ratios in four sweet orange varieties with differing fruit acidity
To compare the fruit sugar/acid ratios in the four varieties of sweet orange, Newhall, Xinhui, Bingtang and Succari, we re-analyzed the fruit sugar and acid accumulation data reported previously [14]. At 45 DPA, the four varieties had sugar/acid ratios of 2.3–3.3, but because of similar increase in sugar levels and different acid levels at 142 DPA, they exhibited different sugar/acid ratios at 142 DPA (Fig. 1a). While Xinhui fruits had the same sugar/acid ratio, Newhall showed a slight increase of the ratio from 3.3 to 5.2. However, the low acid variety Bingtang and the acidless variety Succari had much higher sugar/acid ratios at 142 DPA (13.7 and 35.6, respectively). Therefore, it appears that the four varieties exhibit opposite trends for acidity and sugar/acid ratio.
Fig. 1.
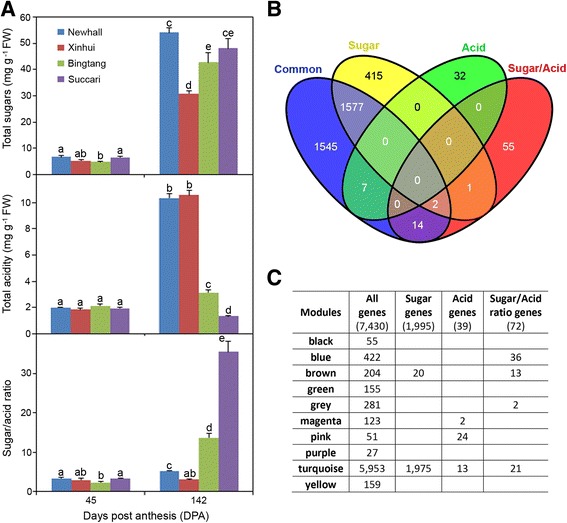
Comparison of sugar/acid ratios in four different sweet orange varieties and of genes and modules associated with sugar, acid and sugar/acid ratios. (a) Comparison of sugar and acid levels and sugar/acid ratios in different sweet orange varieties. Sugar and acid levels were adapted from a prior manuscript [14], and the sugar/acid ratios were calculated using the sugar and acid levels in each fruit for each variety. Values are means and SE of three fruits. Different letters above the column indicates a significant difference (p < 0.05). (b) Venn diagram of overlapping genes correlated with sugar and acid levels and sugar/acid ratios. “Common” refers to the genes commonly regulated from 45 to 142 DPA in all varieties. (c) Comparison of the number of genes associated with sugar, acid and sugar/acid ratio in each of the gene co-expression modules derived from WGCNA
Identification and analysis of genes correlated with the sugar/acid ratio
To identify the candidate genes that are strongly correlated with the fruit sugar/acid ratio in sweet orange fruits, we performed a correlation study between sugar/acid ratio and expression level of 7430 genes which are differentially expressed between 45 and 142 DPA in any of the four varieties reported previously [14]. As a result, we identified a total of 72 genes which showed strong correlations with sugar/acid ratio, with Pcc > = 0.80 or <= −0.80 and a minimal false discovery rate (FDR) of 3.5E-04 (Table 1). Among those 72 genes, only 10 (14%) are negatively correlated with sugar/acid ratio, while all others have positive correlations. The three genes with the strongest correlation (Pcc = 0.90 or 0.91) are Cs1g14800 encoding a Leucine-rich repeat (LRR) receptor-like serine/threonine protein kinase, Cs1g03610 predicted to encode a malate dehydrogenase and Cs7g06285 with no Arabidopsis homolog revealed. Results of qPCR-based expression for six genes showed that gene expression levels measured by qPCR and RNA seq are highly correlated, with Pcc ranging from 0.93 to 0.99 and p-values of <0.001 (Table 2). This indicates that our RNA sequencing data is reliable for use in analysis of genes involved in the control of sugar/acid ratio.
Table 1.
A list of genes strongly correlated with the sugar/acid ratio in orange fruits
| CsGID | Pcc | FDR | Module | AtGID | Arabidopsis gene description |
|---|---|---|---|---|---|
| Transport (9) | |||||
| Cs1g13320 | 0.87 | 1.3E-05 | turquoise | At1g15460 | BOR4, Requires High Boron 4 |
| Cs1g25820 | 0.87 | 1.4E-05 | blue | At5g66110 | HIPP27, heavy metal associated isoprenylated plant protein 27 |
| Cs2g06990 | 0.89 | 8.7E-06 | blue | At4g05120 | FUR1, FUDR Resistant 1 |
| Cs4g20090 | 0.89 | 6.7E-06 | brown | At4g32650 | KAT3, potassium channel in Arabidopsis thaliana 3 |
| Cs5g17210 | 0.81 | 2.1E-04 | blue | At1g17810 | Beta-TIP, beta-tonoplast intrinsic protein |
| Cs5g24670 | −0.87 | 1.4E-05 | blue | At5g48490 | Putative lipid transfer/seed storage protein |
| Cs5g31010 | 0.83 | 1.5E-04 | blue | At3g22600 | Non-specific lipid-transfer/seed storage protein |
| Cs9g18100 | 0.80 | 3.2E-04 | turquoise | At5g53130 | CNGC1, cyclic nucleotide gated channel 1 |
| orange1.1 t01769 | 0.82 | 1.7E-04 | blue | At3g62150 | ABCB21, ATP-binding cassette B21 |
| Signaling (6) | |||||
| Cs1g01170 | 0.86 | 3.0E-05 | blue | At4g23160 | CRK8, cysteine-rich RLK (RECEPTOR-like protein kinase) 8 |
| Cs1g14800 | 0.90 | 5.0E-06 | blue | At3g47570 | probable LRR receptor-like serine/threonine protein kinase |
| Cs2g02680 | 0.88 | 8.7E-06 | brown | At3g21630 | CERK1, chitin elicitor receptor kinase 1 |
| Cs3g07510 | 0.80 | 3.2E-04 | blue | At1g03430 | AHP5, histidine-containing phosphotransfer factor 5 |
| Cs6g02710 | 0.81 | 2.3E-04 | turquoise | At1g16120 | WAKL1, wall associated kinase-like 1 |
| Cs6g05010 | 0.80 | 3.5E-04 | blue | At3g47570 | probable LRR receptorlike serine/threonineprotein kinase |
| Transcription (7) | |||||
| Cs3g01830 | 0.86 | 3.0E-05 | brown | At3g54460 | F-box family protein |
| Cs5g03630 | 0.83 | 1.4E-04 | brown | At5g23750 | remorin like |
| Cs6g03950 | −0.80 | 3.2E-04 | turquoise | At2g37260 | TTG2, Transparent testa Glabra 2, WRKY |
