

Abstract
S -adenosylmethionine (SAM) has a sulfonium ion with three distinct C-S bonds. Conventional radical SAM enzymes use a [4Fe–4S] cluster to homolytically cleave the C5′,adenosine-S bond of SAM to generate a 5′-deoxyadenosyl radical, which catalyzes various downstream chemical reactions. Radical SAM enzymes involved in diphthamide biosynthesis, such as Pyrococcus horikoshii Dph2 (PhDph2) and yeast Dph1-Dph2 instead cleave the Cγ,Met–S bond of methionine to generate the 3-amino-3-carboxylpropyl radical. We here show that radical SAM enzymes can be tuned to cleave the third C-S bond to the sulfonium sulfur by changing the structure of SAM. With a decarboxyl SAM analogue (dc-SAM), PhDph2 cleaves the Cmethyl–S bond, forming 5′-deoxy-5′-(3-aminopropylthio) adenosine (dAPTA, 1). The methyl cleavage activity, like the cleavage of the other two C-S bonds, is dependent on the presence of a [4Fe-4S]+ cluster. Electron-nuclear double resonance (ENDOR) and mass spectroscopy data suggests that mechanistically, one of the S atoms in the [4Fe-4S] cluster captures the methyl group from dc-SAM, forming a distinct EPR-active intermediate, which can transfer the methyl group to nucleophiles such as dithiothreitol. This reveals that the [4Fe-4S] cluster in a radical SAM enzyme can be tuned to cleave any one of the three bonds to the sulfonium sulfur of SAM or analogues, and is the first demonstration that a radical SAM enzyme could switch from an Fe-based one electron transfer reaction to a S-based two electron transfer reaction in a substrate-dependent manner. This study thus provides a remarkable illustration of the versatile reactivity of Fe-S clusters.
Graphical Abstract
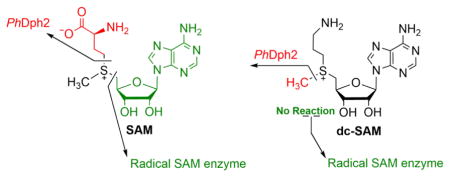
S-Adenosylmethionine (SAM) is a versatile molecule in biology (Figure 1).1,2 In addition to being a well-known methyl donor in many biological reactions, SAM is cleaved homolytically to generate methionine and a 5′-deoxyadenosyl (5′-dA) radical (Figure 1), which can initiate numerous reactions. Proteins that catalyze these reactions are known as radical SAM enzymes3,4 and comprise a superfamily of ~113,000 members5 that share a conserved three cysteine motif (mostly CxxxCxxC) that binds a [4Fe–4S] cluster. SAM can also be homolytically cleaved by unconventional radical SAM enzymes to generate a 3-amino-3-carboxylpropyl (ACP) radical, which is used in diphthamide biosynthesis in archaeal and eukaryotic species.6 For example, the thermophilic archaeon Pyrococcus horikoshii diphthamide biosynthesis protein, PhDph2, transfers the ACP group from SAM to the substrate protein, P. horikoshii translation elongation factor 2 (PhEF2).7,8 PhDph2 forms a homodimer and each monomer binds a [4Fe–4S] cluster. The yeast Dph1-Dph2 heterodimer complex, an ortholog of the PhDph2 homodimer, can also cleave SAM and transfer the ACP group to yeast EF2 via a same radical mechanism.9
Figure 1.
