

Abstract
Isocyanides are exceptional building blocks whose wide deployment in multi-component and metal insertion reactions belies their limited availability. Addressing this deficiency is the first conjugate addition-alkylation method. An array of organolithiums, magnesiates, enolates, and metalated nitriles, add conjugately to β- and β, β-disubstituted arylsulfonyl alkenyl isocyanides to rapidly assemble diverse isocyanide scaffolds. The intermediate metalated isocyanides are efficiently trapped with electrophiles to generate substituted isocyanides incorporating contiguous tri- and tetra-substituted centers. The substituted isocyanides are ideally functionalized for elaboration into synthetic targets as illustrated by the three-step synthesis of the γ-carboline, N-methyl ingenine B.
Keywords: isocyanide, conjugate addition, organometallics, γ-carboline, N-methyl ingenine B
Graphical Abstract
Conjugate addition of diverse organometallics to sulfonyl-substituted alkenyl isocyanides overcomes the historical challenge of rapidly assembling complex isocyanides that retain the isocyanide functionality. The strategy affords complex isocyanides that are poised for elaboration into heterocycles as illustrated with the three-step synthesis of N-methyl ingenine B.
Isocyanides are unusual carbon-based functional groups in formally containing a carbon atom with a free electron pair.[1] The carbene-like structure (Figure 1)[2] confers ambiphilic reactivity on the isocyanide carbon that manifests an exceptionally diverse reactivity for one functional group: metal insertion,[3] radical additions,[4] nucleophilic additions,[1] and electrophilic alkylations.[5] The high reactivity toward disparate reagents is particularly valuable for multicomponent reactions,[5] heterocycle synthesis,[6] and accessing acyclic nitrogenous scaffolds.[6]
Figure 1.
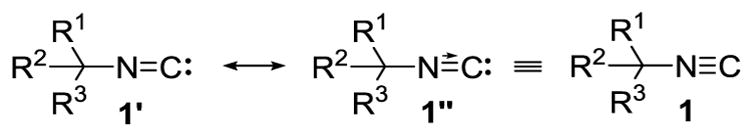
Isocyanide representations.
Isocyanide-containing metabolites, mostly from marine sources,[7] epitomize the challenge in working with isocyanides; the reactive carbene-like carbon confers biological activity through the same type of bonding that creates a susceptability of the R-NC unit toward irreversable complexation to transition metals, hydrolysis, and oxidation.[1] The tenacity of isocyanides to ligate to transition metals results from a strong σ-donation of the electron pair on carbon coupled with a symbiotic removal of electron density from the metal into the RN≡C π*-orbitals.[8]
The high reactivity and delicate nature of isocyanides, combined with the propensity to coordinate transition metals, has caused a severe deficiency of methods for manipulating the carbon scaffold while retaining the isocyanide functionality.[1] Most isocyanides are installed through a late-stage, three-step sequence: amine deprotection, formylation, and dehydration.[9] Described below is the first conjugate addition-alkylation of alkenyl isocyanides employing main group organometallics which effectively expands the limited repertoire of isocyanide-based bond constructions.
The viability of using main group organometallics to develop a general conjugate addition to unsaturated isocyanides was predicated on sporadic additions of Grignards[10] and sulfur ylides[11] to isocyanoacrylates. Initial forays to develop the conjugate addition employed the alkenyl isocyanide 3a, derived from TosMIC (2a, Scheme 1).[12] Exploratory additions of BuMgCl, Me2CuLi, and Et2BuZnLi[13] to 3a afforded a complex mixture of products, suggesting that the process involves more than a simple addition to a vinyl sulfone.[14] Further screening led to promising additions with BuLi (24%) and the magnesiate Bu3MgLi[15] (62%, Scheme 1).
Scheme 1.
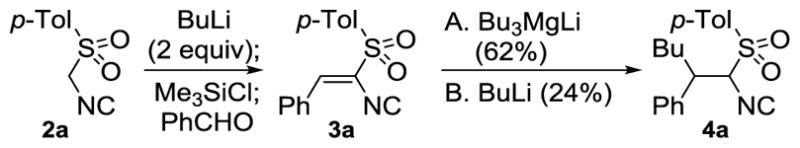
Synthesis and addition to sulfonylisocyanide 3a.
