

Abstract
Intellectual disabilities are genetically heterogeneous and can be associated with congenital anomalies. Using whole-exome sequencing (WES), we identified five different de novo missense variants in the protein phosphatase-1 catalytic subunit beta (PPP1CB) gene in eight unrelated individuals who share an overlapping phenotype of dysmorphic features, macrocephaly, developmental delay or intellectual disability (ID), congenital heart disease, short stature, and skeletal and connective tissue abnormalities. Protein phosphatase-1 (PP1) is a serine/threonine-specific protein phosphatase involved in the dephosphorylation of a variety of proteins. The PPP1CB gene encodes a PP1 subunit that regulates the level of protein phosphorylation. All five altered amino acids we observed are highly conserved among the PP1 subunit family, and all are predicted to disrupt PP1 subunit binding and impair dephosphorylation. Our data suggest that our heterozygous de novo PPP1CB pathogenic variants are associated with syndromic intellectual disability.
Introduction
Developmental delay and intellectual disability are observed in ~1–3 % of the population, and congenital heart defects are present in ~1 % of all live births (Zablotsky et al. 2015; van der Linde et al. 2011). De novo predicted pathogenic sequence variants are more frequently identified in individuals with congenital heart disease who also have neurodevelopmental disorders and/or other congenital anomalies compared to individuals with isolated congenital heart disease (Homsy et al. 2015). The underlying etiology for most individuals with neurodevelopmental disorders and/or congenital heart disease is unclear due to the heterogeneous causes of both conditions (Homsy et al. 2015; Vissers et al. 2016). Whole-exome sequencing (WES) has proven to be a successful strategy to identify disorders due to de novo pathogenic variants in a significant portion of individuals with disorders that affect reproductive fitness (Shang et al. 2016).
Reversible protein serine/threonine phosphorylation, catalyzed by kinases and phosphatases, is a common mechanism for the regulation of many cellular functions. De novo heterozygous pathogenic variants in protein phosphatase-2 regulatory subunit B delta (PPP2R5D) were recently identified using WES in 7 out of 2790 (0.25 %) patients with neurodevelopmental disorders and additional features of dysmorphic facial features, developmental delay, autistic features, macrocephaly, hypotonia, and seizures (Shang et al. 2016). This finding highlights the importance of protein phosphatases in the brain development. There are seven major serine/threonine-specific protein phosphatases, including PPP1-7, in eukaryotic cells. Protein phosphatase 1 is the major serine/threonine phosphatase that regulates diverse cellular processes, such as cell-cycle progression, protein synthesis, muscle contraction, carbohydrate metabolism, transcription, and neuronal signaling (Aggen et al. 2000). Protein phosphatase 1 regulates neurotransmitters and synaptic activity and is regulated by multiple factors (Bausen et al. 2010; Kanematsu et al. 2006; Pribiag and Stellwagen 2013). Protein phosphatase 1 catalytic subunit (PP1C) mediates dephosphorylation of dishevelled (Dvl) protein, a key component in Wnt signaling, via its C-terminal domain. PP1C cooperates with homeodomain-interacting protein kinase 2 (Hipk2) to stabilize Dvl and form Dvl-Hipk2-PP1c complex to sustain Wnt signaling. In the absence of PP1c catalytic activity, Dvl proteins are hyperphosphorylated and consequently degraded by the proteasome (Shimizu et al. 2014). Protein phosphatase type 1 beta regulates and forms a complex with the tumor suppressor genes Merlin and Moesin (Yang et al. 2012). Protein phosphatase-1 catalytic subunit beta (PPP1CB) interacts with the myosin-binding subunit, which directs the phosphatase activity of the enzyme to specifically dephosphorylate the myosin, troponin complex, and myosin light chain in smooth and skeletal muscles (Pereira et al. 2011). A de novo heterozygous frameshift pathogenic variant c.909dupA, p.Tyr304Ilefs*19 in protein phosphatase 1 catalytic subunit beta (PPP1CB), was identified by Hamdan et al. in 1 out of 41 subjects with intellectual disability, developmental delay, postnatal growth deficiency, and some minor dysmorphic features (Hamdan et al. 2014). Another study reported two de novo missense pathogenic variants in PPP1CB in four patients; three of these individuals had a recurrent PPP1CB c.146G>C, p.Pro49Arg variant, while the fourth had a c.166G>C, p.Ala56Pro variant (Gripp et al. 2016). These four patients were initially considered to have a recognizable phenotype closely resembling Noonan syndrome with loose anagen hair (Gripp et al. 2016). In this study, we present eight additional unrelated individuals with de novo variants in PPP1CB, four of them with the recurrent p.Pro49Arg variant, with common features of dysmorphic features, macrocephaly, neurodevelopmental delay, congenital heart disease, short stature, and skeletal and connective tissue abnormalities. We present data to further show that de novo variants in PPP1CB, a catalytic subunit of PP1, cause a distinct disorder related to intellectual disability.
