

Abstract
The need for smart materials in the area of biotechnology has fueled the development of numerous stimuli-responsive polymers. Many of these polymers are responsive to pH, light, temperature, or oxidative stress, and yet very few are responsive toward multiple stimuli. Here we report on the synthesis of a novel dual-stimuli-responsive poly(ethylene glycol)-based polymer capable of changing its hydrophilic properties upon treatment with UV light (exogenous stimulus) and markers of oxidative stress (endogenous stimulus). From this polymer, smart microparticles and fibers were fabricated and their responses to either stimulus separately and in conjunction were examined. Comparison of the degradation kinetics demonstrated that the polymer became water-soluble only after both oxidation and irradiation with UV light, which resulted in selective degradation of the corresponding particles. Furthermore, in vitro experiments demonstrated successful uptake of these particles by Raw 264.7 cells. Such dual-stimuli-responsive particles could have potential applications in drug delivery, imaging, and tissue engineering.
Keywords: electrohydrodynamic cojetting, microparticles, stimuli responsive materials, multifunctional poly(ethylene glycol), anisotropic particles
Graphical abstract
INTRODUCTION
Materials capable of changing their properties in a controlled and reversible manner in response to changes in their environment or a specific trigger, such as light, are often referred to as smart materials.1 Such stimuli-responsive materials have been designed in various forms including switchable surfaces,1–4 responsive hydrogels,5 or anisotropic particles.1,6,7 Compared to many known triggers, switching triggered by light can be performed in mild conditions and in a more specific manner due to exogenous control over light intensity and wavelength.8 For this reason, a wide range of photoresponsive systems has been developed thus far.5,9,10 For example, azobenzene derivatives and spiropyrans were successfully used for fabrication of photoresponsive surfaces.4 In the field of drug delivery, photocleavable protecting groups are used to switch the solubility of carrier polymers. For example, amphiphilic block copolymers with a hydrophobic block bearing photosensitive groups, which render the block hydrophilic upon light exposure and consequently make the copolymer completely water-soluble, have been used for generation of photodegradable micelles.11 Similarly, light-induced degradation of polyester particles has been shown to be useful for controlled release of material.12 In principle, a variety of photolabile protecting groups are available for different functionalities.13,14 In particular, o-nitrobenzyl (NB) groups have found a range of different applications.15 Originally designed for use in organic synthesis,16 these groups were successfully applied to biochemistry.12 When they are incorporated into a copolymer, the photocleavage of the NB groups takes place readily upon illumination with UV light, releasing alcohol or acid and a nitroso derivative.15
Besides light, potential triggers for stimuli-responsive materials include changes in temperature, pH, salt content, presence of specific molecules, and electrical or magnetic fields.9,10,17 Most stimuli-responsive materials for controlled release applications rely on the abnormally low pH of endosomes or tumor tissues.17,18 In comparison, there exist many fewer examples of stimuli-responsive materials triggered by changes in the local oxidative environment, as it is commonly observed in inflammatory tissues.17 However, the local change in the redox environment of a cell due to oxidative stress can be an important indicator of many diseases including cancer,19 atherosclerosis,20 neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease,21 or heart failure.22
Oxidative stress may originate from the overproduction of reactive oxygen and nitrogen species. The latter are generated as byproducts of aerobic metabolic cell processes involving reduction of oxygen to superoxide anions followed by formation of hydrogen peroxide, hydroxyl radicals, hypochlorite, peroxynitrite, and other molecules.23,24 At low oxidant concentrations in cells, a range of enzymes can act as antioxidants and maintain a tight redox balance. In fact, oxidants even play a positive role, because they work as cellular messengers in different signaling pathways and are used by the immune system to kill pathogens.25 However, at higher dosages, oxidants can cause significant damage to proteins, lipids, and DNA and finally lead to cell death. Therefore, the presence of oxidants can be used as a marker for inflamed tissues and can activate a specific action of a biomaterial, such as drug release.26 For example, a natural polymer for drug delivery triggered by oxidative stress was published by Frechet and coworkers.27 The authors prepared nanoparticles from dextran modified with arylboronic ester in order to render the polymer water insoluble. In an oxidative environment, the arylboronic ester groups were cleaved, thus allowing for fast release of the drug due to the resorption of the particles. An alternative route was pursued by Hubbell and co-workers,28–31 who used poly(propylene sulfide) (PPS) as the base polymer material. Thioethers are known to undergo facile oxidation to sulfoxides or sulfones upon exposure to hydrogen peroxide.25 Due to the difference in hydrophilic properties of PPS and its oxidation products (polysulfoxides), PPS can be used for fabrication of drug delivery systems triggered by oxidative stress. Similarly, Mahmoud et al.32 prepared nanoparticles from a polymer that combined redox-active thioether groups and pH-sensitive moieties. The authors argued that dissolution of the particles due to the oxidation of polysufide was followed by degradation of the polymer to ensure rapid and complete release.