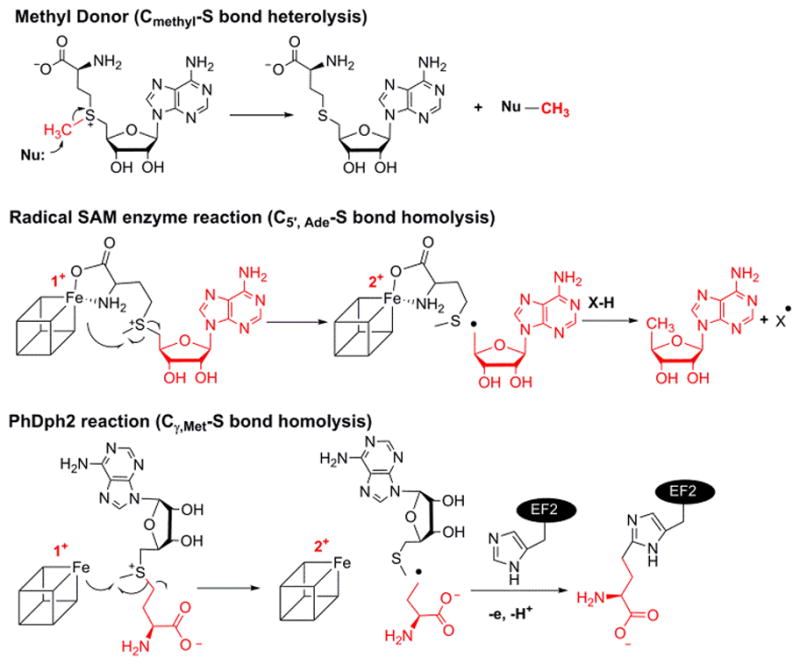
Three categories of C–S bond cleavage reactions of SAM.
When SAM is used as a methyl donor, the C–S bond between the methyl group and sulfonium (Cmethyl–S bond) normally is cleaved heterolytically, through nucleophilic attack by the acceptor atom, without involving a [4Fe–4S] cluster (Figure 1). Although there are several methylases that transfer methyl group from SAM to inert carbon atoms through generation of a 5′-dA radical to initiate the reaction,10 no radical SAM enzyme has been found to homolytically cleave the Cmethyl–S bond.
A longstanding question in the radical SAM enzyme community is how the radical SAM enzymes control which C–S bond in SAM is cleaved. In this study we find that PhDph2 cleaves the CMethyl–S bond of a SAM analogue, decarboxyl SAM (dc-SAM), forming 5′-deoxy-5′-(3-aminopropylthio) adenosine (dAPTA, 1), thus demonstrating for the first time that radical SAM enzymes can cleave all three sulfonium C-S bonds, and in so doing providing important insight into how different enzymes control the cleavage of SAM.
In conventional radical SAM enzymes, the amino and carboxyl groups of SAM coordinate the unique iron in the cluster.11 The interaction of SAM with the iron sulfur cluster is critical for activity. We began this study by synthesizing two SAM analogues to test whether the amino and carboxyl groups of SAM are also important in PhDph2-catalyzed reaction: deamino SAM (da-SAM) and decarboxyl-SAM (dc-SAM) (Figure 2). Although SAM analogues with the adenine changed to other bases have been used to study a radical SAM enzyme NosL,12 da-SAM and dc-SAM had not been used. The da-SAM was not a substrate of PhDph2; we could only detect non-enzymatic decomposition of da-SAM but not PhDph2-catalyzed cleavage (Figure S1). In contrast, based on HPLC analysis, incubation of dc-SAM with PhDph2 produced a new compound that was different from 5′-deoxy-5′-methylthioadenosine (MTA), the product generated in the reaction with natural substrate SAM (Figure 3B). This compound was only produced when PhDph2 and dithionite were present, suggesting that it was an enzymatic reaction product and required the reduced [4Fe-4S] cluster. The rate of this reaction was comparable to that of the natural substrate SAM (Figure S2). The new compound was isolated and characterized by electrospray ionization mass spectrometry (ESI-MS) to be dAPTA, 1, the demethyl product of dc-SAM (Figure 3C).
Figure 2.