Materials and methods
Consent
Informed consent was obtained from all parents of participants included in this study. This study was approved by the Institutional Review Boards of Columbia University and Baylor College of Medicine.
Whole-exome sequencing
Patients 1, 2, and 5 through 8 had whole-exome sequencing (WES) at GeneDx along with their parents. The testing was performed and data were analyzed on the HiSeq 2000, 2500, or 4000 sequencing system, as described previously (Shang et al. 2016). Identified de novo PPP1CB variants were confirmed in all affected probands, and their absence confirmed in their parents with a second independent DNA preparation by dideoxy Sanger sequencing using an ABI3730 DNA Sequencer (Life Technologies, Carlsbad, CA, USA).
Patient 3 had WES as a singleton at the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC). Briefly, DNA sample was prepared into Illumina paired-end libraries. Capture was performed using BCM-HGSC core design (52 Mb, Roche NimbleGen, Inc) and sequenced on the Illumina HiSeq 2000 platform (Illumina, Inc) with an ~150× depth of coverage. Data produced were aligned and mapped to the human genome reference sequence (hg19) using the Mercury pipeline. Variants were called using the ATLAS variant calling method and SAMtools (The Sequence Alignment/Map) and annotated using the in-house-developed “Cassandra” annotation pipeline that uses ANNOVAR (Li et al. 2009; Wang et al. 2010; Bainbridge et al. 2011; Lupski et al. 2013). Potentially pathogenic variants were confirmed in the proband and evaluated in the parents by Sanger sequencing.
Patient 4 underwent WES at Ambry Genetics along with both parents. Exome library preparation, sequencing, bioinformatics, and data analysis were performed as previously described (Farwell et al. 2015). Briefly, samples were prepared using the IDT xGen Exome Research Panel V1.0 and sequenced using paired-end, 150-cycle chemistry on the Illumina HiSeq 2500 (Illumina, San Diego, CA, USA). Identified candidate alterations were confirmed using automated fluorescence dideoxy sequencing. Co-segregation analysis was performed using each available family member.
Results
WES was performed in 5964 probands at GeneDx with developmental delay and/or ID, with or without additional clinical features. 4624 cases were submitted as complete trios with both parents. Ten individuals (0.17 %) were identified to have de novo predicted pathogenic variants in PPP1CB that were all confirmed with Sanger sequencing. The probability of identifying 10 individuals with de novo variants in PPP1CB among 4624 trios is 3.2 × 10−14 by a Poisson test and 1.7 × 10−9 by TADA (He et al. 2013). Given the mutation rate for PPP1CB, the probability of identifying 10 de novo variants is 8.6 × 10−6. Four of those ten individuals declined to be included in this paper, since they were already involved in another publication. Of the remaining six samples tested at GeneDx, WES produced an average of ~10 GB of sequence per sample. Mean coverage of captured regions was ~110X per sample, with >95 % covered with at least 10× coverage, an average of 91 % of base call quality of Q30 or greater, and an overall average mean quality score of >Q36.
At Ambry, WES was performed in 1798 probands with developmental delay and/or ID, with or without additional clinical features. 1358 cases were submitted as complete trios with both parents. Three individuals (0.22 %) were identified to have de novo predicted pathogenic variants in PPP1CB that were all confirmed with Sanger sequencing. Two of those three individuals declined to be included in this paper. Of the remaining sample tested at Ambry, WES produced an average of ~11 GB of sequence per sample. Mean coverage of captured regions was ~133× per sample, with >95 % covered with at least 10× coverage, an average of 91 % of base call quality of Q30 or greater, and an overall average mean quality score of >Q36.
In combining cases from both GeneDx and Ambry, the probability of identifying 13 individuals with de novo variants in PPP1CB among 5982 trios is 2.49 × 10−13 by a Poisson test and 2.22 × 10−16 by TADA (He et al. 2013).