Here, we report the synthesis of a new multifunctional poly(ethylene glycol) (PEG) -based polymer particle that contains both redox-responsive thioether moieties and light-sensitive NB groups. Specifically, this design takes advantage of the cooperative effects of hydrogen peroxide-reactive moieties (endogenous stimulus) and photoresponsive groups (exogenous stimulus).
RESULTS AND DISCUSSION
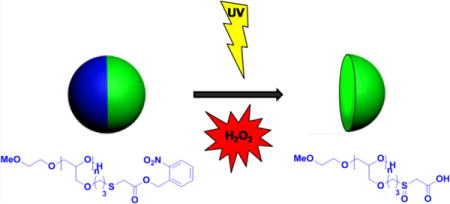
We used electrohydrodynamic (EHD) cojetting to formulate anisotropic microparticles containing a dual-stimuli-responsive polymer in one compartment. Oxidation of the thioether into sulfoxide groups under conditions of oxidative stress (endogenous stimulus) and photocleavage of NB groups (exogenous stimulus) can then act cooperatively to increase the polymer solubility in water and lead to the rapid resorption of one compartment in these microparticles. If the particles are placed in an oxidative environment in the absence of UV light, no resorption will occur. The same is true for a particle that is exposed to light in the absence of the oxidative trigger (Scheme 1B).
Scheme 1.
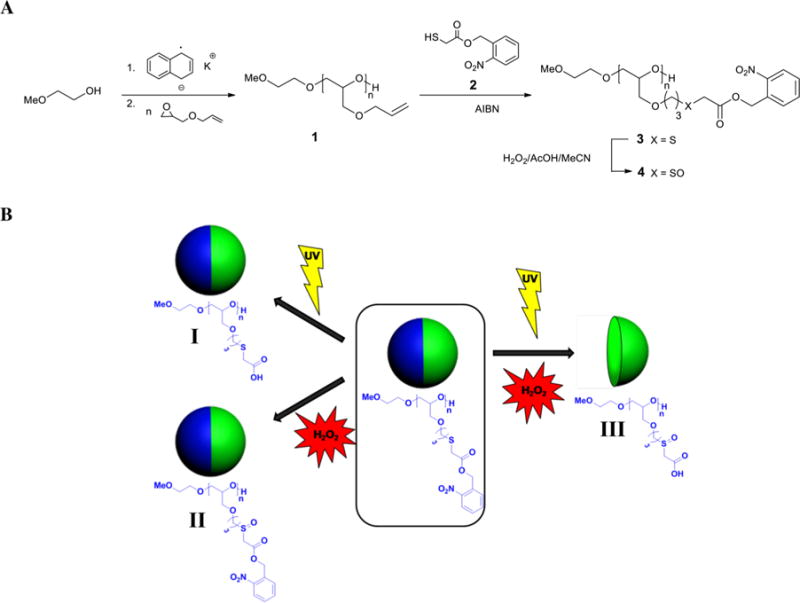
(A) Polymerization of AGE and Synthesis of Multifunctional o-Nitrobenzyl PEGs and (B) Bicompartmental Particles with Dual-Stimuli-Responsive Polymer in One Compartmenta
aThe polymer contains NB groups that are cleaved upon exposure to UV light, releasing carboxylic acid groups (I) and thioether moieties, which undergo oxidation into sulfoxide groups upon hydrogen peroxide action (II). Only upon action of both light and oxidant does the polymer become completely water-soluble due to conversion into the corresponding polyacid with sulfoxide moieties, leading to degradation of the corresponding compartment of the particle (III).
We chose PEG as the main-chain polymer, because PEG is known to have outstanding properties such as biocompatibility, hydrophilicity, protein resistance, and nontoxicity, which are particularly important for biomedical applications and have already made PEG one of the most attractive polymers in biomaterial synthesis.33–35 Moreover, the hydrophilicity of the PEG backbone dramatically enhances the solubility of the polymer but only after combined exposure to endogenous and exogenous stimuli. Typically, PEG polymers are functionalized via their end groups, which limits the number of functional groups per PEG chain. This disadvantage can be overcome by the synthesis of multifunctional derivatives via anionic ring-opening polymerization of functional epoxides and subsequent postpolymerization modifications.36,37 Thus, we first prepared poly(allyl glycidyl ether) (PAGE) as a starting polymer for synthesis of the stimuli-responsive polymer. Subsequently, polymer-analogue modification via thiol–ene “click” reaction yielded the polyether 3.
