
Figure 1.
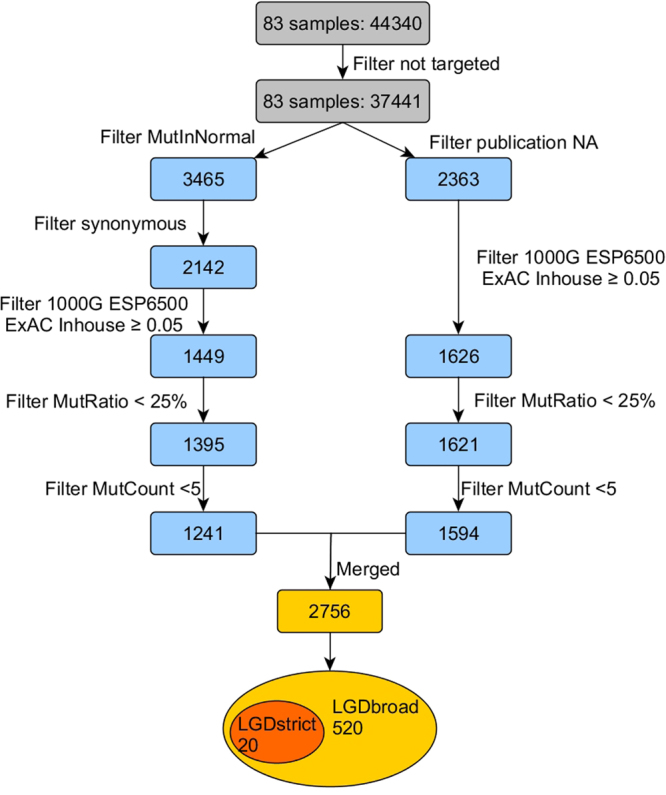
Schematic of the analytical workflow. A two-step variant filtering and prioritization approach was used to select candidate variants for disease-causing mutations. In the first step (on the left), all variants present in the in-house control cohort (MutInNormal) and all synonymous variants were excluded; while in the alternative step (on the right), all variants that had ever been reported were retained, regardless of their effects. Later, variants left from either step were further filtered against high frequency (variants with an alternative allele frequency ≥0.05 in either the 1000 Genome Project, the NHLBI Exome Sequencing Project, the Exome Aggregation Consortium data and an internal exome database of ~500 individuals) and low quality or potential false positives (MutRatio <25% and MutCount <5), before being merged into a non-redundant, integrated list of 2756. Two categories of variants were further extracted with the following definitions: LGDstrict refers to stop-gain, canonical splice-site (±2) variants and frameshift InDels. LGDbroad refers to stop-gain, splice region (±5) variants, frameshift InDels, in-frame InDels, and non-synonymous (missense) variants predicted to be damaging by at least three bioinformatics tools.