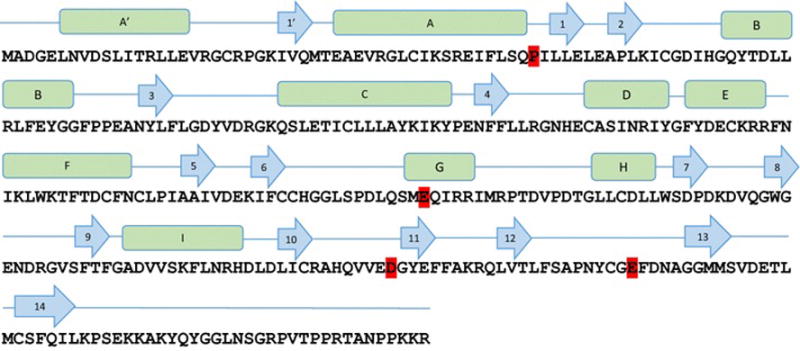
In addition to the six patients analyzed at GeneDx, one patient underwent WES at BCM-HGSC and Sanger sequencing confirmed the presence of the PPP1CB variant in the proband and absence in the unaffected parents and siblings, thus confirming it was de novo. One additional unrelated patient was evaluated by WES at Ambry and found to have a de novo predicted pathogenic variant in PPP1CB that was confirmed with Sanger sequencing. Five novel de novo heterozygous missense variants in PPP1CB were identified in a total of eight unrelated individuals. One variant was previously reported (Gripp et al. 2016), four variants are novel and have not been reported in the Database of Single Nucleotide Polymorphisms (dbSNP, http://www-ncbi-nlm-nih-gov.ezproxyhost.library.tmc.edu/SNP/), 1000 Genomes (http://www.1000genomes.org/), Exome Variant Server (ESP), or in GeneDx’s internal database of over 15,000 control exomes. Although 17 loss-of-function variants were expected in PPP1CB, no loss-of-function variants were observed in exome sequencing data from control subjects catalogued in the ExAC Browser (http://exac.broadinstitute.org/). Hence, PPP1CB’s probability of loss-of-function intolerance (pLI) is 1.0. All variants are predicted to be deleterious by SIFT (http://sift.jcvi.org/), Mutation Taster (http://www.mutationtaster.org/), and CADD (http://cadd.gs.washington.edu/). PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) and MetaSVM (https://sites-google-com.ezproxyhost.library.tmc.edu/site/jpopgen/dbNSFP) predict two of the variants (Pro49Arg and Asp252Tyr) to be deleterious (Table 1). The five variants occur in amino acids that are highly conserved across species (Fig. 1). Four of the eight patients have the same recurrent de novo variant, Pro49Arg, and two variants alter amino acid Glu183 to either Valine to Alanine. The catalytic subunit of PP1 is composed of 9 alpha helices and 14 beta strands comprising 3 beta sheets (Maynes et al. 2001; Peti et al. 2013). According to the secondary structure of the protein, Pro49 is located between alpha helices A and beta loop 1. Glu183 is located in an alpha helix, while Asp252 is in the beta 10–11 loop (Fig. 2). All three variants are located in catalytic core (amino acids 41–269) of PP1 (Ansai et al. 1996) which possesses phosphatase activity. Deletion of ten amino-acid residues from either the N or the C termini of the PP1 catalytic core, in which Pro49 and Asp252 are located, destroyed the phosphatase activity, indicating the critical role of these amino acids to the catalytic function (Ansai et al. 1996). Glu274 is in the active site region in beta 12–13 loop, and changing Glu274 to Lys may affect the intramolecular hydrogen bond with toxin Okadaic acid (Maynes et al. 2001).
Table 1.
Novel variants in PPP1CB and pathogenicity predictions
| Chr | Position | Ref | Alt | Amino Acid Change | AAChange.refgene | SIFT_pred | Polyphe n2_pred | Mutation Taster_pred | MetaSVM_pred | CADD_phred | GERP++_RS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 28999810 | C | G | p.Pro49Arg | PPP1CB:NM_002709:exo n2:c.C146G:p.P49R,PPP1 CB:NM_206876:exon3:c. C146G:p.P49R | D | D | D | D | 24.7 | 4.49 |
| 2 | 29006800 | A | C | p.Glu183Ala | PPP1CB:NM_002709:exo n5:c.A548C:p.E183A,PPP 1CB:NM_206876:exon6: c.A548C:p.E183A | D | B | D | T | 24.5 | 5.87 |
| 2 | 29006800 | A | T | p.Glu183Val | PPP1CB:NM_002709:exo n5:c.A548T:p.E183V,PPP 1CB:NM_206876:exon6: c.A548T:p.E183V | D | B | D | T | 29.8 | 5.87 |
| 2 | 29016738 | G | T | p.Asp252Tyr | PPP1CB:NM_002709:exo n7:c.G754T:p.D252Y,PPP 1CB:NM_206876:exon8: c.G754T:p.D252Y | D | D | D | D | 25.8 | 5.13 |
| 2 | 29016804 | G | A | p.Glu274Lys | PPP1CB:NM_002709:exo n7:c.G820A:p.E274K,PPP 1CB:NM_206876:exon8: c.G820A:p.E274K | D | P | D | T | 26.1 | 5.31 |
Figure 1.