| Cs7g27620 | 0.85 | 4.1E-05 | brown | At1g07900 | LBD1, LOB domain-containing protein 1 |
| Cs9g08500 | 0.80 | 3.2E-04 | turquoise | At5g43630 | TZP, tandem zinc knuckle protein |
| orange1.1 t00185 | 0.83 | 1.2E-04 | blue | At4g00050 | UNE10, unfertilized embryo sac 10, bHLH |
| orange1.1 t00294 | 0.80 | 3.2E-04 | brown | At4g16110 | RR2/ARR2, response regulator 2 |
| Protein degradation (3) | |||||
| Cs8g05200 | 0.80 | 3.2E-04 | blue | At1g47128 | RD21A, responsive to dehydration 21A |
| orange1.1 t00281 | 0.88 | 8.7E-06 | brown | At4g00230 | XSP1, xylem serine peptidase 1 |
| orange1.1 t02370 | 0.83 | 1.2E-04 | blue | At2g31980 | CYS2, Phytocystatin 2 |
| Development (2) | |||||
| Cs4g15700 | 0.80 | 3.0E-04 | turquoise | At4g29860 | EMB2757, Embryo Deffective 2757 |
| orange1.1 t02243 | 0.83 | 1.4E-04 | blue | At5g06760 | LEA4–5, Late Embryogenesis Abundant 4–5 |
| Stress response (4) | |||||
| Cs5g18450 | 0.82 | 1.7E-04 | turquoise | At5g17680 | Leucine rich repeat-containing protein |
| Cs5g19950 | 0.85 | 4.3E-05 | turquoise | At5g17680 | TMV resistance protein N-like |
| Cs7g32260 | 0.80 | 3.5E-04 | turquoise | At5g59720 | HSP18.2, heat shock protein 18.2 |
| orange1.1 t01829 | 0.84 | 7.8E-05 | turquoise | At5g17680 | TMV resistance protein N-like |
| Metabolism (20) | |||||
| Cs1g03610 | 0.91 | 4.9E-06 | blue | At5g43330 | c-NAD-MDH2, cytosolic-NAD-dependent malate dehydrogenase 2 |
| Cs1g10530 | 0.81 | 2.3E-04 | turquoise | At2g18950 | HPT1, homogentisate phytyltransferase 1 |
| Cs1g25210 | −0.84 | 9.1E-05 | brown | At2g18020 | EMB2296, embryo defective 2296, ribosomal protein |
| Cs2g27550 | 0.80 | 3.2E-04 | blue | At3g01570 | Oleosin family protein |
| Cs3g12560 | 0.81 | 2.7E-04 | blue | At1g54740 | ribosomal protein |
| Cs3g24700 | 0.81 | 2.3E-04 | brown | At5g42740 | PGI, glucose-6-phosphate isomerase, cytosolic 1 |
| Cs4g14130 | 0.81 | 2.3E-04 | blue | At2g45550 | CYP76C4, cytochrome P450, family 76, subfamily C, polypeptide 4 |
| Cs5g18850 | 0.82 | 1.5E-04 | blue | At1g71250 | GDSL-motif esterase/acyltransferase/lipase |
| Cs5g20010 | 0.83 | 1.2E-04 | turquoise | At1g35190 | Hyoscyamine 6-dioxygenase |
| Cs7g13310 | −0.83 | 1.2E-04 | turquoise | At1g68530 | KCS6, 3-ketoacyl-CoA synthase 6 |
| Cs7g30920 | 0.81 | 2.2E-04 | blue | At2g40170 | GEA6/Em6, Late embryogenesis abundant 6 |
| Cs7g31620 | 0.80 | 3.0E-04 | blue | At2g38080 | IRX12, Irregular Xylem 12 |
| Cs8g06880 | −0.84 | 1.1E-04 | grey | At4g36750 | Minor allergen Alt a, lipid metabolism |
| Cs9g06700 | 0.86 | 3.0E-05 | brown | At1g31690 | Copper amine oxidase family protein |
| Cs9g07750 | −0.89 | 7.0E-06 | brown | At3g22890 | APS1, ATP sulfurylase 1 |
| Cs9g13750 | 0.81 | 2.3E-04 | blue | At5g07475 | Cupredoxin superfamily protein; copper ion binding |
| Cs9g17670 | 0.81 | 2.3E-04 | grey | At4g23420 | short chain dehydrogenase/reductase (SDR) |
| orange1.1 t00612 | 0.82 | 2.0E-04 | turquoise | At5g66460 | MAN7, endo-beta-mannase 7 |
| orange1.1 t02858 | 0.81 | 2.7E-04 | blue | At1g79640 | serine/threonine protein kinase, putative |
| orange1.1 t03587 | 0.83 | 1.2E-04 | turquoise | At1g17020 | SRG1, senescence-related gene 1 |
| Unknown function (11) | |||||
| Cs1g20290 | 0.80 | 3.0E-04 | blue | ||
| Cs1g20300 | 0.80 | 3.2E-04 | blue | ||
| Cs1g23800 | 0.80 | 3.2E-04 | blue | At2g18540 | vicilin GC72A like, cupin |
| Cs1g24590 | 0.81 | 2.3E-04 | blue | ||
| Cs2g06500 | −0.82 | 2.0E-04 | turquoise | At1g11090 | MGL, Monoglyceride lipase |
| Cs2g07220 | −0.88 | 1.2E-05 | blue | At3g13160 | pentatricopeptide repeat-containing protein |
| Cs2g20110 | 0.88 | 8.7E-06 | blue | ||
| Cs3g17860 | 0.81 | 2.1E-04 | brown | queuine tRNA ribosyltransferase like | |
| Cs5g05940 | 0.80 | 3.2E-04 | blue | ||
| Cs5g06080 | 0.88 | 9.3E-06 | turquoise | At5g50170 | C2 and GRAM domain-containing protein |
| Cs5g20400 | 0.81 | 2.2E-04 | turquoise | ||
| Cs5g23250 | 0.81 | 2.1E-04 | turquoise | At5g67550 | |
| Cs6g16160 | −0.89 | 7.0E-06 | blue | At4g24380 | hypothetical protein SORBIDRAFT_02g043510 |
| Cs7g06285 | 0.90 | 4.9E-06 | brown | ||
| Cs7g23240 | 0.86 | 2.9E-05 | blue | At5g04830 | hypothetical protein ARALYDRAFT_487267 |
| Cs7g25170 | −0.82 | 1.5E-04 | turquoise | ||
| Cs7g30380 | 0.83 | 1.5E-04 | blue | At2g40390 | |
| Cs8g10300 | 0.84 | 1.1E-04 | blue | ||
| Cs8g18390 | −0.80 | 3.0E-04 | turquoise | ||
| Cs8g19210 | 0.82 | 2.0E-04 | blue | ||
| orange1.1 t01135 | 0.84 | 7.8E-05 | blue | hypothetical protein ARALYDRAFT_487267 | |
A total of 72 citrus genes are highly correlated with the sugar/acid ratios in orange fruits of four varieties, with a Pearson correlation coefficient (Pcc) of above 0.80 or below −0.80 and an adjusted p-value (FDR, false discovery rate) of larger than 1.0E-04. CsGID, Cs gene ID. The number of genes for individual biological process is indicated in parenthesis and the module to which the genes belong is inidcated. The most closely related homologs of Arabidopsis gene for each citrus gene is presented as AtGID (At gene ID), with Arabidopsis gene description shown. Absence of AtGID indicates no Arabidopsis homolog for CsGID identified
Table 2.