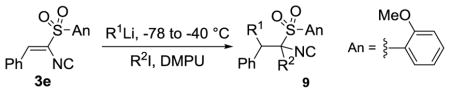
Attempts to expand the conjugate additions to 3a with additional organometallics led to an effective reaction with lithiated dithiane (Table 1, entry 1) but identified two limitations: poor tolerance of structural diversity in the organometallic and a pronounced instability of the resulting isocyanides toward storage and purification.[16] The limitations stimulated tuning the electronic and steric nature of the arylsulfone substituent to maximize reactivity and stability (Table 1).[17] Electron deficient arylsulfonyl-substituents were anticipated to facilitate the conjugate addition but the adduct from addition of Bu3MgLi to the trifluoromethyl-substituted isocyanide 3b was particularly unstable (Table 1, entry 2). Incorporating o-CF3 and o-OMe substituents (3c) improved the conjugate addition with PhLi and lithiated dithiane but the resulting isocyanides 6a and 6b were prone to decompose during purification (Table 1, entries 3 and 4).[16] Speculating that the efficacy of 3c was due to precomplexation between the organometallic and the o-OMe,[18] led to evaluation of di-o-methoxyphenylisocyanide 3d and o-anisyl (An) isocyanide 3e. While the addition of Bu3MgLi to 3d afforded a modest yield of 7a, the o-anisyl isocyanide 3e efficiently reacted with Bu3MgLi and BuLi to afford stable 8a in 54% and 53% yield, respectively (Table 1, entry 6).
Table 1.
Dependence of conjugate addition efficiency on sulfone structure.

|
The diastereomeric ratios were determined by 1H NMR integration of diagnostic methine signals.
The isocyanide was unable to be purified for characterization.
The generality of the conjugate additon to (o-anisyl)sulfonyl alkenyl isocyanides was probed with a variety of organometallics (Table 2). Diverse organolithiums with sp3, sp2- and sp-hybridization readily added to (o-anisyl)sulfonyl alkenyl isocyanides 3 (Table 2, 8b–8d, 8e–8i, and 8j, respectively).[19] The conjugate addition of MeLi to afford 8b, initially performed at temperatures between −100 to −95 °C (62% yield), was found to be more readily performed, and in higher yield, with MeLi·LiBr at −78 °C (72% yield). Selective allyl and benzyl additions[20] were performed from the mixed organomagnesiates allylMgBu2Li and BnMgBu2Li to afford isocyanides 8k and 8l, respectively. Lithiated acetonitrile and lithiated cyclohexanecarbonitrile afforded the nitrile-containing isocyanides 8m–8o, two of which contain quaternary centers. The lithium enolate derived from ethyl 2-methylpropionate afforded ester-isocyanide 8p, also installing a quaternary center. Conjugate reduction with NaBH4 afforded the alkylisocyanides 8q to 8s, which provides a valuable route to branched isocyanides.[21]
Table 2.
Conjugate additions to o-anisylsulfonyl alkenisocyanides 3.

|
The conjugate additions generated metalated isocyanides that were effectively intercepted by electrophiles (Table 3). Initial optimization focused on the methylation of the lithiated isocyanide derived from addition of PhLi to 3e. Methylation afforded 9a (53%) with incomplete conversion; addition of HMPA or DMPU (4 equiv) improved the reaction efficiency to 74% and 79%, respectively (Table 3, entry 1). DMPU-promoted electrophilic capture led to efficient addition-alkylations of 3e with PhLi and PrI (Table 3, entry 2) and addition-methylations with lithiated N-methyl indole and lithiated benzofuran (Table 3, entries 3–4). The conjugate addition-alkylation provides a valuable method to sterically encumbered alkylisocyanides that are challenging to prepare by direct alkylation of sulfinylmethylisocyanides such as TosMIC (2a).[24]
Table 3.