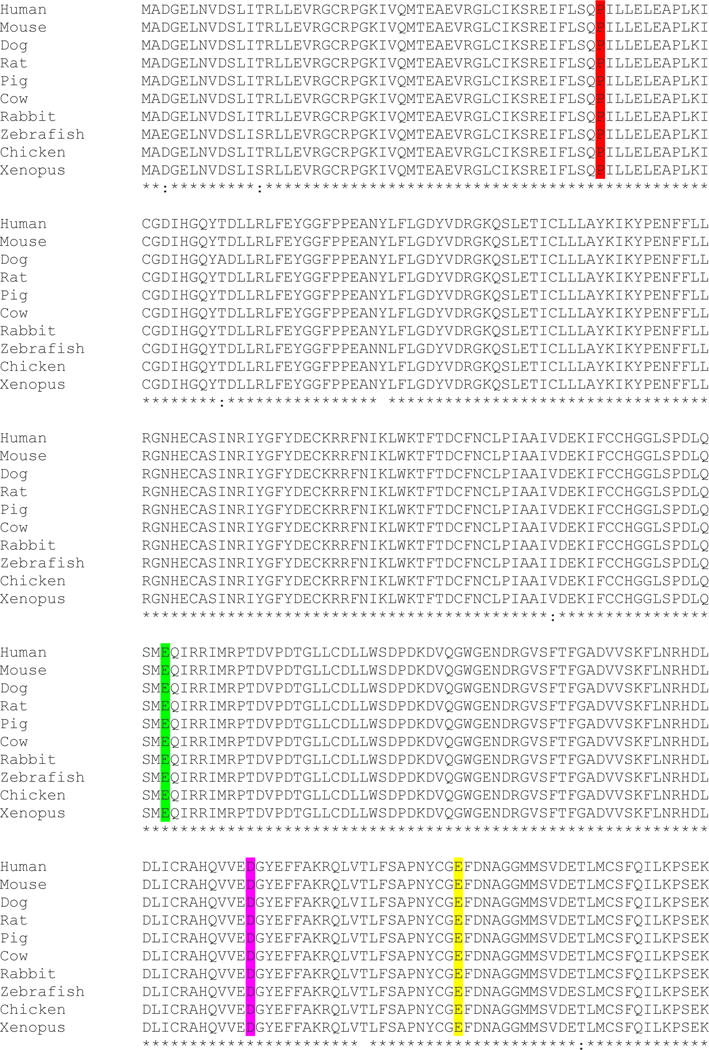

Conservation of amino acids of PPP1CB and de novo variants identified in patients. A. Conservation of amino acid sequence of PPP1CB among human, dog, rat, pig, cow, mouse, chicken, xenopus, and zebrafish indicated. Variants identified in our patients are highlighted at amino acids 49, 183, 252 and 274.
Figure 2.

Conservation of PPP1CB with isoforms of the catalytic subunit of PPP1C. Sequence alignment of PPP1CB with two other members of PPP1C-PPP1CA and PPP1CC show sequence conservation across the three proteins. Variants identified in this study are high-lighted in red.
Discussion
We identified five de novo missense variants in PPP1CB in eight independent individuals with a common clinical phenotype of developmental delay and/or ID, congenital heart disease, macrocephaly, short stature, and dysmorphic features. The gene is highly intolerable to variation, and the frequency of missense variants in PPP1CB in ExAC is 8.5e–06. Heterozygous predicted pathogenic variants in PPP1CB account for approximately 0.2 % of individuals referred for WES with ID or developmental delay.
PPP1CB is one of the three isoforms of catalytic subunits of PP1: PP1α (PPP1CA), PP1β (PPP1CB), and PP1γ (PPP1CC). There is conservation of these proteins throughout evolution (Cohen and Cohen 1989), and the isoforms of PP1 are >85 % identical and expressed in every tissue examined (Cohen and Cohen 1989). PPP1CB protein contains a calcineurin-like phosphoesterase domain, a metallophosphatase domain, and a serine/threonine protein phosphatase domain, where all five de novo variants identified in this study are located. Four unrelated individuals all have the recurrent Pro49Arg missense variant, and two variants replaced Glu183 with a small neutral amino acid, suggesting that amino acids Pro49 and Glu183 may play an important role in PPP1CB function. Pro49 is highly conserved among proteins in PPP1C family (PPP1CA, PPP1CB, and PPP1CC). Recently, Gripp et al. identified the de novo PPP1CB missense variant Pro49R in three unrelated individuals. In comparing patients who carry Pro49Arg in our current study with those previously reported, all individuals had dysmorphic features and developmental delay, but there is some phenotypic variation in cardiac and skeletal anomalies and growth. Functional studies suggest that one of the functions of PP1 is to dephosphorylate substrates required for the completion of mitosis, and the regulatory subunit(s) associated with this function of PP1 is SDS22, also called PPP1R7 (Ceulemans et al. 2002). SDS22 interacts with PP1 and is part of a complex with PP1. Essential SDS22-binding sites are in the region of residues 43–173 of PPP1CC, and Pro49 is one of the highly conserved amino acids in this region. The missense variant Pro49Arg may alter the interaction of PPP1CB and SDS22 to regulate PPP1CB function. The Glu274Lys mutation is in the beta12-beta13 loop, which has been proposed to confer inhibitor specificity in PP1 (Kelker et al. 2009). Beta12-beta13 loop is important for toxin binding, including microcystin and okadaic acid, and is in the active site of PP1 (Maynes et al. 2001). Amino acid 274 in PP1 participates in PP1: toxin interactions through PP1: toxin hydrogen-bonds (Kelker et al. 2009), and substitution of a Lys for Glu at amino acid 274 may affect the interaction of PPP1CB with toxin or other proteins.