Polymer 1 was synthesized by anionic ring-opening polymerization of allyl glycidyl ether with 2-methoxyethanol and potassium naphthalenide as the initiating system. The crude polymer was purified chromatographically over silica gel (Scheme 1A). Synthesis of the target PEG-based polymer containing both thioether and NB moieties was performed by postpolymerization modification of poly(allyl glycidyl ether) via thiol–ene reaction with a thiol bearing NB-protected acid groups (Scheme 1A). This method allowed the introduction of both stimuli-reactive moieties in a single modification step. The thiol 2 bearing NB-protected carboxylic acid group was then synthesized by esterification of thioglycolic acid with o-nitrobenzyl alcohol. Reaction of polymer 1 with thiol 2 was performed in the presence of azobis(isobutyronitrile) (AIBN) with an excess of thiol to avoid undesired cross-linking reactions. Complete conversion of allyl moieties was confirmed by 1H NMR spectrometry (Figure S1A,B, Supporting Information). The resulting polymer 3 was further purified chromatographically over silica gel.
To compare the hydrophilic properties of polymer 3 and its sulfoxide analogue, oxidation of polymer 3 with hydrogen peroxide into the corresponding polymer 4 was performed (Scheme 1A). To ensure that oxidation of thioether moieties proceeded just until the formation of sulfoxide groups and to avoid generation of sulfone groups in the polymer product, the oxidation reaction was allowed to proceed at 0 °C. Oxidation to the sulfone is highly undesirable, not only because of the difference in solubility of sulfoxides and sulfones but also because of the low stability of sulfones, which can undergo degradation upon irradiation.24 However, detailed characterization of polymer 4 by 1H NMR and IR spectroscopy revealed the absence of sulfone groups (Figures S1C and S2A, Supporting Information). Moreover, complete oxidation of the thioether groups was confirmed by the disappearance of a signal at 3.28 ppm in the NMR spectrum corresponding to the sulfur neighboring methylene protons (d′) and the appearance of a new signal at 3.81 ppm from the corresponding protons (d″) in polymer 4 (Figure S1B,C, Supporting Information). This was also confirmed by IR spectroscopy, which revealed the appearance of a characteristic band at 1010 cm−1 corresponding to the S=O stretching bands in sulfoxides (Figure S2A, Supporting Information). Gel-permeation chromatographic (GPC) analysis indicated only a minor shift of the elution band of polymer 4 toward shorter elution times. We concluded that this shift is too small to be indicative of appreciable levels of degradation of polymer 3 during oxidation (Figure S2B, Supporting Information). Since the in vivo concentration of hydrogen peroxide under conditions of oxidative stress is relatively low,33 we expect the in vivo oxidation of polysulfides to predominantly result in sulfoxides, not sulfones.
Due to the combined redox responsiveness and photo-reactivity of the polymer, cleavage of the NB protecting groups of polymer 4 resulted in formation of the more polar polymer 5 (Figure S3A, Supporting Information). To validate this aspect systematically, deprotection of polymer 4 was monitored by 1H NMR spectrometry in DMSO-d6 solution through various exposures to UV light with a maximum wavelength of 312 nm. Disappearance of signals corresponding to aromatic (e–h) and benzylic (d) protons of the NB groups explicitly confirmed the cleavage of NB esters (Figure S4, Supporting Information). On the other hand, methylene protons (a–c) were still present in the NMR spectrum, showing the expected shifts (a′–c′) after light exposure of the polymer. This aspect provides further evidence that degradation of the polymer can be ruled out (Figure S4, Supporting Information). Dependence of the deprotection degree of the polymer on UV exposure time is displayed in Figure S3B (Supporting Information). A somewhat longer than expected exposure period was needed for complete cleavage of NB groups, which was attributed to the solvent used (dimethyl sulfoxide, DMSO) and high concentration of the polymer (5 mg/mL). However, these conditions were necessary for proper detection and assignment of signals in the NMR spectra. To identify the optimum UV exposure time in bulk, polymer 4 was spin-coated on gold wafers and exposed to light at a wavelength of 365 nm for different periods of time. Cleavage of the NB groups was then monitored by IR spectroscopy (Figure S5, Supporting Information). Deprotection of NB groups was directly correlated to disappearance of the characteristic nitro bands at 1530 and 1346 cm−1, as well as the shift of carbonyl band from 1742 to 1729 cm−1. Complete cleavage of NB protection was finally detected after light exposure for 30 min.