Conjugate addition-alkylations with alkenyl isocyanide 3e.
| |||
|---|---|---|---|
| Entry | R1Li | R2I | Alkaneisocyanide yield [%] (dr)[a] |
| 1 | PhLi | MeI |
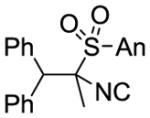 9a 79% |
| 2 | PhLi | PrI |
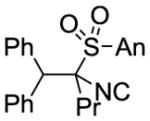 9b 82% |
| 3 |
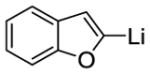
|
MeI |
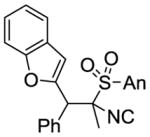 9c 82% (3:2:1) |
| 4 |
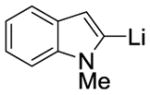
|
MeI |
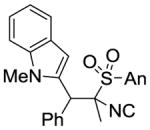 9d 63%[b] (4:0:1) |
The diastereomeric ratios were determined by 1H NMR integration of diagnostic methine signals in the crude reaction mixture.
The configuration was unambiguously secured by x-ray diffraction.
Mechanistically the conjugate addition likely proceeds through a preassociation of the organometallic with the sulfone and anisyl oxygens (Scheme 2).[18] Close proximity between the nucleophilic organometallic and the alkenyl isocyanide would facilitate intramolecular delivery of the alkyl group to the β-carbon (10) while preventing attack on the isocyanide. Complexation within the resultant lithiated isocyanide 11 may retard the alkylation which DMPU assists by solvation to a more nucleophilic, and accessible, solvent-separated ion pair (12 → 13).
Scheme 2.
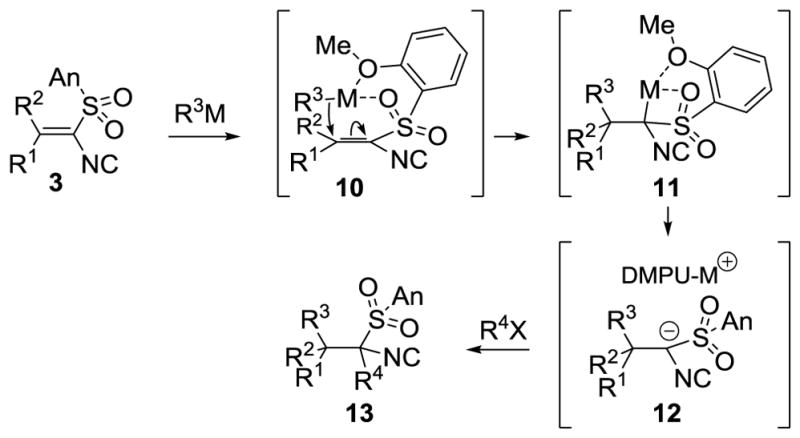
Conjugate addition-alkylation mechanism.
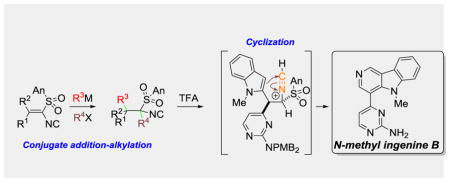
The conjugate addition affords substituted isocyanides that are ideally functionalized for heterocyclic synthesis. Rapid access to the indole-containing isocyanides 8h and 9d prompted cyclization to the γ-carboline scaffold,[25] an emerging pharmacophore.[26] Optimization experiments revealed that substoichiometric TFA triggered an efficient cyclization with (1S*, 2R*)-8h or 9d (Scheme 3).[27] Generation of the nitrilium ion 14 likely triggers cyclization to imine 15 which eliminates sulfinic acid to afford γ-carboline 16.[28] Rapid access to the γ-carboline 16a[29] is significant because of their potential as hepatitis C inhibitors.[30]
Scheme 3.
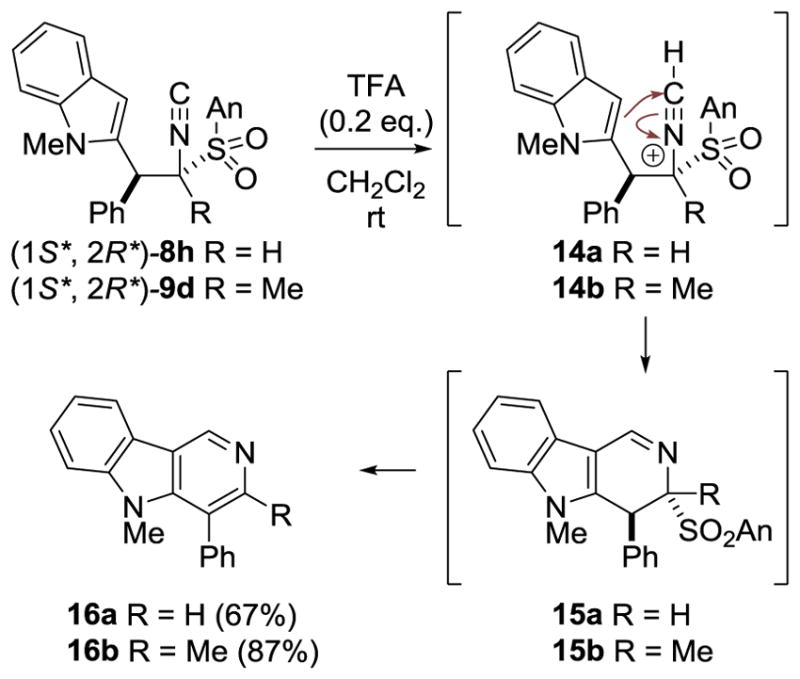
Isocyanide cyclization to γ-carbolines.