Pearson correlation analysis of sugar/acid ratio-related gene expression levels detected by RNA sequencing and qPCR
| Genes | Methods | 45 DPA | 142 DPA | Pcc | p-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Newhall | Xinhui | Bingtang | Succari | Newhall | Xinhui | Bingtang | Succari | ||||
| Cs1g03610 (malate dehydrogenase) | RNAseq | 0.4 | 0.4 | 0.3 | 0.6 | 0.6 | 0.4 | 0.6 | 3.8 | 0.99 | 1.3E-08 |
| qPCR | 1.0 | 1.4 | 0.8 | 1.6 | 1.3 | 1.4 | 1.4 | 10.3 | |||
| Cs5g03630 (remorin like) | RNAseq | 0.8 | 1.3 | 0.5 | 1.3 | 1.8 | 1.8 | 1.3 | 7.1 | 0.99 | 1.6E-07 |
| qPCR | 1.0 | 1.3 | 0.8 | 1.4 | 2.3 | 2.6 | 2.2 | 11.2 | |||
| Cs5g20010 (Hyoscyamine 6dioxygenase) | RNAseq | 0.3 | 0.2 | 0.1 | 0.1 | 0.8 | 0.3 | 1.5 | 2.0 | 0.99 | 3.1E-07 |
| qPCR | 1.0 | 0.8 | 0.6 | 0.4 | 4.2 | 1.0 | 7.8 | 11.8 | |||
| Cs5g24670 (lipid transfer) | RNAseq | 47.2 | 45.9 | 58.2 | 93.3 | 39.4 | 42.8 | 25.5 | 4.6 | 0.93 | 7.1E-04 |
| qPCR | 1.0 | 1.0 | 1.0 | 2.1 | 1.2 | 1.2 | 0.6 | 0.2 | |||
| Cs6g16160 (unknown) | RNAseq | 78.3 | 76.9 | 73.4 | 29.8 | 148.0 | 63.9 | 17.3 | 2.3 | 0.97 | 4.6E-05 |
| qPCR | 0.8 | 1.3 | 0.5 | 1.3 | 1.8 | 1.8 | 1.3 | 7.1 | |||
| Cs7g32260 (HSP18.2) | RNAseq | 0.4 | 0.4 | 0.4 | 0.1 | 5.3 | 5.7 | 4.5 | 109.3 | 0.99 | 6.5E-11 |
| qPCR | 1.0 | 0.8 | 0.6 | 0.6 | 32.9 | 24.0 | 16.9 | 776.8 | |||
Expression levels for six genes which were determined to be differentially regulated between different varieties by RNA sequencing (RNA seq) were validated by using quantitative PCR (qPCR) analysis. Values are the means of RNA seq data (RPKM) or qPCR data (with the value for Newhall at 45 DPA set as 1 after normalization to the Actin control) from three biological replicates. DPA, days post anthesis; Pcc, Pearson correlation coefficient
Gene ontology (GO) analysis and Arabidopsis gene homolog prediction result showed that 11 of 72 sugar/acid ratio-correlated genes have unknown functions, 20 likely function in metabolism, and the remaining 41 genes are involved in regulatory functions including transport, signaling, transcription, degradation, stress response and development (Table 1).
The 20 genes in the metabolism GO category encode a wide array of enzymes involved in various aspects of metabolism, including secondary metabolism (Cs5g20010/ hyoscyamine 6-dioxygenase, Cs7g13310/KCS6, Cs7g31620/IRX12 and orange1.1 t03587/SRG1), lipid metabolism (Cs2g27550 and Cs8g06880), protein synthesis (Cs1g25210/EMB2296, and Cs3g12560, both being ribosomal proteins), primary carbon metabolism (Cs3g24700/PGI, and Cs1g03610/MDH2), and sulfate metabolism (Cs9g07750/APS1). Of particular note, Cs3g24700 is orthologous to At5g42740-encoded cytosolic glucose-6-phosphate isomerase (PGI) involved in glycolysis [26], and Cs1g03610, the most strongly sugar/acid ratio-correlated gene, is most closely related to the Arabidopsis cytosolic-NAD-dependent malate dehydrogenase (c-NAD-MDH2), raising the possible involvement of malate metabolism and glycolysis in the fruit sugar/acid ratio control. The role of Cs9g07750, which is predicted to encode APS1, an enzyme involved in sulfate assimilation to Cys [27], in fruit sugar and acid metabolism remains unclear. The observation that sulfur metabolism cross-talks with the carbohydrate and nitrogen status [28] might indicate a possible role for modulation of Cs9g07750/APS1 expression in sensing of or response to sugar level.
As in the case of acid-related genes [14], a considerable proportion (19) of the sugar/acid ratio-correlated genes are involved in transport, transcription or protein degradation. Among nine genes in the transport category, two (Cs5g24670 and Cs5g31010) are predicted to act in lipid transfer in the seed storage process. Cs2g06990 is closely related to FUR1, a nucleoside transporter [29], while orange1.1 t01769 is similar to ATP-binding cassette B21, which is involved in auxin efflux and influx [30]. Cs1g13320 is similar to an efflux-type borate transmembrane transporter [31], Cs4g20090 is closely related to potassium channel protein KAT3, Cs9g18100 is predicted to encode a cyclic nucleotide gated channel (CNGC1), and both Cs1g25820 and Cs5g17210 show high similarity to Arabidopsis metal ion transporter. Regarding the three genes involved in protein degradation, two of them (Cs8g05200 and orange1.1 t02370) are putative cysteine proteases and the other one (orange1.1 t00281) may act as a serine peptidase. With regard to the seven genes in the category of transcription, they belong to distinct family of transcriptional regulators, such as F-box family (Cs3g01830), WRKY family (Cs6g03950), bHLH family (orange1.1 t00185), tandem zinc family (Cs9g08500), remornin-like (Cs5g03630), LBD family (Cs7g27620), and ARR family (orange1.1 t00294). In Arabidopsis, TTG2 is involved in proanthocyanidin synthesis and cell size control [32]. ARR2 has been shown to act in response to cytokinin and ethylene and in the expression of nuclear genes encoding components of mitochondrial complex I [33, 34]. Overall, none of these three categories of proteins has been shown to be involved in regulation of sugar or acid synthesis, transport or response.
The sugar/acid ratio-correlated genes also have two GO categories that were not enriched for the acid-correlated genes: signaling and stress response. Interestingly, five of six signaling-related genes encode receptor-like kinases (RLK), such as Cs1g01170/CRK8 and Cs6g02710/WAKL1, and the remaining one is Cs3g07510/AHP5. In Arabidopsis, CRK8 and CERK1 have been shown to act in defense response [35, 36] and expression of CRK8 could be modulated by hormones [35]. AHP5, one of the six Arabidopsis histidine phosphotransfer proteins (AHPs), has a redundant function as positive regulators of cytokinin signaling [37]. Concerning the category of stress response, three of four genes (Cs5g18450, Cs5g19950, orange1.1 t01829) are involved in biotic stress response, while the fourth one, Cs7g32260/HSP18.2, is predicted to act in abiotic stress response.