Anisotropic bicompartmental microspheres made of poly-(lactic-co-glycolic acid) (PLGA) and containing the stimuli-responsive polymer 4 in one compartment only were prepared by EHD cojetting (Figure 1).38 During EHD cojetting, polymer solutions are transported through a side-by-side configuration under a laminar flow (Figure 1A).39 Application of an appropriate electric potential causes the droplet at the tip of the needles to form a Taylor cone,40 which releases a very thin polymer thread from the appendix of the droplet. Due to the increase in surface area, evaporation of the solvent occurs very quickly and results in rapid precipitation of the polymers dissolved in the jetting solutions. Depending on the jetting conditions (e.g., concentration of the polymer in solution, flow rate, and applied electrical field), the ejected polymeric thread can break into particles or be collected as a continuous fiber.41 To ensure the exclusive formation of microspheres, the total concentration of all polymers dissolved in the jetting solutions was below 15% (w/v). For bicompartmental particles, one jetting solution contained PLGA only, while the second solution contained PLGA/polymer 4 mixtures at a ratio of 1:1 (w/w). Both jetting solutions were loaded with fluorescent dyes, which allowed for confirmation of the bicompartmental structure of the microspheres via confocal laser scanning microscopy (CLSM; Figure 1B). The spherical shape of the particles was confirmed by scanning electron microscopy (SEM; Figure 1C). While our group has previously shown that nanoparticles can be fabricated via the EHD cojetting process,42 we have used microparticles in these studies for their larger size and ease in imaging analysis.
Figure 1.
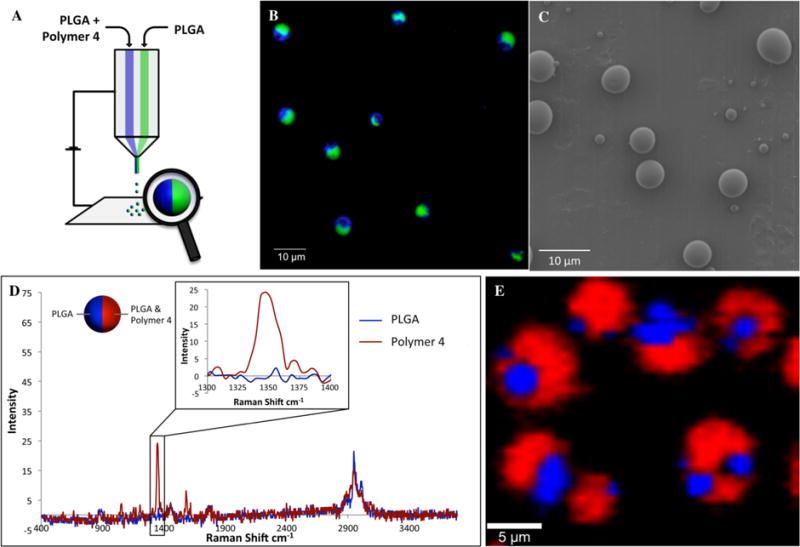
(A) Schematic representation of bicompartamental microparticles containing polymer 4 localized in one compartment. (B) CLSM image of particles, where green and blue dyes indicate PLGA and polymer 4, respectively. (C) SEM image of particles. (D) Confocal Raman microscopy spectra of bicompartmental PLGA/polymer 4 microspheres. (E) Two-dimensional (2D) reconstruction of confocal Raman spectra, confirming the selective localization of polymer 4 (red) in one compartment (the ratio PLGA:polymer 4 in one compartment was 3:1).
Furthermore, the selective localization of polymer 4 in one compartment only was verified by confocal Raman microscopy (Figure 1D,E). The characteristic signal of the nitro group at 1348 cm−1 was used as the reference band for assessing spatial distribution of polymer 4 within the particle. Confocal Raman microscopy revealed the selective location of polymer 4 in only one hemisphere of the microsphere as indicated by the red signal. For comparison, the PLGA polymer is indicated by the blue color.
For comparison, bicompartmental particles with PLGA and polymer 3 were also prepared by EHD cojetting and were characterized by SEM and CLSM. On the basis of combined analysis of these results, both microspheres were found to be bicompartmental and structurally identical. Next, the resorb-ability of PLGA/polymer 3 and PLGA/polymer 4 particles upon UV irradiation was evaluated. For this purpose, dry particles were exposed to 365 nm light and washed for 30 min before being imaged by SEM. The SEM images of particles with and without UV exposure are shown in Figure 2B–E.
Figure 2.