PPP1CB maps to chromosome 2p23.2 (Barker et al. 1994; Saadat et al. 1994). A 4.8 Mb microdeletion of 2p23.2, which contains 29 genes, including PPP1CB, was found in one affected subject who had overlapping features with our patients, including dysmorphic features and developmental delay. A recurrent microduplication at 2p23.2 was found in ten individuals who had overlapping features with the patients included in our study (https://decipher.sanger.ac.uk), suggesting that dosage of PPP1CB or nearby genes is critical to brain function. PPP1CB forms heterodimers with various PP1 regulatory subunits (Cohen and Cohen 1989). In the cardiovascular system, PPP1CB affects angiogenesis and endothelial migration in an endothelial tube formation assay (Iacobazzi et al. 2015). Cardiac-specific deletion of Ppp1cb (PP1β) in mice resulted in concentric remodeling of the heart, interstitial fibrosis, and contractile dysregulation without congenital heart disease (Liu et al. 2015).
PPP1CB is involved in several biological processes, including cell adhesion, cell cycle, small GTPase-mediated signal transduction, protein dephosphorylation, negative regulation of transforming growth factor, regulation of glycogen catabolic process, and striated muscle tissue development (Gibbons et al. 2007; Flores-Delgado et al. 2007; Bianchi et al. 2005; Moorhead et al. 1998). PPP1CB also regulates protein kinase C and protein kinase B (AKT), which are implicated in the regulation of cell growth, proliferation, survival, differentiation, and cytoskeletal changes. In primates, PPP1CB is expressed in spines, dendrites, axon terminals, axons, and glia in the prefrontal cortex, and is enriched in dendrites (Bordelon et al. 2005). PPP1CB is abundant in the brain and regulates synaptic plasticity (Jouvenceau et al. 2006). In mice, partial inhibition of PPP1CB in forebrain neurons improved learning efficacy, suggesting that PPP1CB is important in brain function (Genoux et al. 2002; Gräff et al. 2010). Suppressor of clear, C. elegans, homolog of (SHOC2), also called RAS-binding protein SUR8, is a regulatory subunit of PP1c in the Ras pathway and muscle Ras viral oncogene homolog (M-Ras) interacts with a Shoc2-PP1c dimer to form a ternary complex (Rodriguez-Viciana et al. 2006). Several studies identified a missense pathogenic variant p.S2G in SHOC2 in Noonan syndrome or a Noonan-like syndrome with loose anagen hair (Cordeddu et al. 2009; Hoban et al. 2012; Gripp et al. 2013). The phenotype in patients with heterozygous pathogenic variants in PPP1CB clinically resembles the Noonan syndrome (Gripp et al. 2016), suggesting that mechanisms of PPP1CB in disease could be related to SHOC2 gene function.
In summary, we identified five de novo heterozygous missense variants in PPP1CB in eight individuals, with the recurrent Pro49Arg variant in four of the individuals. Missense variants in PPP1CB may define a novel genetic syndrome associated with dysmorphic features, developmental delay/intellectual disability, congenital heart disease, macrocephaly, and short stature. PPP1CB is one of a growing number of phosphatases associated with neurodevelopmental disorders.
Supplementary Material
Figure 3.
PPP1CB protein modeling.
Figure 4.
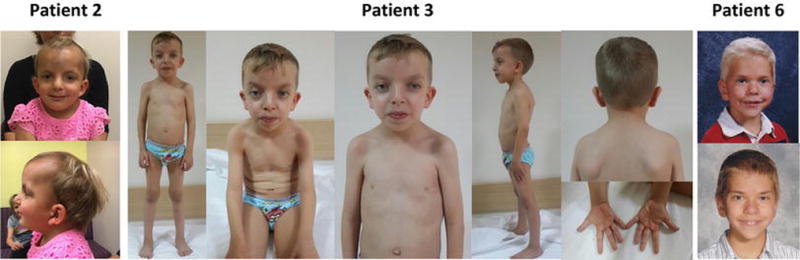
Facial features of individuals with the Pro49Arg (Patients 2 and 3) and Glu183Val PPP1CB (Patient 6) de novo variants.