Comparison of sugar/acid ratio-correlated genes with sugar- or acid-associated genes
As mathematically the sugar/acid ratio could be affected by changes in levels of sugar alone, acid alone or both, we decided to assess whether any of the 72 sugar/acid ratio-correlated genes has overlap with the set of 39 acid-correlated genes. Surprisingly, analysis using Venn diagram showed that none of the acid- and sugar/acid ratio-correlated genes overlaps (Fig. 1b).
We then analyzed the degree of overlap between the sugar/acid ratio-correlated genes and the sugar-correlated genes. We were surprised that 4166 out of 7430 differentially expressed genes exhibited strong correlations with sugar level, with Pcc above 0.8 or below −0.8 (Additional file 1). Thus, we used a higher Pcc cutoff (+/−0.9) for the prediction of sugar-correlated genes, with a total of 1995 genes (Additional file 1). Comparison of these 1995 genes and 72 sugar/acid ratio-related genes revealed that only three genes (Cs1g10530, Cs8g18390 and Cs9g08500) are strongly correlated with both sugar level and sugar/acid ratio (Fig. 1b). Nine additional sugar/acid ratio-related genes also had Pcc with sugar between 0.8 and 0.9 (Additional file 2). This indicates that the majority of the sugar/acid ratio-correlated genes do not strongly correlate with sugar level. Taken together, our comparative analysis of sugar-, acid- and sugar/acid ratio-correlated genes strongly indicates that at least in these four sweet orange varieties a unique subset of genes might be involved in the control of fruit sugar/acid ratio independent of sugar and acid accumulation alone.
To reveal whether the control of sugar/acid ratio involves distinct gene coexpression modules, we respectively mapped the subsets of acid-, sugar- and sugar/acid-correlated genes into the 10 modules present in the gene coexpression network reported in our prior work [14]. Results showed that among the 1995 sugar-correlated genes, 1975 belong to the Turquoise module and 20 are classified to the Brown module; however, the sugar/acid ratio-correlated genes can be grouped into four modules, Blue, Grey, Brown and Turquoise (Fig. 1c). Compared to the acid-related gene modules, the sugar/acid ratio-correlated genes contain three distinct modules, Blue, Brown and Grey. Taken together, the sugar/acid ratio-related genes possess two unique modules, Blue and Grey. The Blue module is the largest one, containing 36 out of 72 sugar/acid ratio genes, while the Grey module has only two genes (Fig. 1c; Table 1). Thus, the Blue module might represent a distinct subnetwork for the control of fruit sugar/ acid ratio.
Construction and analysis of distinct module-based sugar/acid ratio gene subnetworks
To provide a systems view of gene networks for the control of sugar/acid ratio in orange fruits, we constructed the subnetworks based on the four distinct gene coexpression modules, Blue, Brown, Grey and Turquoise (Fig. 1c). The subnetworks were built using the module-specific subset of sugar/acid ratio-related genes as seed nodes to extract the gene coexpression network previously constructed by WGCNA analysis of 7430 genes [14]. Except for the Grey module with two genes, all other modules have genes present in the network and are analyzed below.
The Blue module-based subnetwork contains 16 of 36 sugar/acid ratio-correlated genes (coded in yellow and green), with a total of 142 nodes and 689 edges (Fig. 2). In this module, nine of sugar/acid ratio genes represent large hubs, with five of them having more than 90 edges or interactions, including Cs3g07510/AHP5, Cs1g24590 (unknown function), Cs7g30920/GEA6, Cs3g12560 (a ribosomal protein involved in protein synthesis) and Cs5g05940 (unknown function). Four other mid-size hubs are Cs1g23800 (a functionally unknown Cupin-like protein), orange 1.1 t02370/CYS2 (involved in protein degradation), Cs1g20300 (unknown function), and Cs2g27550/Oleosin (involved in lipid metabolism). The Arabidopsis homologs for most of these hub genes have not been tested regarding their physiological functions. Cs7g30920 is most similar to Arabidopsis GEA6 or Em6, a group 1 LEA gene that is involved in response to abscisic acid and is predicted to act in metabolism during seed development [38]. Cs3g07510/AHP5 and a RLK gene encoded by Cs1g14800 are involved in signal transduction. The finding that Cs3g07510/AHP5 acts as a large hub indicates a potentially crucial role of cytokinin signaling in the sugar/acid ratio subnetwork.
Fig. 2.
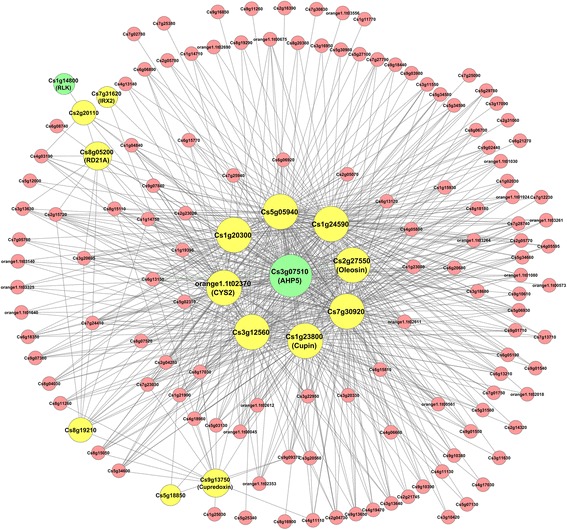
The Blue module-based sugar/acid ratio-associated gene subnetwork. The subnetwork constructed by using the sugar/acid ratio-correlated genes from the blue module as seed nodes to extract the weighted gene coexpression network of all 7430 differentially expressed genes with an edge weight cutoff of 0.6 is visualized by Cytoscape. Two seed node genes involved in signal transduction are coded in green, and all other seed node genes coded in yellow
The Brown module-based subnetwork has 45 nodes, including eight of the 13 sugar/acid ratio-correlated genes, and 57 edges (Fig. 3). In this module, Cs5g03630/remorin and Cs4g20090/KAT3 only interact with each other. It remains to be determined whether the interaction between remornin-like transcriptional regulator and the potassium channel KAT3 has any physiological or metabolic relevance in the sugar/acid ratio control. In the mid-sized subnetwork which contains all other nodes and edges, two hubs, Cs3g17860 and Cs7g06285, do not have their orthologs identified in Arabidopsis. The other hubs, Cs3g24700/PGI and Cs1g25210/EMB2296, together with Cs9g07750/APS1, are involved in metabolism, indicating a potential role of metabolic regulation in the Brown module-based sugar/acid ratio control subnetwork.
Fig. 3.