The versatility of the isocyanide conjugate addition-cyclization is illustrated in the synthesis of N-methyl ingenine B (20, Scheme 4).[31] In situ silylation-olefination of 2b[12] with aldehyde 17[32] afforded alkenyl isocyanide 18 that participated in a smooth conjugate addition with lithiated N-methylindole to provide 19 (Scheme 4). (1S*, 2R*)-19[33] was efficiently converted to N-methyl ingenine B (20)[34] on exposure to TFA, a process involving cyclization, sulfinate elimination, and debenzylation.
Scheme 4.
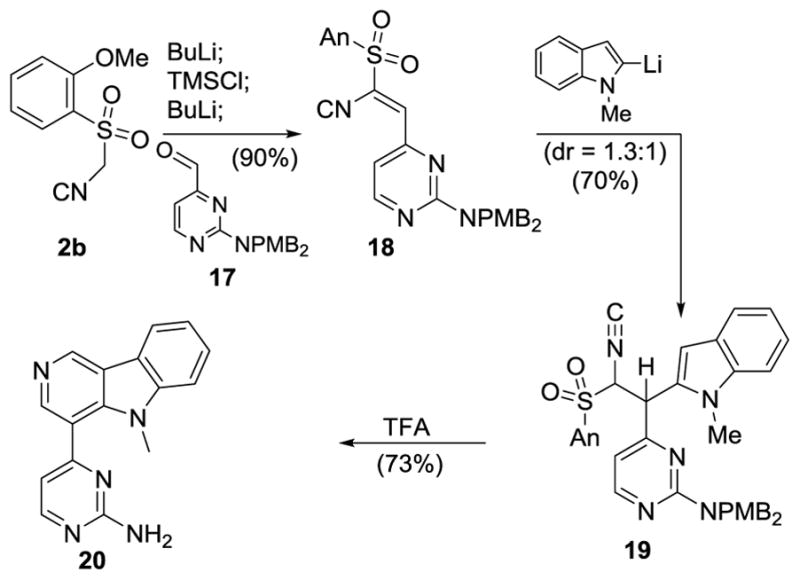
Synthesis of N-Methyl ingenine B (20).
Main group organometallics react with (o-anisyl)sulfonyl al-keneisocyanides in the first conjugate addition-alkylation of alkenyl isocyanides. Diverse organolithiums, magnesiates, enolates, and metalated nitriles, afford metalated isocyanides that are readily intercepted by electrophiles to efficiently install contiguous tri- and tetra-substituted centers. The highly-substituted isocyanides are ideally functionalized for elaboration into synthetic targets as illustrated by the three-step synthesis of the γ-carboline, N-methyl ingenine B (20).
Supplementary Material
Acknowledgments
Financial support for this research initially from NIH (2R15AI051352-04) and later from Drexel University, is gratefully acknowledged. Crystallographic analyses performed by Patrick J. Carroll and HRMS analysis conducted by Timothy P. Wade are gratefully acknowledged.
Footnotes
Supporting information for this article can be found under: http://dx.doi.org/10.1002/anie.XXXXXXXXX.