Table 2.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | |
|---|---|---|---|---|---|---|---|---|
| PPP1CB Variant | c.146C>G; p.Pro49Arg |
c.146C>G; p.Pro49Arg | c.146C>G; p.Pro49Arg | c.146C>G; p.Pro49Arg | c.548A>C; p.Glu183Ala | c.548A>T; p.Glu183Val | c.754G>T; p.Asp252Tyr | c.820G>A; p.Glu274Lys |
| [Clinician] | Dr. Turner | Dr. Lindstrom | Dr. Gulec | Dr. Zadeh | JHU/CNMC | Dr. Bupp | Dr. Lalani | Dr. Scott |
| Sex | M | F | M | M | F | M | F | M |
| Current Age | 2 | 6 | 8 | 2 | 4 | 20 | 2 | 6 |
| Growth Parameters | At 6 years: HT 105 cm (2%ile) WT 18.9 kg (28%) OFC 54 cm (99%) |
At 8 years: HT 112 cm (<1%ile) WT 18 kg (<1%ile) OFC 55 cm (98%ile) |
At birth: HT 48.3 cm (30%ile) WT 32 kg (50%ile) OFC 36 cm (90%ille) |
At 3 years: HT 95.8 cm (53%ile) WT 14.8 kg (67%ile) OFC 52.0 cm (99%ile) |
At 17 years: HT 163.8 cm (9%ile) WT 54.98 kg (17%ile) OFC 57 cm (91%ile) |
At 2 years: HT 76.7 cm (<1%ile) WT 8.2 kg (<1%ile) OFC 46.9 cm (34%ile) |
At 4 years: HT 87.6 cm (<1%ile) WT 11.5 kg (<1%ile) OFC 48.5 cm (10%ile) |
|
| Dysmorphism | Hypertelorism Bifid uvula |
Arched eyebrows Downslanting palpebral fissures Hypertelorism Low set and posteriorly rotated ears Thickened helices Anterior ear pit Broad, pear-shaped nose Bulbous nasal tip Hypoplastic alae nasi Thin upper lip Small, widely spaced teeth Prominent chin Broad neck High anterior hairline Widely spaced nipples |
Frontal bossing Hypertelorism Prominent, low-set ears Triangular mouth Webbed neck Low posterior hairline |
Prominent forehead Large anterior fontanelle Low nasal bridge Small nose with upturned tip Smooth philtrum Microstomia Narrow chin Overfolded helices Bilateral anterior ear lobe dimples |
Prominent forehead Broad eyebrows Broad nose Thin upper lip |
Broad nasal tip | Hypertelorism Upslanted palpebral fissures Depressed nasal bridge Long philtrum Small posteriorly rotated ears Preauricular pit Ptosis Triangular mouth Thin upper lip |
Prominent forehead Short palpebral fissures Hypertelorism Broad nasal root |
| Hypotonia | Y | N | Y | Y | N | Y | N | |
| Development | DD, SD | DD, SD | DD, SD | DD, ID | DD, SD | DD, SD | DD, SD | DD, SD |
| Cardiovascular | Dilated aortic root | N/A | Mild mitral and tricuspid valve insufficiency | Patent foramen ovale | Atrial septal defect | Aortic coarctation | Peripheral pulmonary stenosis Aortic coarctation, dilation, and tortuosity Patent foramen ovale |
Hypoplastic left aortic arch |
| Skeletal | Cervical fusion Pectus excavatum Clinodactyly Delayed bone age |
Pectus excavatum Delayed bone age |
N/A | N/A | Delayed bone age | Short limbs Brachydactyly Clinodactyly Pectus excavatum Spinal stenosis |
2,3 toe syndactyly | |
| Connective tissue | High-arched palate Joint hypermobility |
Joint hypermobility Thin and loose skin |
High-arched palate | Joint hypermobility Doughy skin |
N/A | Joint hypermobility Translucent skin |
High-arched palate | |
| Other | Bilateral hydronephrosis Slow growing hair Prominent subarachnoid spaces Gastrointestinal reflux Constipation Reduced sweating |
Brittle nails Bilateral inguinal hernia Cryptorchidism |
N/A | Constipation Dysphagia |
Abnormal sleep patterns Dental crowding Gliosis versus migrational abnormality Kidney cysts Epistaxis Heat intolerance Slow growing hair and nails |
Feeding difficulties Iris coloboma Vesicoureteral reflux Thin, slow growing, bright orange hair Neonatal teeth |
Septo-optic dysplasia Feeding difficulties Constipation Myopia Unsteady gait Inguinal hernia |
M = male, F = female, HT = height, WT = weight, Y = yes, N = no, N/A = not applicable, DD = developmental delay, SD = speech delay
Acknowledgments
We thank the patients and their families for their generous participation. This work was supported in part by a grant from the Simons Foundation and from the NIH (GM030518), NINDS (NS058529), and NHGRI/NHLBI (HG006542).
Heather McLaughlin, Megan Cho, Kyle Retterer, Rhonda Schnur, Ingrid Wentzensen, and Sherri Bale are employees of GeneDx. Wendy Chung is a former employee of GeneDx and a paid consultant for Regeneron Pharmaceuticals. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, has stock options in Lasergen, Inc., serves on the Scientific Advisory Board of the Baylor Miraca Genetics Laboratory, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting.
Footnotes
Conflicts of Interest
The other authors declare that they have no conflict of interest.