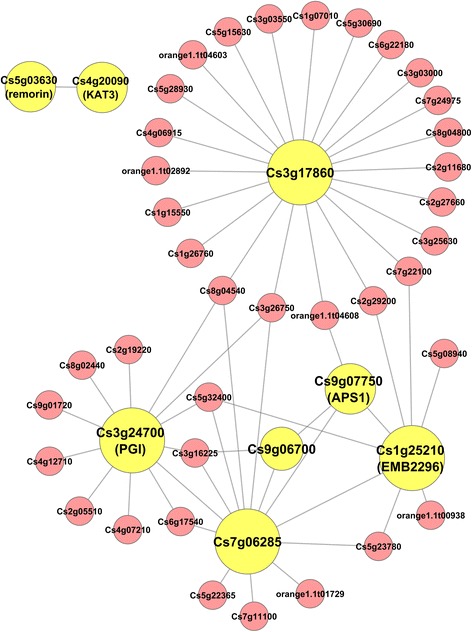
The Brown module-based sugar/acid ratio gene subnetwork. The sugar/acid ratio-correlated genes from the brown module are used as seed nodes to extract the weighted gene coexpression network as described in Fig. 2, resulting in the subnetwork with an edge weight cutoff of 0.2. The seed node genes present in the subnetwork are coded in yellow
For the Turquoise module which contains 21 sugar/acid ratio-correlated genes, 18 of them are present in the subnetwork. This subnetwork has 137 nodes and 472 edges (Fig. 4). The largest hub Cs8g18390 does not have any Arabidopsis homolog, while all other hubs have their orthologs present in Arabidopsis, including a transcription factor gene Cs9g08500/TZP, two genes involved in metabolism (Cs1g10530/HPT1 and Cs7g13310/KCS6), a heat shock protein HSP18.2/Cs7g32260, an embryo development-related gene Cs4g15700/EMB2757, and the Cs2g06500-encoded putative monoglyceride lipase gene MGL. Besides HPT1 and KCS6, the two hub genes involved in metabolism, three other genes (Cs5g20010, orange1.1 t00612 and orange1.1 t03587) in the subnetwork are also related to metabolism, indicating that metabolism might have a critical role in in this Turquoise module-based sugar/acid ratio subnetwork. In addition, two genes related to transport (Cs1g13320/BOR4 and Cs9g18100/CNGC1) are also present in the subnetwork.
Fig. 4.
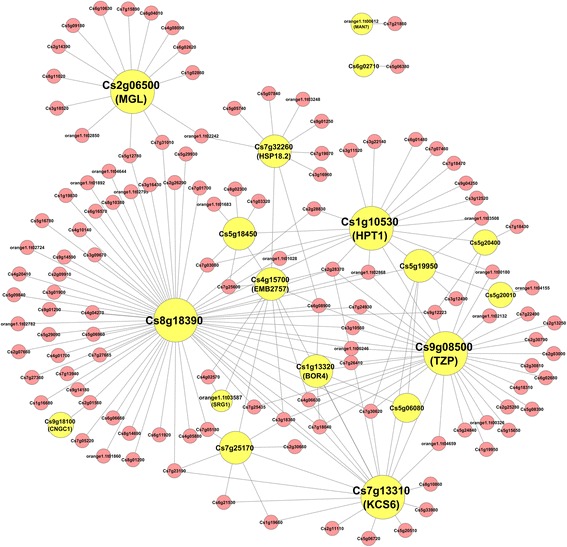
The Turquoise module-based sugar/acid ratio gene subnetwork. The subnetwork is constructed by extracting the weighted gene coexpression network using the sugar/acid ratio-correlated genes belonging to the Turquoise module as seed nodes, with an edge weight cutoff of 0.3. The seed node genes present in the subnetwork are coded in yellow
Discussion
Despite the well-recognized contribution of the sugar/acid ratio in determination of the fruit sweetness trait, except for one genetic study in melon [19], none of the prior large-scale studies in both gene-trait association mapping or gene expression-trait correlation in tomato and citrus fruits [7–9, 11, 13–15] has attempted to address the possible relationship between genes and the sugar/acid ratio. Using the four sweet orange varieties with dramatic variations in fruit acidity and very small difference in sugar levels at 142 DPA, together with gene coexpression analysis, we have identified a subset of genes belonging to distinct modules that are strongly associated with the sugar/acid ratio.
Among 72 genes correlated with the sugar/acid ratio, two groups of genes are worth of further discussion here. The first group includes malate metabolism- and glycolysis-related genes, Cs1g03610/MDH2 and Cs3g24700/PGI. Given the predicted role of the cytosolic NAD-MDH2/Cs1g03610 in converting malate to pyruvate in the cytosol followed by glycolysis, perhaps this pathway is involved in slightly reducing citrate accumulation, contributing to the alteration of sugar/acid ratio. Alternatively, malate metabolism has been shown to be involved in the control of sugar accumulation [4]. Therefore, it remains a possibility that increasing orange MDH2 gene expression would lead to enhanced malate metabolism, and subsequently sugar level may be slightly increased and acid level slightly reduced, leading to the increase of sugar/acid ratio. Future research directly testing this MDH gene, which exhibits the strongest correlation with the sugar/acid ratio in expanding orange fruits, will provide genetic evidence regarding the determination of sugar/acid ratio.
The second group includes the genes related to signaling and transcription, in particular Cs3g07510/AHP5 and orange1.1 t00294/ARR2 which are likely involved in cytokinin signaling. Cytokinin has been demonstrated to act in sink regulation of photosynthesis [39]. Specifically, cytokinin acts as both root-to-shoot and shoot-to-root signals and interacts with carbohydrate and nitrogen supplies to control the expression of photosynthesis genes. Given that expanding fruits can serve as a strong sink, it is conceivable that Cs3g07510 and orange1.1 t00294 may act to regulate cytokinin response, which then directs various cellular activities including driving the whole plant carbon and nitrogen metabolism so as to transport carbohydrates and amino acids to the developing fruits for sugar and acid metabolism.
Although our study has led to the identification of sugar-correlated genes in sweet orange fruits, this result on the large number of sugar-related genes needs to be interpreted with caution. With a Pcc cutoff +/−0.8, 56% of 7430 genes differentially expressed from 45 to 142 DPA have strong correlation with sugar (Additional file 1). Even with a higher Pcc cutoff (+/−0.9), we still obtained approximately 27% of genes that are highly correlated with sugar level (Additional file 1). Considering an early finding that more than 2500 genes are responsive to sugar in Arabidopsis [40], the number of 1995 sugar-correlated genes might be a closer approximation of genes potentially involved in either controlling sugar accumulation or responding to the increasing sugar status in fruit cells. Despite this, we should also consider the possibility that the large proportion (approximately 56% for Pcc of +/−0.8 or 27% for Pcc of +/−0.9) of genes showing strong correlations with sugar levels might be coincident with the similar pace in sugar accumulation and fruit growth and development. Our prior work has shown that among 7430 genes differentially regulated in any of the four varieties, 3145 genes are commonly regulated in all four varieties at 142 DPA compared to 45 DPA [14]. It is anticipated that these commonly regulated genes likely have conserved functions in fruit growth, development and metabolism in all four varieties during Stage II, such as in cell expansion and various aspects of metabolism. Indeed, analysis using Venn diagram showed that 1577 out of 1995 sugar-correlated genes are also commonly regulated (Fig. 1b). Therefore, it is conceivable that a considerable portion of 1995 sugar-correlated genes may function in cellular activities other than sugar and acid accumulation.