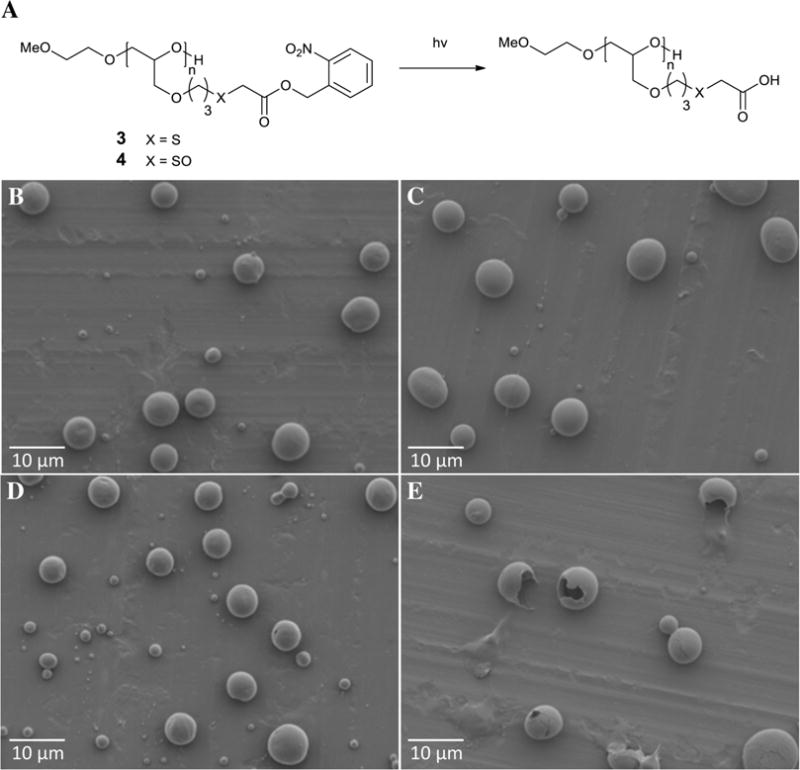
(A) Deprotection of polymers 3 and 4 into corresponding polyacids upon exposure to light at 365 nm. (B–E) SEM images of bicompartmental microparticles without light exposure [(B) PLGA/polymer 3 and (C) PLGA/polymer 4] and after 30 min of light exposure [(D) PLGA/polymer 3 and (E) PLGA/polymer 4].
Without UV exposure, no resorption was detected for either PLGA/polymer 3 or PLGA/polymer 4 particles (Figure 2B,C). After exposure to markers of oxidative stress, the thioether moieties of polymer 3 underwent oxidation into sulfoxide groups, which make polymer 4 more hydrophilic than polymer 3. However, due to the presence of hydrophobic NB groups in both polymers, the resorbability of both polymers was still negligible. Similarly, particles composed of PLGA/polymer 3 showed no signs of resorption, even after exposure to UV light (Figure 2D). Even though photocleavage of NB protection in polymer 3 yielded the corresponding polyacid (Figure 2A), the absence of sulfoxide moieties was sufficient to prevent resorption of the deprotected polymer 3 (Figure 2D). In contrast, incubation of UV-treated PLGA/polymer 4 particles led to fast resorption of polymer 4 (Figure 2E). Taken together, the results confirmed that only simultaneous exposure to both stimuli, light and markers of oxidative stress, allowed for the rapid resorption of polymer 3, whereas each stimulus on its own did not have sufficient effects to induce polymer resorption (Figure 2C,D). This suggests that PLGA/polymer 3 particles are a promising multi-stimuli-responsive system by the dual actions of oxidants and UV exposure.
To further prove that the particles containing polymer 3 and PLGA are responsive to two stimuli, particles with polymer 3 were fabricated and their resorbability was examined by combined exposure to oxidative stress and UV light. As displayed in Figure 3, the particles were first exposed to hydrogen peroxide, which resulted in the more hydrophilic polymer 4. The particles were then exposed to UV light at 365 nm for 30 min, washed, and analyzed by SEM. The results clearly demonstrate that specific degradation of one hemisphere can be achieved. Further UV duration led to complete degradation of one side, as displayed in Figure S6B (Supporting Information).
Figure 3.
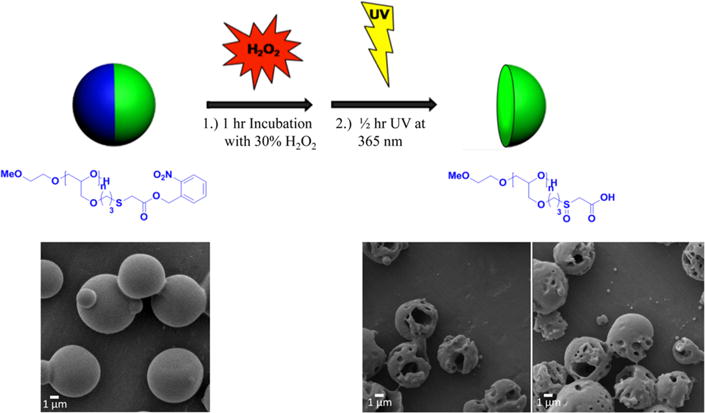
Degradation of one compartment through oxidation and UV illumination of bicompartmental microparticles. Here, microparticles composed of PLGA in one compartment (green) and polymer 3 and PLGA in the second compartment (blue) were incubated in 30% (v/v) hydrogen peroxide for 1 h, followed by UV illumination at 365 nm for 30 min. Similar to the treatment of the neat polymer, hydrogen peroxide oxidizes polymer 3 to polymer 4, which is then water-soluble upon UV illumination and cleavage of the nitrobenzyl group. Upon oxidative and UV treatments, the particles were washed with deiionized (DI) water several times before imaging by SEM to visualize the degradation of the particles.