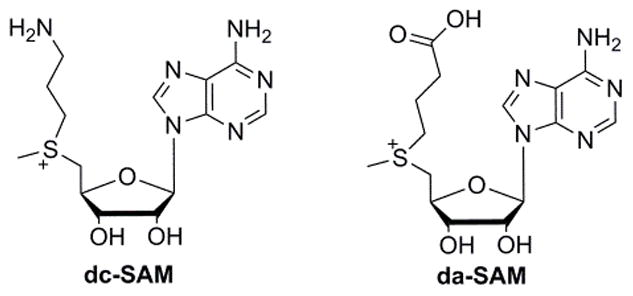
Structure of dc-SAM and da-SAM
Figure 3.
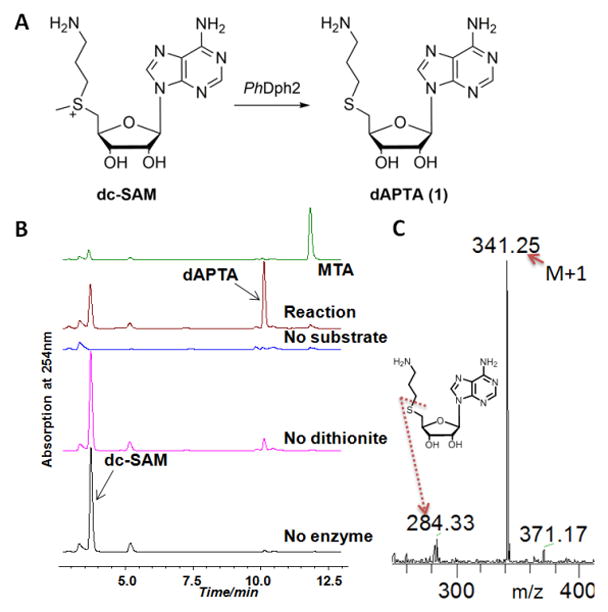
The reaction of dc-SAM with PhDph2. A) PhDph2 can convert dc-SAM to dAPTA, 1. B) High-performance liquid chromatography analysis showing a new product, different with the standard MTA, was generated in the reaction. C) ESI-MS of the isolated new peak in B. The [M+1]+ of the demethyl product 1 and the fragment peak can be detected.
The fact that without the carboxyl group of SAM, PhDph2 could cleave a different C-S bond was very intriguing and surprising. Mechanistically, this showed that the amino and carboxyl groups in SAM are important for positioning SAM in the right orientation in the active site of PhDph2 for the natural enzymatic reaction to happen.
It is known that the amino and carboxyl groups of SAM are crucial in conventional radical SAM enzymes.13 The bidentate interaction of the amino and carboxyl group of SAM with the unique iron in the [4Fe–4S] cluster facilitates the electron transfer from the unique iron to the sulfonium of SAM resulting in the reductive cleavage of SAM and the generation of the 5′-dA radical. We thus tested whether conventional radical SAM enzymes would recognize dc-SAM or da-SAM. We incubated dc-SAM or da-SAM with BtrN,14,15 a radical SAM dehydrogenase involved in the biosynthesis of antibiotic butirosin B. No dc-SAM or da-SAM cleavage product was detected (Figure S3). The result highlighted the different reactivities of PhDph2 and conventional radical SAM enzymes, but also showed that the amino and carboxyl groups of SAM are important for the reactivities of both types of enzymes.
We further investigated whether the amino group in dc-SAM was important for the methyl cleavage by PhDPh2. We synthesized two other SAM analogues (2 and 3, Figure 4), with an m-dimethylamino-phenoxyl and phenoxyl groups substituting the amino group of dc-SAM. When analogue 2 was incubated with PhDph2, we detected the formation of the demethyl product compound 4. Similarly, when analogue 3 was incubated with PhDph2, the demethyl product 5 was also produced (Figure S4). These results suggested that the amino group in dc-SAM is not important for this novel cleavage activity.
Figure 4.