Given that mathematically the sugar/acid ratio is determined by sugar alone, acid alone and their relative levels, we had expected that many of the sugar/acid ratio-related genes should also strongly correlate with sugar or acid alone. However, to our surprise, we found that the majority of sugar/acid ratio-correlated genes are not correlated with either sugar or acid levels. There are at least two possibilities to explain this surprising finding. The first one is that most of these genes, if not all, might represent a unique group of genes which are involved in the control or sensing of sugar/acid ratio independently of controlling sugar alone or acid alone. Although the sugar/acid ratio seemed to exhibit an opposite pattern to acid content and a similar pattern with sugar content (Fig. 1A), none of the pairwise correlations among sugar level, acid level and the sugar/acid ratio in these four sweet orange varieties was statistically significant. Thus, this phenotypic variation among the four varieties seems to be consistent with this possibility. The second possibility is raised because mathematically the ratio can also be altered to a bigger difference even if sugar and acid levels are slightly altered in an opposite direction. In this case, some of the sugar/acid ratio-correlated genes would be expected to show weak correlations with the sugar or acid levels. Indeed, 20 such genes exhibit weak correlations with sugar (p-value <0.05), but only two genes have statistically significant (p-value < 0.05) weak correlations with the acid level (Additional file 2). If this is true, some of the sugar/acid ratio-related genes might have a minor role in micro-tuning the accumulation of either sugars or acids, in addition to their more promising roles in determining the sugar/acid ratio. Clearly, functional evidence will be critical for us to distinguish these two possibilities. It will also help to determine whether any of the sugar/acid ratio-related genes identified in this study is actually involved in the control of sugar/acid ratio rather than merely a gene expression response to the alterations in sugar/acid ratios during Stage II of fruit development. Whichever the possible mechanisms will be to explain our finding, work reported here has provided intriguing hypotheses for future testing using transgenic approaches.
Conclusions
Our comparative transcriptome analysis has revealed a total of 72 genes that are highly correlated with the fruit sugar/acid ratio. However, very few of them correlate with sugar or acid contents, indicating that these genes may potentially be involved in maintaining fruit sugar/acid ratios and/or responding to the cellular sugar/acid ratio status. The majority of these sugar/acid ratio-related genes are predicted to be involved in regulatory functions such as transport, signaling and transcription or encode enzymes involved in metabolism. Results from gene coexpression network analysis indicate that half of these genes are organized into two regulatory modules unique to the sugar/acid ratio control. In summary, our analysis of orange transcriptomes provides an intriguing insight into the potentially novel genetic or molecular mechanisms controlling the sugar/acid ratio in fruits.
Methods
Data analysis
The sugar/acid ratios at 45 and 142 DPA for each of the four sweet orange (C. sinensis L. Osbeck) varieties with differing fruit acidity, Newhall, Xinhui, Bingtang and Succari, were derived from the sugar and acid levels reported in a prior manuscript [14]. Pairwise t-test was performed to determine the significant differences in sugar content, acid content and sugar/acid ratio between four varieties at 45 and 142 DPA. RNA sequencing-based transcriptome data deposited in the NCBI GEO database (Accession number GSE78046) was used for correlation study. RNA seq data has been processed and analyzed using the methods implemented in EdgeR, resulting in a total of 7430 genes that are differentially expressed between 45 and 142 DPA in any of the four varieties, with at least two-fold difference and an FDR cutoff of 0.05 as previously described [14].
Pearson correlation analysis of sugar/acid ratios and gene expression levels
To identify the genes that show significant correlation between their expression levels and sugar/acid ratios, only those 7430 genes differentially expressed from 45 to 142 DPA in any of the four sweet orange varieties [14] were used for Pearson correlation coefficients (Pcc). Pcc are calculated between sugar/acid ratios and gene expression levels (which were first log2 transformed to normalize the data distribution) in a total of 24 samples (three biological replicates at each developmental stage for each variety), using an FDR cutoff of 0.05.
GO analysis and sugar/acid ratio subnetwork construction and visualization
As described elsewhere [14], GO analysis for citrus genes was done by first predicting their most closely related Arabidopsis orthologs (based on TAIR Release 10) using the functional annotation website Mercator with a BLAST-Cutoff of 80 and then by assigning the GO terms for those Arabidopsis orthologs to the corresponding citrus genes. The gene coexpression network using 7430 differentially expressed genes was constructed using the WGCNA package in R [41], and a total of 10 different modules were derived as described elsewhere [14]. To construct various sugar/acid ratio subnetworks, the sugar/acid-correlated genes belonging to different modules were used as seed nodes to extract the gene coexpression network and the resulting gene-gene interactions were used to visualize the subnetworks using Cytoscape.
Quantitative reverse transcription-PCR (qRT-PCR) analysis
qRT-PCR analysis was performed as described previously [14], using Cs1g05000-encoded actin gene (sense primer HZP14-L: TCCGTGACATGAAGGAGAAG; antisense primer HZP14-R: GCTCCAATGGTGATGATCTG) as a reference and specific primers for the following six genes: Cs1g03610 (QZQP201:TCTCGTACCGATGATTGCTC; QZQP202: GGCTGCTGGTTCAATATCAA), Cs5g03630 (QZQP205: TAATTGAAGCACAGCGAGGT; QZQP206: AGCCAAAGCAAGAGAGGAAC), Cs5g20010 (QZQP207: GAGAAACCGAGGCTACAAGC; QZQP208: TCTTCAGTTTCGGGACCAA), Cs5g24670 (QZQP209: CATGCTGACTTGGAATGCTT; QZQP210: AGGAAGCTTGCACTTATCCG), Cs6g16160 (QZQP211: GGAGAATGGGCTAATCGAAA; QZQP212: CTGTCTTCCCTGCATTAGCA), Cs7g32260 (QZQP215: GAGGAGCAACGTGTTCGAT; QZQP216: CGGAATATTAGCGACTGACG). The relative mRNA levels for each gene in Newhall at 45 DPA is set as 1.
Additional files
A list of genes correlated with the sugar level with Pcc = +/−0.8 and 0.9, respectively. (XLSX 286 kb)
Correlation of sugar/acid raio-correlated genes with sugar or acid levels. (XLSX 17 kb)
Acknowledgments
We appreciate Dong Jiang, Taisheng Li and Li Cao for providing sweet orange varieties in the original fruit transcriptome analysis.
Funding
This research is supported by a grant from Chongqing Science and Technology Commission (Grant No. cstc2012gg-yyjsB80004) and in part by a grant from the Chinese Ministry of Agriculture “Program 948” [Grant No. 2011-G21(2)]. The funders have no role in the study design, data analysis and interpretation, and manuscript writing.
Availability of data and materials
The RNA sequencing RAW data can be found in the NCBI GEO database (Accession number GSE78046). All data analyzed in this study are included in this published article and its supplementary information files.