References
- Aggen JB, Nairn AC, Chamberlin R. Regulation of protein phosphatase-1. Chem Biol. 2000;7(1):R13–R23. doi: 10.1016/s1074-5521(00)00069-7. [DOI] [PubMed] [Google Scholar]
- Ansai T, Dupuy LC, Barik S. Interactions between a minimal protein serine/threonine phosphatase and its phosphopeptide substrate sequence. J Biol Chem. 1996;271(40):24401–24407. doi: 10.1074/jbc.271.40.24401. [DOI] [PubMed] [Google Scholar]
- Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;3(87):87re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker HM, Brewis ND, Street AJ, Spurr NK, Cohen PT. Three genes for protein phosphatase 1 map to different human chromosomes: sequence, expression and gene localisation of protein serine/threonine phosphatase 1 beta (PPP1CB) Biochim Biophys Acta. 1994;1220(2):212–218. doi: 10.1016/0167-4889(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Bausen M, Weltzien F, Betz H, O’Sullivan GA. Regulation of postsynaptic gephyrin cluster size by protein phosphatase 1. Mol Cell Neurosci. 2010;44:201–209. doi: 10.1016/j.mcn.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Bianchi M, De LS, Vietri M, Vietri M, Villa-Moruzzi E. Reciprocally interacting domains of protein phosphatase 1 and focal adhesion kinase. Mol Cell Biochem. 2005;272:85–90. doi: 10.1007/s11010-005-7639-z. [DOI] [PubMed] [Google Scholar]
- Bordelon JR, Smith Y, Nairn AC, Colbran RJ, Greengard P, et al. Differential localization of protein phosphatase-1alpha, beta and gamma1 isoforms in primate prefrontal cortex. Cereb Cortex. 2005;15(12):1928–1937. doi: 10.1093/cercor/bhi070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceulemans H, Vulsteke V, De Maeyer M, Tatchell K, Stalmans W, et al. Binding of the concave surface of the Sds22 superhelix to the alpha 4/alpha 5/alpha 6-triangle of protein phosphatase-1. J Biol Chem. 2002;277(49):47331–47337. doi: 10.1074/jbc.M206838200. [DOI] [PubMed] [Google Scholar]
- Cohen P, Cohen PT. Protein phosphatases come of age. J Biol Chem. 1989;264(36):21435–21438. [PubMed] [Google Scholar]
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nature Genet. 2009;41:1022–1026. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17(7):578–586. doi: 10.1038/gim.2014.154. [DOI] [PubMed] [Google Scholar]
- Flores-Delgado G, Liu CW, Sposto R, Berndt N. A limited screen for protein interactions reveals new roles for protein phosphatase 1 in cell cycle control and apoptosis. J Proteome Res. 2007;6(3):1165–1175. doi: 10.1021/pr060504h. [DOI] [PubMed] [Google Scholar]
- Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D, et al. Protein phosphatase 1 is a molecular constraint on learning and memory. Nature. 2002;418(6901):970–975. doi: 10.1038/nature00928. [DOI] [PubMed] [Google Scholar]
- Gibbons JA, Kozubowski L, Tatchell K, Shenolikar S. Expression of human protein phosphatase-1 in Saccharomyces cerevisiae highlights the role of phosphatase isoforms in regulating eukaryotic functions. J Biol Chem. 2007;282(30):21838–21847. doi: 10.1074/jbc.M701272200. [DOI] [PubMed] [Google Scholar]
- Gräff J, Koshibu K, Jouvenceau A, Dutar P, Mansuy IM. Protein phosphatase 1-dependent transcriptional programs for long-term memory and plasticity. Learn Mem. 2010;17(7):355–363. doi: 10.1101/lm.1766510. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Zand DJ, Demmer L, Anderson CE, Dobyns WB, et al. Expanding the SHOC2 mutation associated phenotype of Noonan syndrome with loose anagen hair: structural brain anomalies and myelofibrosis. Am J Med Genet. 2013;161A:2420–2430. doi: 10.1002/ajmg.a.36098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Aldinger KA, Bennett JT, Baker L, Tusi J, et al. A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair. Am J Med Genet A. 2016;170(9):2237–2247. doi: 10.1002/ajmg.a.37781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10(10):e1004772. doi: 10.1371/journal.pgen.1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013;9(8):e1003671. doi: 10.1371/journal.pgen.1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban R, Roberts AE, Demmer L, Jethva R, Shephard B. Noonan syndrome due to a SHOC2 mutation presenting with fetal distress and fatal hypertrophic cardiomyopathy in a premature infant. Am J Med Genet. 2012;158A:1411–1413. doi: 10.1002/ajmg.a.35318. [DOI] [PubMed] [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350(6265):1262–1266. doi: 10.1126/science.aac9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobazzi D, Garaeva I, Albertario A, Cherif M, Angelini GD, et al. Protein Phosphatase 1 Beta is Modulated by Chronic Hypoxia and Involved in the Angiogenic Endothelial Cell Migration. Cell Physiol Biochem. 2015;36(1):384–394. doi: 10.1159/000430257. [DOI] [PubMed] [Google Scholar]