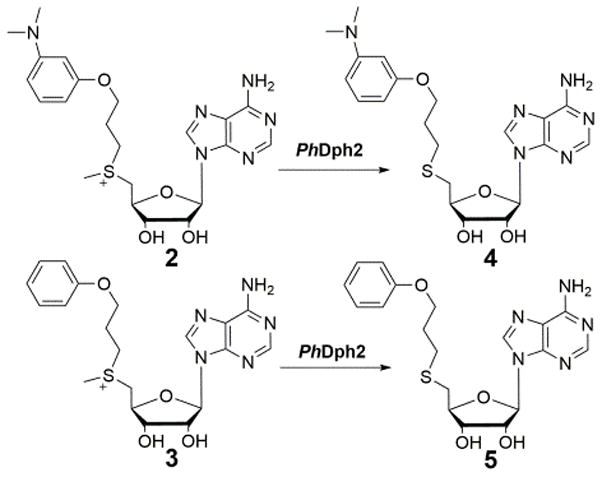
PhDph2 catalyzes demethylation reactions on SAM analogues 2 and 3.
To determine the fate of the cleaved methyl group from dc-SAM, we used 13Cmethyl-dc-SAM and 13C NMR to monitor the reaction. A new peak around 15 ppm was detected in the reaction, which was absent in the control reaction without dithionite (Figure S5A). The chemical shift indicated a methylated thiol group, likely resulting from the reaction with the DTT molecules in the buffer. This was further confirmed with the detection of Badan (6-bromoacetyl-2-dimethylaminonaphthalene)-derivatized methylated DTT by LC-MS (Figure S5B). When we carried out the same reaction with PhDph2 and dc-SAM in the absence of DTT, demethylation still occurred, but was much slower. We hypothesized that proteins in the reaction could be the methyl acceptor in the absence of DTT. To test this, we tried the reaction of PhDph2 and 14Cmethyl-dc-SAM without DTT in the buffer. 14Cmethyl-dc-SAM was prepared in situ by using SAM decarboxylase (SAMDC) and 14Cmethyl-SAM before adding PhDph2. Interestingly, the SAM decarbxylase in the reaction was strongly labeled (Figure 5). The labeling of SAM decarboxylase was dependent on dithionite, suggesting that the methylation event was dependent on an active PhDph2 with the reduced [4Fe-4S]+ cluster. These results showed that both small molecules and proteins could serve as the methyl acceptor in the reaction with PhDph2 and dc-SAM.
Figure 5.
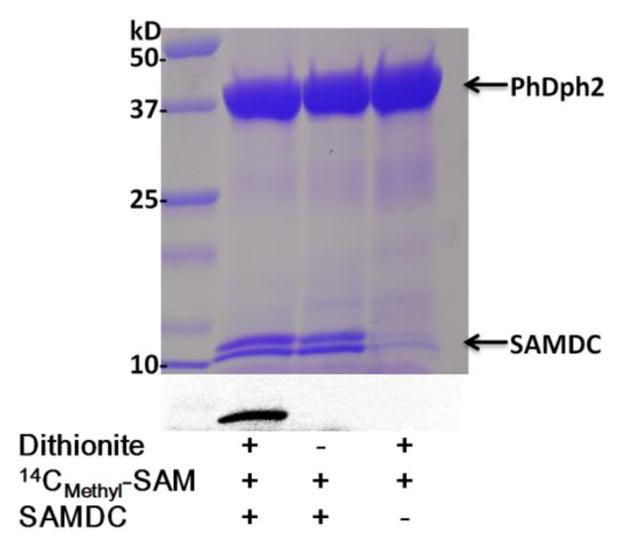
PhDph2 transfers 14C-methyl group to SAM decarboxylase (SAMDC) from 14Cmethyl-dc-SAM. The top panel shows the Coomassie blue-stained gel; the bottom panel shows the autoradiography. All the reactions in lanes 1–3 contained PhDph2. The presence of other reagents is indicated below each lane.