Abbreviations
- AHP5
Arabidopsis histidine phosphotransfer protein 5
- ARR2
Arabidopsis response regulator 2
- CNGC1
Cyclic nucleotide gated channel 1
- DPA
Days post anthesis
- FDR
False discovery rate
- GO
Gene ontology
- MDH2
Malate dehydrogenase 2
- Pcc
Pearson correlation coefficients
- PGI
Glucose-6-phosphate isomerase or phosphoglucose isomerase
- qRT-PCR
Quantitative reverse transcription-PCR
- RLK
Receptor-like kinases
- WGCNA
Weighted Gene Coexpression Network Analysis
Authors’ contributions
Z-LZ conceived of the project, designed the experiments, performed bioinformatic analysis, analyzed the data, and wrote the manuscript; LQ, MC and JZ performed the RNA extraction and qPCR validation experiments and assisted in manuscript writing; and YH performed bioinformatic and statistical analyses and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The sweet orange varieties used for transcriptome data collection were grown in China National Citrus Germplasm Repository located in Citrus Research Institute of Southwest University, Chongqing, China. No specific permits were required for plant collection. The study did not require ethical consent or approval as no human subjects or endangered or protected plant species were involved.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12870-017-1138-8) contains supplementary material, which is available to authorized users.
Contributor Information
Liang Qiao, Email: ql651304217@163.com.
Minghao Cao, Email: deserteagle1982@163.com.
Jian Zheng, Email: leozheng91@163.com.
Yihong Zhao, Email: zhaoy05@nyumc.org.
Zhi-Liang Zheng, Email: zhiliang.zheng@lehman.cuny.edu.
References
- 1.Albertini MV, Carcouet E, Pailly O, Gambotti C, Luro F, Berti L. Changes in organic acids and sugars during early stages of development of acidic and acidless citrus fruit. J Agric Food Chem. 2006;54(21):8335–8339. doi: 10.1021/jf061648j. [DOI] [PubMed] [Google Scholar]
- 2.Etienne A, Genard M, Lobit P, Mbeguie AMD, Bugaud C. What controls fleshy fruit acidity? A review of malate and citrate accumulation in fruit cells. J Exp Bot. 2013;64(6):1451–1469. doi: 10.1093/jxb/ert035. [DOI] [PubMed] [Google Scholar]
- 3.Osorio S, Scossa F, Fernie AR. Molecular regulation of fruit ripening. Front Plant Sci. 2013;4:198. doi: 10.3389/fpls.2013.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centeno DC, Osorio S, Nunes-Nesi A, Bertolo AL, Carneiro RT, Araujo WL, Steinhauser MC, Michalska J, Rohrmann J, Geigenberger P, et al. Malate plays a crucial role in starch metabolism, ripening, and soluble solid content of tomato fruit and affects postharvest softening. Plant Cell. 2011;23(1):162–184. doi: 10.1105/tpc.109.072231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan MJ, Osorio S, Gehl B, Baxter CJ, Kruger NJ, Ratcliffe RG, Fernie AR, Sweetlove LJ. Metabolic engineering of tomato fruit organic acid content guided by biochemical analysis of an introgression line. Plant Physiol. 2013;161(1):397–407. doi: 10.1104/pp.112.209619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klee HJ, Giovannoni JJ. Genetics and control of tomato fruit ripening and quality attributes. Annu Rev Genet. 2011;45:41–59. doi: 10.1146/annurev-genet-110410-132507. [DOI] [PubMed] [Google Scholar]
- 7.Osorio S, Alba R, Nikoloski Z, Kochevenko A, Fernie AR, Giovannoni JJ. Integrative comparative analyses of transcript and metabolite profiles from pepper and tomato ripening and development stages uncovers species-specific patterns of network regulatory behavior. Plant Physiol. 2012;159(4):1713–1729. doi: 10.1104/pp.112.199711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauvage C, Segura V, Bauchet G, Stevens R, Do PT, Nikoloski Z, Fernie AR, Causse M. Genome-wide Association in Tomato Reveals 44 candidate loci for fruit metabolic traits. Plant Physiol. 2014;165(3):1120–1132. doi: 10.1104/pp.114.241521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tieman D, Zhu G, Resende MF, Jr., Lin T, Nguyen C, Bies D, Rambla JL, Beltran KS, Taylor M, Zhang B et al: A chemical genetic roadmap to improved tomato flavor. Science 2017, 355(6323):391–394. [DOI] [PubMed]
- 10.Fang DQ, Federici CT, Roose ML. Development of molecular markers linked to a gene controlling fruit acidity in citrus. Genome. 1997;40(6):841–849. doi: 10.1139/g97-809. [DOI] [PubMed] [Google Scholar]
- 11.Cercos M, Soler G, Iglesias DJ, Gadea J, Forment J, Talon M. Global analysis of gene expression during development and ripening of citrus fruit flesh. A proposed mechanism for citric acid utilization. Plant Mol Biol. 2006;62(4–5):513–527. doi: 10.1007/s11103-006-9037-7. [DOI] [PubMed] [Google Scholar]
- 12.Katz E, Fon M, Lee YJ, Phinney BS, Sadka A, Blumwald E. The citrus fruit proteome: insights into citrus fruit metabolism. Planta. 2007;226(4):989–1005. doi: 10.1007/s00425-007-0545-8. [DOI] [PubMed] [Google Scholar]
- 13.Aprile A, Federici C, Close TJ, De Bellis L, Cattivelli L, Roose ML. Expression of the H+−ATPase AHA10 proton pump is associated with citric acid accumulation in lemon juice sac cells. Funct Integr Genomics. 2011;11(4):551–563. doi: 10.1007/s10142-011-0226-3. [DOI] [PubMed] [Google Scholar]
- 14.Huang D, Zhao Y, Cao M, Qiao L, Zheng Z-L. Integrated systems biology analysis of transcriptomes reveals candidate genes for acidity control in developing fruits of sweet orange (Citrus sinensis L. Osbeck) Front Plant Sci. 2016;7:486. doi: 10.3389/fpls.2016.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheng L, Shen D, Yang W, Zhang M, Zeng Y, Xu J, Deng X, Cheng Y. GABA pathway rate-limit citrate degradation in postharvest citrus fruit evidence from HB pumelo (Citrus Grandis) x Fairchild (Citrus Reticulata) hybrid population. J Agric Food Chem. 2017;65(8):1669–1676. doi: 10.1021/acs.jafc.6b05237. [DOI] [PubMed] [Google Scholar]
- 16.Bai Y, Dougherty L, Li M, Fazio G, Cheng L, Xu K. A natural mutation-led truncation in one of the two aluminum-activated malate transporter-like genes at the ma locus is associated with low fruit acidity in apple. Mol Genet Genomics. 2012;287(8):663–678. doi: 10.1007/s00438-012-0707-7. [DOI] [PubMed] [Google Scholar]
- 17.Cohen S, Itkin M, Yeselson Y, Tzuri G, Portnoy V, Harel-Baja R, Lev S, Sa’ar U, Davidovitz-Rikanati R, Baranes N, et al. The PH gene determines fruit acidity and contributes to the evolution of sweet melons. Nat Commun. 2014;5:4026. doi: 10.1038/ncomms5026. [DOI] [PubMed] [Google Scholar]
- 18.DG H, Sun CH, Ma QJ, You CX, Cheng L, Hao YJ. MdMYB1 regulates anthocyanin and malate accumulation by directly facilitating their transport into vacuoles in apples. Plant Physiol. 2016;170(3):1315–1330. doi: 10.1104/pp.15.01333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Wang HS, He CX, Zhang ZB, Zhang X, Yi HP, Wu MZ: Genetic study on sugar and sour traits of melon (Cucumis melo L.) Acta Horticulturae Sinica 2009, 36(7):989–996.