- Jouvenceau A, Hédou G, Potier B, Kollen M, Dutar P, et al. Partial inhibition of PP1 alters bidirectional synaptic plasticity in the hippocampus. Eur J Neurosci. 2006;24(2):564–572. doi: 10.1111/j.1460-9568.2006.04938.x. [DOI] [PubMed] [Google Scholar]
- Kanematsu T, Yasunaga A, Mizoguchi Y, Kuratani A, Kittler JT, Jovanovic JN, Takenaka K, Nakayama KI, Fukami K, Takenawa T, Moss SJ, Nabekura J, Hirata M. Modulation of GABA(A) receptor phosphorylation and membrane trafficking by phospholipase C-related inactive protein/protein phosphatase 1 and 2A signaling complex underlying brain-derived neurotrophic factor-dependent regulation of GABAergic inhibition. J Biol Chem. 2006;281(31):22180–22189. doi: 10.1074/jbc.M603118200. [DOI] [PubMed] [Google Scholar]
- Kelker MS, Page R, Peti W. Crystal structures of protein phosphatase-1 bound to nodularin-R and tautomycin: a novel scaffold for structure-based drug design of serine/threonine phosphatase inhibitors. J Mol Biol. 2009;385(1):11–21. doi: 10.1016/j.jmb.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. The sequence alignment/map format and SAM tools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Correll RN, Davis J, Vagnozzi RJ, York AJ, et al. Cardiac-specific deletion of protein phosphatase 1β promotes increased myofilament protein phosphorylation and contractile alterations. J Mol Cell Cardiol. 2015;87:204–213. doi: 10.1016/j.yjmcc.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge MN, Jhangiani S, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Med. 2013;5(6):57. doi: 10.1186/gm461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynes JT, Bateman KS, Cherney MM, Das AK, Luu HA, et al. Structal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J Biol Chem. 2001;276(47):44078–44082. doi: 10.1074/jbc.M107656200. [DOI] [PubMed] [Google Scholar]
- Moorhead G, Johnson D, Morrice N, Cohen P. The major myosin phosphatase in skeletal muscle is a complex between the β isoform of protein phosphatase 1 and the MYPT2 gene product. FEBS Lett. 1998;438:141–144. doi: 10.1016/s0014-5793(98)01276-9. [DOI] [PubMed] [Google Scholar]
- Pereira SR, Vasconcelos VM, Antunes A. The phosphoprotein phosphatase family of Ser/Thr phosphatases as principal targets of naturally occurring toxins. Crit Rev Toxicol. 2011;41(2):83–110. doi: 10.3109/10408444.2010.515564. [DOI] [PubMed] [Google Scholar]
- Peti W, Nairn AC, Page R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 2013;280(2):596–611. doi: 10.1111/j.1742-4658.2012.08509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribiag H, Stellwagen D. TNF-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J Neurosci. 2013;33(40):15879–15893. doi: 10.1523/JNEUROSCI.0530-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol Cell. 2006;22(2):217–230. doi: 10.1016/j.molcel.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Saadat M, Kakinoki Y, Mizuno Y, Kikuchi K, Yoshida MC. Chromosomal localization of human, rat, and mouse protein phosphatase type 1 beta catalytic subunit genes (PPP1CB) by fluorescence in situ hybridization. Jpn J Genet. 1994;69(6):697–700. doi: 10.1266/jjg.69.697. [DOI] [PubMed] [Google Scholar]
- Shang L, Henderson LB, Cho MT, Petrey DS, Fong CT, et al. De novo missense variants in PPP2R5D are associated with intellectual disability, macrocephaly, hypotonia, and autism. Neurogenetics. 2016;17(1):43–49. doi: 10.1007/s10048-015-0466-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Ishitani S, Sato A, Shibuya H, Ishitani T. Hipk2 and PP1c cooperate to maintain Dvl protein levels required for Wnt signal transduction. Cell Rep. 2014;8(5):1391–1404. doi: 10.1016/j.celrep.2014.07.040. [DOI] [PubMed] [Google Scholar]
- van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58(21):2241–2247. doi: 10.1016/j.jacc.2011.08.025. [DOI] [PubMed] [Google Scholar]
- Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17(1):9–18. doi: 10.1038/nrg3999. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Primrose DA, Leung AC, Fitzsimmons RB, McDermand MC, Missellbrook A, Haskins J, Smylie AS, Hughes SC. The PP1 phosphatase flapwing regulates the activity of Merlin and Moesin in Drosophila. Dev Biol. 2012;361(2):412–426. doi: 10.1016/j.ydbio.2011.11.007. [DOI] [PubMed] [Google Scholar]
- Zablotsky B, Black LI, Maenner MJ, Schieve LA, Blumberg SJ. Estimated prevalence of autism and other developmental disabilities following questionnaire changes in the 2014 national health interview survey. Natl Health Stat Report. 2015;87:1–20. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.