To explore the mechanism and search for intermediates in the methyltransfer reaction, we switched to the yeast ortholog Dph1-Dph2 system because the enzyme was active at room temperature, which facilitated experiments that were designed to capture potential reaction intermediate. Similar to PhDph2, Dph1-Dph2 cleaved dc-SAM to generate dAPTA and methylated SAMDC (Figure S6). We set up Dph1-Dph2 reaction with dc-SAM without DTT, collected samples at different time points by manually freezing the samples in liquid nitrogen, and examined them by eletron paramagnetic resonance (EPR) spectroscopy. A new species with g = [2.03, 1.98, 1.95] accumulated while the original [4Fe-4S]+ signal diminished at longer time points (Figure 6A). The signal broadened beyond detection at 35 K (Figure S7). The g-values and temperature dependence of the new species indicated that it involved the cluster, and was not an organic radical. This new species dissappeared when we added DTT, which indicated it was a reactive intermediate (Figure S8).
Figure 6.
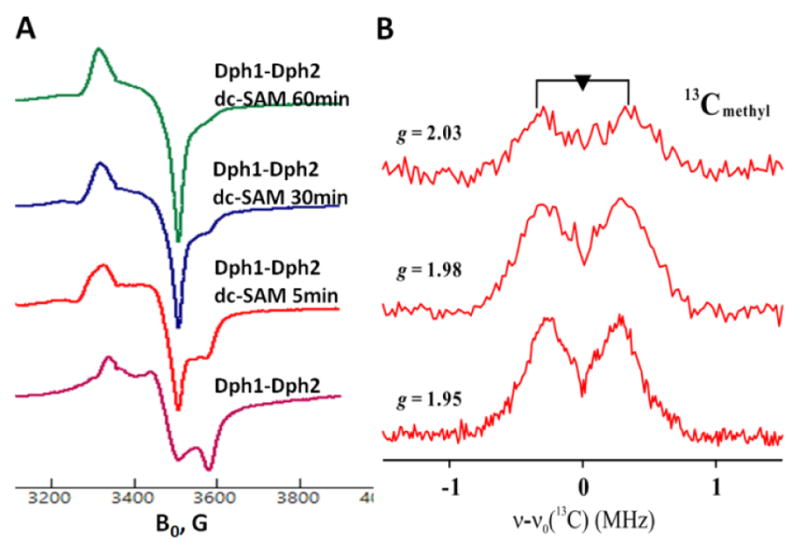
A) X-band CW EPR spectra of Dph1-Dph2 in the absence and presence of dc-SAM at 12 K. B) 2D field-frequency 35 GHz 13C Mims ENDOR of the 60 min sample of Dph1-2 with 13Cmethyl-dc SAM. Experimental fields are as indicated. Conditions: MW frequency = 34.8 GHz, MW pulse length (π/2) = 50 ns, τ = 500 ns and T = 2 K.
We further characterized this intermediate by preparing it with 13Cmethyl-dc SAM and using Mims pulsed electron-nuclear double resonance (ENDOR) spectroscopy. A small, apparently near-isotropic coupling, aiso ≈ 0.6 MHz, was detected (Figure 6B). Such a small coupling indicated that the 13C-methyl group was in the vicinity of the cluster but only weakly interacting with its spin. In previous studies of PFL-AE, the nonlocal through-space coupling constant is 0.33MHz for 13C-Me SAM,16 and the isotropic hyperfine coupling constant of 13C-carboxyl SAM is 0.71 MHz.13 The methyl group could have been transferred to a molecule adjacent, but we propose that it bound to a sulfur atom of the [4Fe-4S] cluster. Although without precedent, the spin density on such a sulfur would be small, and would plausibly lead to the small observed coupling for a cluster S-methyl. Such a species could arise if, instead of the reduced cluster causing homolytic cleavage with the natural substrate SAM, a SN2 displacement mechanism occurred: a sulfur atom in the reduced [4Fe-4S] cluster attacked the sulfonium center of dc-SAM, forming a S-methyl-[4Fe-4S] intermediate. Other nucleophiles, such as the thiol groups on DTT or nucleophilic residues on proteins, would subsequently attack the S-methyl-[4Fe-4S] intermediate and complete the methyl transfer reaction (Figure 7). This intermediate, could be compared to the catalytic organometallic intermediate in pyruvate formate lyase-activate enzyme (PFL-AE).17 Both intermediates are formed by substrate (SAM or SAM analogue) cleavage with bonding of the nascent radical/cleavage product to the [4Fe-4S] cluster, with the difference being whether the Fe or the S atom in the cluster is directly bonded.