- 20.Xu Q, Chen LL, Ruan X, Chen D, Zhu A, Chen C, Bertrand D, Jiao WB, Hao BH, Lyon MP, et al. The draft genome of sweet orange (Citrus Sinensis) Nat Genet. 2013;45(1):59–66. doi: 10.1038/ng.2472. [DOI] [PubMed] [Google Scholar]
- 21.GA W, Prochnik S, Jenkins J, Salse J, Hellsten U, Murat F, Perrier X, Ruiz M, Scalabrin S, Terol J, et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat Biotechnol. 2014;32(7):656–662. doi: 10.1038/nbt.2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng ZL, Zhao Y. Transcriptome comparison and gene coexpression network analysis provide a systems view of citrus response to 'Candidatus Liberibacter asiaticus' infection. BMC Genomics. 2013;14:27. doi: 10.1186/1471-2164-14-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong DC, Sweetman C, Ford CM. Annotation of gene function in citrus using gene expression information and co-expression networks. BMC Plant Biol. 2014;14:186. doi: 10.1186/1471-2229-14-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du D, Rawat N, Deng Z, Gmitter FG., Jr Construction of citrus gene coexpression networks from microarray data using random matrix theory. Horticulture research. 2015;2:15026. doi: 10.1038/hortres.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rawat N, Kiran SP, Du D, Gmitter FG, Jr, Deng Z. Comprehensive meta-analysis, co-expression, and miRNA nested network analysis identifies gene candidates in citrus against Huanglongbing disease. BMC Plant Biol. 2015;15:184. doi: 10.1186/s12870-015-0568-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keurentjes JJ, Sulpice R, Gibon Y, Steinhauser MC, Fu J, Koornneef M, Stitt M, Vreugdenhil D. Integrative analyses of genetic variation in enzyme activities of primary carbohydrate metabolism reveal distinct modes of regulation in Arabidopsis Thaliana. Genome Biol. 2008;9(8):R129. doi: 10.1186/gb-2008-9-8-r129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koprivova A, Giovannetti M, Baraniecka P, Lee BR, Grondin C, Loudet O, Kopriva S. Natural variation in the ATPS1 isoform of ATP sulfurylase contributes to the control of sulfate levels in Arabidopsis. Plant Physiol. 2013;163(3):1133–1141. doi: 10.1104/pp.113.225748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dan H, Yang G, Zheng ZL. A negative regulatory role for auxin in sulphate deficiency response in Arabidopsis thaliana. Plant Mol Biol. 2007;63(2):221–235. doi: 10.1007/s11103-006-9084-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Traub M, Florchinger M, Piecuch J, Kunz HH, Weise-Steinmetz A, Deitmer JW, Ekkehard Neuhaus H, Mohlmann T. The fluorouridine insensitive 1 (fur1) mutant is defective in equilibrative nucleoside transporter 3 (ENT3), and thus represents an important pyrimidine nucleoside uptake system in Arabidopsis Thaliana. Plant J. 2007;49(5):855–864. doi: 10.1111/j.1365-313X.2006.02998.x. [DOI] [PubMed] [Google Scholar]
- 30.Kamimoto Y, Terasaka K, Hamamoto M, Takanashi K, Fukuda S, Shitan N, Sugiyama A, Suzuki H, Shibata D, Wang B, et al. Arabidopsis ABCB21 is a facultative auxin importer/exporter regulated by cytoplasmic auxin concentration. Plant Cell Physiol. 2012;53(12):2090–2100. doi: 10.1093/pcp/pcs149. [DOI] [PubMed] [Google Scholar]
- 31.Kajikawa M, Fujibe T, Uraguchi S, Miwa K, Fujiwara T. Expression of the Arabidopsis borate efflux transporter gene, AtBOR4, in rice affects the xylem loading of boron and tolerance to excess boron. Biosci Biotechnol Biochem. 2011;75(12):2421–2423. doi: 10.1271/bbb.110629. [DOI] [PubMed] [Google Scholar]
- 32.Johnson CS, Kolevski B, Smyth DR. TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell. 2002;14(6):1359–1375. doi: 10.1105/tpc.001404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hass C, Lohrmann J, Albrecht V, Sweere U, Hummel F, Yoo SD, Hwang I, Zhu T, Schafer E, Kudla J, et al. The response regulator 2 mediates ethylene signalling and hormone signal integration in Arabidopsis. EMBO J. 2004;23(16):3290–3302. doi: 10.1038/sj.emboj.7600337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lohrmann J, Sweere U, Zabaleta E, Baurle I, Keitel C, Kozma-Bognar L, Brennicke A, Schafer E, Kudla J, Harter K. The response regulator ARR2: a pollen-specific transcription factor involved in the expression of nuclear genes for components of mitochondrial complex I in Arabidopsis. Mol Genet Genomics. 2001;265(1):2–13. doi: 10.1007/s004380000400. [DOI] [PubMed] [Google Scholar]
- 35.Wrzaczek M, Brosche M, Salojarvi J, Kangasjarvi S, Idanheimo N, Mersmann S, Robatzek S, Karpinski S, Karpinska B, Kangasjarvi J. Transcriptional regulation of the CRK/DUF26 group of receptor-like protein kinases by ozone and plant hormones in Arabidopsis. BMC Plant Biol. 2010;10:95. doi: 10.1186/1471-2229-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gimenez-Ibanez S, Ntoukakis V, Rathjen JP. The LysM receptor kinase CERK1 mediates bacterial perception in Arabidopsis. Plant Signal Behav. 2009;4(6):539–541. doi: 10.4161/psb.4.6.8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hutchison CE, Li J, Argueso C, Gonzalez M, Lee E, Lewis MW, Maxwell BB, Perdue TD, Schaller GE, Alonso JM, et al. The Arabidopsis histidine phosphotransfer proteins are redundant positive regulators of cytokinin signaling. Plant Cell. 2006;18(11):3073–3087. doi: 10.1105/tpc.106.045674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopez-Molina L, Mongrand S, McLachlin DT, Chait BT, Chua NH. ABI5 acts downstream of ABI3 to execute an ABA-dependent growth arrest during germination. Plant J. 2002;32(3):317–328. doi: 10.1046/j.1365-313X.2002.01430.x. [DOI] [PubMed] [Google Scholar]
- 39.Paul MJ, Foyer CH. Sink regulation of photosynthesis. J Exp Bot. 2001;52(360):1383–1400. doi: 10.1093/jexbot/52.360.1383. [DOI] [PubMed] [Google Scholar]
- 40.Price J, Laxmi A, St Martin SK, Jang JC. Global transcription profiling reveals multiple sugar signal transduction mechanisms in Arabidopsis. Plant Cell. 2004;16(8):2128–2150. doi: 10.1105/tpc.104.022616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A list of genes correlated with the sugar level with Pcc = +/−0.8 and 0.9, respectively. (XLSX 286 kb)
Correlation of sugar/acid raio-correlated genes with sugar or acid levels. (XLSX 17 kb)
Data Availability Statement
The RNA sequencing RAW data can be found in the NCBI GEO database (Accession number GSE78046). All data analyzed in this study are included in this published article and its supplementary information files.