To generalize the approach and to demonstrate other material architectures, bicompartmental fibers containing the stimuli-responsive PEG polymer in one compartment were prepared. The resulting microfibers displayed a clear bicompartmental architecture (Figure S7B, Supporting Information) with polymer 4 localized in just one compartment, as confirmed by confocal Raman microscopy (Figure S7C, Supporting Information). The microfibers were exposed to UV light and incubated in Tris-buffered saline (TBS) under the same conditions as described for the microspheres. This resulted in complete resorption of the compartment, which contained polymer 4, while no changes were detected without exposure to UV light. The possibility to produce fibers with a photosensitive polymer has potential application for the synthesis of degradable scaffolds for tissue engineering.
Because macrophages are directly involved in critical biological processes associated with oxidative stress, this cell type was selected for initial in vitro studies. First, the ability of Raw264.7 cells to internalize PLGA/polymer 4 microparticles was examined. Particles containing green and red dyes in separate compartments were prepared, collected in a buffered solution, counted, and incubated with cells. Several solutions with different concentrations of particles (50 000, 100 000, and 500 000 particles/mL) were incubated with Raw264.7 cells. After 4 h, cellular uptake of the PLGA/polymer 4 particles was analyzed by CLSM (Figure 4).
Figure 4.

(A) PLGA/polymer 4 microparticle uptake by Raw264.7 cells. (Inset) CLSM images of stained Raw264.7 cells after incubation with PLGA/polymer 4 microparticles at various concentrations (all scale bars are 20 μm). (B, C) CLSM images of Raw264.7 cells showing (B) complete uptake and (C) unsuccessful uptake (frustrated phagocytosis) of PLGA/polymer 4 microparticles. (D, E) Three-dimensional (3D) CLSM renderings (from Imaris software) of the same z-stack: (D) transparent projection and (E) shadow projection. The particle indicated by the red arrows is clearly uptaken and therefore is not seen in the shadow projection rendering; the particles indicated by the yellow arrows are slightly protruding out of the cell in the shadow projection, indicating frustrated phagocytosis.
Raw264.7 cells have the ability to phagocytize large microparticles, as shown in the CLSM images (inset, Figure 4A). In this study, both successful phagocytosis and frustrated phagocytosis coexisted in the same sample set (Figure 4B–E). This may be attributed to variability in the size of the microparticles and the inability of Raw264.7 cells to take up the largest particles. Only particles completely phagocytized were considered during the quantification of cellular particle uptake (Figure 4A). An increase in the number of internalized particles was observed at higher particle incubation concentrations (dose-dependence response), indicating nonspecific phagocytosis by Raw264.7 cells. Moreover, the particles were clearly visible and could be imaged after internalization, making them robust carriers for functional imaging or cell tracking experiments.
CONCLUSIONS
A novel dual-stimuli-responsive PEG-based polymer was prepared by postpolymerization modification of PAGE via a thiol–ene click reaction. Only the cooperative action of oxidants and UV light induced rapid resorption of the polymer under physiological conditions. The exogenous stimulus (UV light) acted as an on switch, after which the particles became susceptible to the second, endogenous stimulus, a marker of oxidative stress. Bicompartmental microparticles and fibers containing the dual-stimuli-responsive polymer were prepared by EHD cojetting, and their resorbability after exposure to UV light and oxidative stress was confirmed. These particles can serve as smart drug carriers with controlled drug release triggered by both oxidation stress and UV light. Additionally, in vitro experiments showed successful and dose-dependent uptake of the particles by Raw264.7 cells.
Supplementary Material
Acknowledgments
We thank Dr. Maria Schneider from the Institute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, for GPC measurements. We acknowledge funding from the Biointerfaces Program at KIT, the Multidisciplinary University Research Initiative of the Department of Defense, the Army Research Office (W911NF-10-1-0518), the DOD Idea Award (W81XWH-11-1-0111), and the EU-Project SAVVY (310445).