Figure 7.
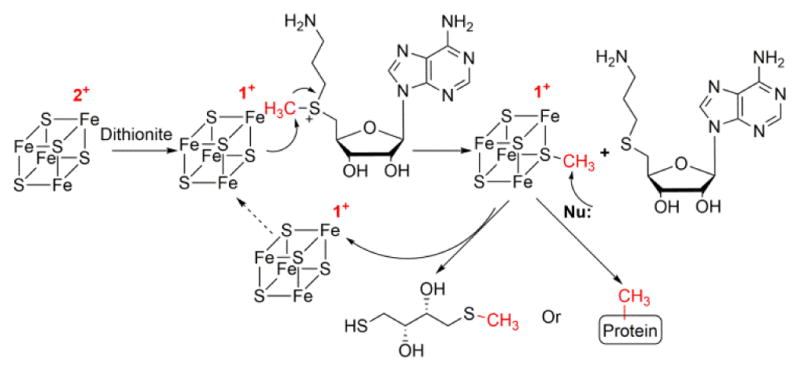
Proposed mechanism of PhDph2 catalyzed methyl transfer reaction with dc-SAM.
As a test of this mechanism, we reasoned that this intermediate should produce methanethiol after denaturation of the protein and Fe-S cluster in acidic conditions. Indeed, we were able to detect the formation of methanethiol in the reaction after denaturation of the protein subjected to turnover, while no methanethiol was detected in the control reaction without dithionite (Figure S9).
Computational studies suggest that the bidentate coordination of the amino and carboxyl groups of SAM to the unique cluster iron of conventional radical SAM enzymes is essential for the generation of the 5′-dA radical. Firstly, the sulfonium ion and unique iron orbitals should have matching energies, which is required for electron transfer between two atoms.18 Secondly, the bidentate structure and the resulting conformation of SAM explain the regioselectivity of C5′, Ade–S bond cleavage.19 PhDph2, representing a different type of radical SAM enzymes, cleaves the Cγ,Met–S bond in SAM and produce an ACP radical. Our results here indicate that PhDph2-catalyzed reaction also requires both the amino group and carboxylate group of SAM. However, unlike classical radical SAM enzymes, PhDph2 can still accept the dc-SAM analogue as a substrate, but catalyzes the cleavage of the Cmethyl-S bond, not the Cγ,Met-S bond cleaved in the natural enzymatic reaction. Instead of a CxxxCxxC motif in traditional radical SAM enzymes, the three conserved cysteine residues of PhDph2 that bind the [4Fe–4S] cluster are located in separate structural domains that separated by more than 100 residues in the primary sequence.7 We believe this structure allows SAM to bind the [4Fe-4S] in PhDph2 different from that in classical radical SAM enzymes. The different binding mode of SAM in PhDph2 (and yeast Dph1-Dph2) likely contribute to the stereoelectronic control of SAM cleavage and the unique reactivity with the SAM analogues described here.