References
- 1.Suginome M, Ito Y. In: Science of Synthesis. Murahashi S-I, editor. Vol. 19. Thieme; Stuttgart: 2004. pp. 445–530. [Google Scholar]
- 2.Ramozzi R, Chéron N, Braïda B, Hiberty PC, Fleurat-Lessard P. New J Chem. 2012;36:1137. [Google Scholar]
- 3.Chakrabarty S, Choudhary S, Doshi A, Liu FQ, Mohan R, Ravindra MP, Shah D, Yang X, Fleming FF. Adv Synth Cat. 2014;356:2135. doi: 10.1002/adsc.201400017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang B, Studer A. Chem Soc Rev. 2015;44:3505. doi: 10.1039/c5cs00083a. [DOI] [PubMed] [Google Scholar]
- 5.a) Zhu J, Wang Q, Wang M-X. Multicomponent Reactions in Organic Synthesis. VCH; Weinheim: 2014. [Google Scholar]; b) Dömling A. Chem Rev. 2006;106:17. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; c) Zhu J. Eur J Org Chem. 2003:1133. [Google Scholar]
- 6.Nenajdenko VG. Isocyanide Chemistry: Applications in Synthesis and Material Science. VCH; Weinheim: 2012. [Google Scholar]
- 7.a) Mancini I, Guella G, Defant A. Mini Rev Med Chem. 2008;8:1265. doi: 10.2174/138955708786141007. [DOI] [PubMed] [Google Scholar]; b) Garson MJ, Simpson JS. Nat Prod Rep. 2004;21:164. doi: 10.1039/b302359c. [DOI] [PubMed] [Google Scholar]; c) Chang CWJ. Prog Chem Org Nat Prod. 2000;80:1. [Google Scholar]; d) Edenborough MS, Herbert RB. Nat Prod Rep. 1988;5:229. doi: 10.1039/np9880500229. [DOI] [PubMed] [Google Scholar]
- 8.Lazar M, Angelici RJ. In: Modern Surface Organometallic Chemistry. Basset J-M, Psaro R, Roberto D, Ugo R, editors. Ch. 13. VCH; Weinheim: 2009. pp. 513–556. [Google Scholar]
- 9.Huters AD, Styduhar ED, Garg NK. Angew Chem Int Ed. 2012;51:3758. doi: 10.1002/anie.201107567. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2012;124:3820. [Google Scholar]
- 10.Schöllkopf U, Meyer R. Angew Chem Int Ed. 1975;14:629. doi: 10.1002/anie.197506292. [DOI] [PubMed] [Google Scholar]; Angew Chem. 1975;87:624. [Google Scholar]
- 11.a) Honma M, Kirihata M, Uchimura Y, Ichimoto I. Biosci Biotech Biochem. 1993;57:659. doi: 10.1271/bbb.57.69. [DOI] [PubMed] [Google Scholar]; b) Kirihata M, Sakamoto A, Ichimoto I, Ueda H, Honma M. Agric Biol Chem. 1990;54:1845. [PubMed] [Google Scholar]; c) Schöllkopf U, Harms R, Hoppe D. Liebigs Ann Chem. 1973:611. doi: 10.1002/jlac.19727630102. [DOI] [PubMed] [Google Scholar]
- 12.Van Leusen AM, Wildeman J. Rec Trav Chim Pays-Bas. 1982;101:202. [Google Scholar]
- 13.a) Hevia E, Chua JZ, García-Álvarez P, Kennedy AR, McCall MD. Proc Natl Acad Sci USA. 2010;107:5294. doi: 10.1073/pnas.0913307107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Merkel S, Stern D, Henn J, Stalke D. Angew Chem Int Ed. 2009;48:6350. doi: 10.1002/anie.200901587. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2009;121:6468. [Google Scholar]
- 14.El-Awa A, Noshi MN, du Jourdin XM, Fuchs PL. Chem Rev. 2009;109:2315. doi: 10.1021/cr800309r. [DOI] [PubMed] [Google Scholar]
- 15.a) Mongin F, Harrison-Marchand A. Chem Rev. 2013;113:7563. doi: 10.1021/cr3002966. [DOI] [PubMed] [Google Scholar]; b) Krasovskiy A, Straub BF, Knochel P. Angew Chem Int Ed. 2006;45:159. doi: 10.1002/anie.200502220. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2005;118:165. [Google Scholar]
- 16.Some isocyanides hydrolyze during silica gel chromatography while others exhibit a pronounced tendency toward irreversible adsorption: Kotha S, Sreenivasachary N. Bioorg Med Chem Lett. 1998;8:257. doi: 10.1016/s0960-894x(98)00002-x.Kotha S, Brahmachary E, Sreenivasachary N. Tetrahedron Lett. 1998;39:4095.