Footnotes
Supporting Information
Additional text of the Experimental Section and seven figures showing 1H NMR spectra of polymers 1, 3, and 4; IR spectra and GPCs of 3 and 4; photocleavage and UV-induced cleavage of NB protecting groups in 4; IR spectra of 4 before and after irradiation; SEM images of microparticles; and CLSM, confocal Raman, and SEM images of 4. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.5b01592.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Yoshida M, Lahann J. Smart nanomaterials. ACS Nano. 2008;2:1101–1107. doi: 10.1021/nn800332g. [DOI] [PubMed] [Google Scholar]
- 2.Stuart MAC, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M, Winnik F, Zauscher S, Luzinov I, Minko S. Emerging applications of stimuli-responsive polymer materials. Nat Mater. 2010;9:101–113. doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- 3.Sun A, Lahann J. Dynamically switchable biointerfaces. Soft Matter. 2009;5:1555–1561. [Google Scholar]
- 4.Nandivada H, Ross AM, Lahann J. Stimuli-responsive monolayers for biotechnology. Prog Polym Sci. 2010;35:141–154. [Google Scholar]
- 5.Tomatsu I, Peng K, Kros A. Photoresponsive hydrogels for biomedical applications. Adv Drug Delivery Rev. 2011;63:1257–1266. doi: 10.1016/j.addr.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 6.Li YY, Dong HQ, Wang K, Shi DL, Zhang XZ, Zhuo RX. Stimulus-responsive polymeric nanoparticles for biomedical applications. Sci China: Chem. 2010;53:447–457. [Google Scholar]
- 7.Hwang S, Lahann J. Differentially degradable Janus particles for controlled release applications. Macromol Rapid Commun. 2012;33:1178–1183. doi: 10.1002/marc.201200054. [DOI] [PubMed] [Google Scholar]
- 8.Mayer G, Heckel A. Biologically active molecules with a “light switch”. Angew Chem, Int Ed. 2006;45:4900–4921. doi: 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]
- 9.Roy D, Cambre JN, Sumerlin BS. Future perspectives and recent advances in stimuli-responsive materials. Prog Polym Sci. 2010;35:278–301. [Google Scholar]
- 10.Kim J, Hayward RC. Mimicking dynamic in vivo environments with stimuli-responsive materials for cell culture. Trends Biotechnol. 2012;30:426–439. doi: 10.1016/j.tibtech.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Schumers JM, Fustin CA, Gohy JF. Light-responsive block copolymers. Macromol Rapid Commun. 2010;31:1588–1607. doi: 10.1002/marc.201000108. [DOI] [PubMed] [Google Scholar]
- 12.Park TH, Eyster TW, Lumley JM, Hwang S, Lee KJ, Misra A, Rahmani S, Lahann J. Photoswitchable particles for on-demand degradation and triggered release. Small. 2013;9:3051–3057. doi: 10.1002/smll.201201921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelliccioli AP, Wirz J. Photoremovable protecting groups: Reaction mechanisms and applications. Photochem Photobiol Sci. 2002;1:441–458. doi: 10.1039/b200777k. [DOI] [PubMed] [Google Scholar]
- 14.Yu HT, Li JB, Wu DD, Qiu ZJ, Zhang Y. Chemistry and biological applications of photo-labile organic molecules. Chem Soc Rev. 2010;39:464–473. doi: 10.1039/b901255a. [DOI] [PubMed] [Google Scholar]
- 15.Zhao H, Sterner ES, Coughlin EB, Theato P. o-Nitrobenzyl alcohol derivatives: Opportunities in polymer and materials science. Macromolecules. 2012;45:1723–1736. [Google Scholar]
- 16.Pillai VNR. Photo-removable protecting groups in organic-synthesis. Synthesis. 1980:1–26. [Google Scholar]
- 17.Colson YL, Grinstaff MW. Biologically responsive polymeric nanoparticles for drug delivery. Adv Mater. 2012;24:3878–3886. doi: 10.1002/adma.201200420. [DOI] [PubMed] [Google Scholar]
- 18.Gao WW, Chan JM, Farokhzad OC. pH-Responsive nanoparticles for drug delivery. Mol Pharmaceutics. 2010;7:1913–1920. doi: 10.1021/mp100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: How are they linked. Free Radical Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondo T, Hirose M, Kageyama K. Roles of oxidative stress and redox regulation in atherosclerosis. J Atheroscler Thromb. 2009;16:532–538. doi: 10.5551/jat.1255. [DOI] [PubMed] [Google Scholar]