Although we cannot totally rule out the possibility of a radical mechanism for formation of the S-methyl-[4Fe-4S] intermediate, the SN2 nucleaphilic mechanism is most likely for the following two reasons: (1) A homolytic cleavage of the Cmethyl-S bond in SAM is difficult due to the high energy of methyl radical;4 (2) If a methyl radical is generated, it should recombine with the closest unique iron and form a C-Fe bond, as the organometallic intermediate in PFL-AE.17 Although recently the formation of low level of 5′-deoxy-5′-thioadenosine, a reaction product of 5′-deoxyadenosyl radical with one sulfur in the oxidized cluster, was reported in NosL study. The iron sulfur cluster was inactivated instead of playing a catalytic role.20 We therefore believe that this is the first case in which a [4Fe-4S]+ cluster in a radical SAM enzyme switches from catalyzing an Fe-based one electron transfer reaction to catalyzing a S-based two electron transfer reaction by simply changing the structure of SAM. This activity of PhDph2 is dependent on the reduced cluster [4Fe-4S]+, which is directly involved in the methyl transfer reaction.
The methyl transfer studied here is different from that in the traditional methyltransferase and other radical SAM enzymes with methyl transfer activity. The well-studied methylsynthases RimN and Cfr,21–23 use conserved cysteine residues to cleave the methyl group from SAM via an SN2 mechanism. RimO and MiaB,24–26 representing a subgroup of methylthio transferase in radical SAM enzyme family, consume two equivalent of SAM: one donates a methyl group to the external sulfur attached to 4Fe-4S cluster, another SAM is reductively cleaved to generate a 5′-dA radical, which abstracts a proton from the substrate to generate a substrate radical. The substrate radical in turn attacks the methylthio group on the 4Fe-4S cluster to complete the methylthio transfer. In our case, PhDph2 or yeast Dph1-Dph2 only consumes one equivalent of the SAM analogue in the catalytic cycle and the methyl acceptor is a sulfur of the [4Fe-4S] cluster. A recent study reported that NosN can use MTA, instead of SAM, as a direct methyl donor.27 The methyltransferase activity of PhDph2 on dc-SAM resembles the cobalamin dependent SN2-based methyltransferase.28 Instead of the reduced cobalt in methylcobalamin, the sulfur in [4Fe-4S]+ serves as an intermediate methyl acceptor to transfer methyl group to other nucleophiles. Our studies here thus have revealed that the [4Fe-4S] cluster in a radical SAM enzyme can be tuned to cleave any one of the three bonds to the sulfonium sulfur of SAM or analogues. Together with recent studies on PFL-AE and PhDph2,17,29 our study provides compelling evidence for the versatility of these [4Fe-4S] clusters.
Supplementary Material
Acknowledgments
This work is supported by NIH/NIGMS (GM 088276 and P41GM103521 to HL and GM 111097 to BMH). We thank Dr. Ivan Keresztes for assistance with NMR, Dr. Tyler L Grove and Squire Booker at Penn State University for providing BtrN protein.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Experimental materials and methods, supporting figures. This material is available free of charge on the ACS Publications website.