- 17.Lujan-Montelongo JA, Estevez AO, Fleming FF. Eur J Org Chem. 2015:1602. doi: 10.1002/ejoc.201403615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whisler MC, MacNeil S, Snieckus V, Beak P. Angew Chem Int Ed. 2004;43:2206. doi: 10.1002/anie.200300590. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2004;116:2256. [Google Scholar]
- 19.Two exceptions are vinyllithium (17% yield of the conjugate adduct) and tBuLi; the main product is an unstable imine resulting from addition to the isocyanide.
- 20.Sośnicki JG. Tetrahedron Lett. 2006;47:6809. [Google Scholar]
- 21.van Leusen D, van Leusen AM. Synthesis. 1991:531. [Google Scholar]
- 22.The diastereomeric ratios were determined by 1H NMR integration of diagnostic methine signals in the crude reaction mixture.
- 23.(1S*, 2S*)-8d was a crystalline solid whose structure was determined by x-ray crystallography. Determination of the relative stereochemistry for (1S*, 2S*)-8d allowed assignment of the diagnostic methine signals that provided a signature for the assignments of the isocyanides 8 in Table 1. Krishna PR, Prapurna YL. Synlett. 2009:2613.
- 24.van Leusen AM, Bouma RJ, Possel O. Tetrahedron Lett. 1975;16:3487. [Google Scholar]
- 25.a) Pilipenko AS, Uchuskin MG, Trushkov IV, Butin AV. Tetrahedron. 2015;71:8786. [Google Scholar]; b) Nissen F, Richard V, Alayrac C, Witulski B. Chem Commun. 2011;47:6656. doi: 10.1039/c1cc11298h. [DOI] [PubMed] [Google Scholar]; c) Ding S, Shi Z, Jiao N. Org Lett. 2010;12:1540. doi: 10.1021/ol100272s. [DOI] [PubMed] [Google Scholar]; d) Alekseyev RS, Kurkin AV, Yurovskaya MA. Chem Heterocycl Comp. 2009;45:889. [Google Scholar]
- 26.a) Salim MTA, Goto Y, Hamasaki T, Okamoto M, Aoyama H, Hashimoto Y, Musiu S, Paeshuyse J, Neyts J, Froeyen M, Herdewijn P, Baba M. Antiviral Res. 2010;88:263. doi: 10.1016/j.antiviral.2010.09.013. [DOI] [PubMed] [Google Scholar]; b) Chen J, Dong X, Liu T, Lou J, Jiang C, Huang W, He Q, Yang B, Hu Y. Bioorg Med Chem. 2009;17:3324. doi: 10.1016/j.bmc.2009.03.037. [DOI] [PubMed] [Google Scholar]
- 27.Although treating the (1S*, 2S*)-diastereomer with TFA afforded a complex mixture, the (1S*, 2S*)-diastereomers were readily equilibrated with t-BuOK.
- 28.Livinghouse T. Tetrahedron. 1999;55:9947. [Google Scholar]
- 29.Zhang H. PhD thesis. Iowa State University (IA); 2003. [Google Scholar]
- 30.Aoyama H, Sako K, Sato S, Nakamura M, Miyachi H, Goto Y, Okamoto M, Baba M, Hashimoto Y. Heterocycles. 2009;77:779. [Google Scholar]
- 31.Ingenine B, des-methyl 20, exhibits pronounced cytotoxicity against murine lymphoma: Ibrahim SRM, Mohamed GA, Zayed MF, Sayed HM. Drug Res. 2015;65:361. doi: 10.1055/s-0034-1384577.
- 32.See the supporting information for synthetic details.
- 33.Equilibration of (1S*, 2S*)-19 with t-BuOK afforded a 1:1 ratio of (1S*, 2S*)- and (1S*,2R*)-19 whose purification afforded pure (1S*, 2R*)-19 (37%), pure (1S*, 2S*)-19 (35%), and a 1:1 mixture of (1S*,2R*)- and (1S*,2S*)-19 (15%).
- 34.An oxidative demethylation was successfully performed on 16a but application of the same sequence to N-methyl ingenine B (20) caused significant degradation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.