- 21.Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discovery. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 22.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol. 2011;301:H2181–H2190. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 23.Giustarini D, Dalle-Donne I, Tsikas D, Rossi R. Oxidative stress and human diseases: Origin, link, measurement, mechanisms, and biomarkers. Crit Rev Clin Lab Sci. 2009;46:241–281. doi: 10.3109/10408360903142326. [DOI] [PubMed] [Google Scholar]
- 24.Vo CD, Kilcher G, Tirelli N. Polymers and sulfur: What are organic polysulfides good for? Preparative strategies and biological applications. Macromol Rapid Commun. 2009;30:299–315. doi: 10.1002/marc.200800740. [DOI] [PubMed] [Google Scholar]
- 25.Jabaut J, Ckless K. In Inflammation, Chronic Diseases and Cancer: Cell and Molecular Biology, Immunology and Clinical Bases. InTechEurope; Rijeka, Croatia: 2012. pp. 145–160. Chapt. 7. [Google Scholar]
- 26.Yui N, Okano T, Sakurai Y. Inflammation responsive degradation of cross-linked hyaluronic-acid gels. J Controlled Release. 1992;22:105–116. [Google Scholar]
- 27.Broaders KE, Grandhe S, Frechet JMJ. A biocompatible oxidation-triggered carrier polymer with potential in therapeutics. J Am Chem Soc. 2011;133:756–758. doi: 10.1021/ja110468v. [DOI] [PubMed] [Google Scholar]
- 28.Rehor A, Hubbell JA, Tirelli N. Oxidation-sensitive polymeric nanoparticles. Langmuir. 2005;21:411–417. doi: 10.1021/la0478043. [DOI] [PubMed] [Google Scholar]
- 29.Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J Controlled Release. 2006;112:26–34. doi: 10.1016/j.jconrel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neill CP, Lee LK, Swartz MA, Hubbell JA. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 31.Thomas SN, van der Vlies AJ, O’Neil CP, Reddy ST, Yu SS, Giorgio TD, Swartz MA, Hubbell JA. Engineering complement activation on polypropylene sulfide vaccine nanoparticles. Biomaterials. 2011;32:2194–2203. doi: 10.1016/j.biomaterials.2010.11.037. [DOI] [PubMed] [Google Scholar]
- 32.Mahmoud EA, Sankaranarayanan J, Morachis JM, Kim G, Almutairi A. Inflammation responsive logic gate nanoparticles for the delivery of proteins. Bioconjugate Chem. 2011;22:1416–1421. doi: 10.1021/bc200141h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasut G, Veronese FM. PEG conjugates in clinical development or use as anticancer agents: An overview. Adv Drug Delivery Rev. 2009;61:1177–1188. doi: 10.1016/j.addr.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Zhang F, Kang ET, Neoh KG, Wang P, Tan KL. Surface modification of stainless steel by grafting of poly(ethylene glycol) for reduction in protein adsorption. Biomaterials. 2001;22:1541–1548. doi: 10.1016/s0142-9612(00)00310-0. [DOI] [PubMed] [Google Scholar]
- 35.Bjugstad KB, Lampe K, Kern DS, Mahoney M. Biocompatibility of poly(ethylene glycol)-based hydrogels in the brain: An analysis of the glial response across space and time. J Biomed Mater Res, Part A. 2010;95A:79–91. doi: 10.1002/jbm.a.32809. [DOI] [PubMed] [Google Scholar]
- 36.Mangold C, Wurm F, Frey H. Functional PEG-based polymers with reactive groups via anionic ROP of tailor-made epoxides. Polym Chem. 2012;3:1714–1721. [Google Scholar]
- 37.Li ZY, Chau Y. Synthesis of linear polyether polyol derivatives as new materials for bioconjugation. Bioconjugate Chem. 2009;20:780–789. doi: 10.1021/bc900036f. [DOI] [PubMed] [Google Scholar]
- 38.Roh KH, Martin DC, Lahann J. Biphasic Janus particles with nanoscale anisotropy. Nat Mater. 2005;4:759–763. doi: 10.1038/nmat1486. [DOI] [PubMed] [Google Scholar]
- 39.Saha S, Copic D, Bhaskar S, Clay N, Donini A, Hart AJ, Lahann J. Chemically controlled bending of compositionally anisotropic microcylinders. Angew Chem, Int Ed. 2012;51:660–665. doi: 10.1002/anie.201105387. [DOI] [PubMed] [Google Scholar]
- 40.Taylor G. Disintegration of water drops in electric field. Proc R Soc London, Ser A. 1964;280:383–397. [Google Scholar]
- 41.Bhaskar S, Pollock KM, Yoshida M, Lahann J. Towards designer microparticles: Simultaneous control of anisotropy, shape, and size. Small. 2010;6:404–411. doi: 10.1002/smll.200901306. [DOI] [PubMed] [Google Scholar]
- 42.Misra AC, Bhaskar S, Clay N, Lahann J. Multi-compartmental particles for combined imaging and siRNA delivery. Adv Mater. 2012;24:3850–3856. doi: 10.1002/adma.201200372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.