References
- 1.Fontecave M, Atta M, Mulliez E. Trends Biochem Sci. 2004;29:243. doi: 10.1016/j.tibs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Lin H. Bioorg Chem. 2011;39:161. doi: 10.1016/j.bioorg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frey PA, Hegeman AD, Ruzicka FJ. Crit Rev Biochem Mol Biol. 2008;43:63. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 4.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akiva E, Brown S, Almonacid DE, Barber AE, Custer AF, Hicks MA, Huang CC, Lauck F, Mashiyama ST, Meng EC, Mischel D, Morris JH, Ojha S, Schnoes AM, Stryke D, Yunes JM, Ferrin TE, Holliday GL, Babbitt PC. Nucleic Acids Research. 2014;42:D521. doi: 10.1093/nar/gkt1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su X, Lin Z, Lin H. Crit Rev Biochem Mol Biol. 2013;48:515. doi: 10.3109/10409238.2013.831023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, Lin H. Nature. 2010;465:891. doi: 10.1038/nature09138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu X, Dzikovski B, Su X, Torelli AT, Zhang Y, Ealick SE, Freed JH, Lin H. Mol Biosyst. 2011;7:74. doi: 10.1039/c0mb00076k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong M, Su X, Dzikovski B, Dando EE, Zhu X, Du J, Freed JH, Lin H. J Am Chem Soc. 2014;136:1754. doi: 10.1021/ja4118957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauerle MR, Schwalm EL, Booker SJ. J Biol Chem. 2015;290:3995. doi: 10.1074/jbc.R114.607044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frey PA, Magnusson OT. Chem Rev. 2003;103:2129. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 12.Ji X, Li Y, Xie L, Lu H, Ding W, Zhang Q. Angew Chem Int Ed Engl. 2016;55:11845. doi: 10.1002/anie.201605917. [DOI] [PubMed] [Google Scholar]
- 13.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:11270. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. J Am Chem Soc. 2007;129:15147. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- 15.Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ. Biochemistry. 2010;49:3783. doi: 10.1021/bi9022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:3143. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 17.Horitani M, Shisler K, Broderick WE, Hutcheson RU, Duschene KS, Marts AR, Hoffman BM, Broderick JB. Science. 2016;352:822. doi: 10.1126/science.aaf5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicolet Y, Amara P, Mouesca JM, Fontecilla-Camps JC. Proc Natl Acad Sci U S A. 2009;106:14867. doi: 10.1073/pnas.0904385106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kampmeier JA. Biochemistry. 2010;49:10770. doi: 10.1021/bi101509u. [DOI] [PubMed] [Google Scholar]
- 20.Bhandari DM, Fedoseyenko D, Begley TP. J Am Chem Soc. 2016;138:16184. doi: 10.1021/jacs.6b06139. [DOI] [PubMed] [Google Scholar]
- 21.Grove TL, Radle MI, Krebs C, Booker SJ. J Am Chem Soc. 2011;133:19586. doi: 10.1021/ja207327v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ. Science. 2011;332:604. doi: 10.1126/science.1200877. [DOI] [PubMed] [Google Scholar]
- 23.Silakov A, Grove TL, Radle MI, Bauerle MR, Green MT, Rosenzweig AC, Boal AK, Booker SJ. J Am Chem Soc. 2014;136:8221. doi: 10.1021/ja410560p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forouhar F, Arragain S, Atta M, Gambarelli S, Mouesca JM, Hussain M, Xiao R, Kieffer-Jaquinod S, Seetharaman J, Acton TB, Montelione GT, Mulliez E, Hunt JF, Fontecave M. Nat Chem Biol. 2013;9:333. doi: 10.1038/nchembio.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molle T, Moreau Y, Clemancey M, Forouhar F, Ravanat JL, Duraffourg N, Fourmond V, Latour JM, Gambarelli S, Mulliez E, Atta M. Biochemistry. 2016;55:5798. doi: 10.1021/acs.biochem.6b00597. [DOI] [PubMed] [Google Scholar]
- 26.Landgraf BJ, Arcinas AJ, Lee KH, Booker SJ. J Am Chem Soc. 2013;135:15404. doi: 10.1021/ja4048448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding W, Li Y, Zhao J, Ji X, Mo T, Qianzhu H, Tu T, Deng Z, Yu Y, Chen F, Zhang Q. Angew Chem Int Ed. 2017;56:3857. doi: 10.1002/anie.201609948. [DOI] [PubMed] [Google Scholar]
- 28.Zydowsky TM, Courtney LF, Frasca V, Kobayashi K, Shimizu H, Yuen LD, Matthews RG, Benkovic SJ, Floss HG. J Am Chem Soc. 1986;108:3152. [Google Scholar]
- 29.Dong M, Horitani M, Dzikovski B, Pandelia ME, Krebs C, Freed JH, Hoffman BM, Lin H. J Am Chem Soc. 2016;138:9755. doi: 10.1021/jacs.6b04155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.