

Abstract
Most neurodegenerative disorders (NDDs) are characterized by cognitive impairment and other neurological defects. The definite cause of and pathways underlying the progression of these NDDs are not well defined. Several mechanisms have been proposed to contribute to the development of NDDs. These mechanisms may proceed concurrently or successively, and they differ among cell types at different developmental stages in distinct brain regions. The endocannabinoid system, which involves cannabinoid receptors type 1 (CB1R) and type 2 (CB2R), endogenous cannabinoids and the enzymes that catabolize these compounds, has been shown to contribute to the development of NDDs in several animal models and human studies. In this review, we discuss the functions of the endocannabinoid (EC) system in NDDs and converse the therapeutic efficacy of targeting the endocannabinoid system to rescue NDDs.
Keywords: Loss of neurons, motor and memory behavior, CB1 receptors, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease
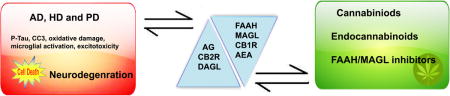
Graphical abstract
In this issue description: The Alzheimer (AD), Huntington (HD) and Parkinson (PD) diseases are characterized by cognitive impairment and other neurological defects. Although the mechanisms underlying the progression of these diseases are not well defined, the endocannabinoid system has been shown to contribute to the neurologic deficits found in AD, HD and PD in several animal models and human studies. The review provides a recent progress and discusses the therapeutic potential of targeting the endocannabinoid system to rescue some of the neurological defects of AD, HD, and PD.
Introduction
Cannabinoids are naturally occurring compounds found in the plant Cannabis sativa. Of over 500 different compounds present in the plant, only approximately 85 are termed cannabinoids (Brenneisen 2007). The most well-known among these compounds is delta-9-tetrahydrocannabinol (Δ9-THC) (Dewey 1986, Hollister 1986). Δ9-THC elicits its psychoactive effects by binding to receptors on the cell membrane called cannabinoid receptors (CBRs) (Howlett et al. 1990). These receptors are present in both the central nervous system (CNS) and the periphery and are classified as type 1 (CB1R) and type 2 (CB2R). Signaling downstream of these CBRs is significantly involved in a variety of standard functions as well as several pathophysiological functions of the CNS. Signaling events of CB1R and CB2R are depicted in Figs. 1 and 2, respectively. Discovery of the first endogenous cannabinoid substance, anandamide (N-arachidonoyl ethanolamine, AEA) (Devane & Axelrod 1994), in the brain emphasized the importance of the cannabinoid receptor and its endogenous ligands in the regulation of a wide variety of biological functions (Pertwee 1997). Later, 2-arachidonoyl glycerol (2-AG) was identified as a second endogenous cannabinoid (Sugiura et al. 2006). These two endogenous cannabinoids are derivatives of arachidonic acid (Fig. 3) and are synthesized and metabolized by different pathways (Fig. 4A and B). Both AEA and 2-AG act on the CBRs to elicit their biological activities; therefore, they are termed endocannabinoids (ECs). ECs are lipophilic in nature and are synthesized on demand from membrane phospholipids, and they can readily partition into and diffuse throughout cellular membranes without storage in vesicles. After their release from the postsynaptic neuron, ECs bind to CB1Rs located on the presynaptic membrane to inhibit neurotransmitter release (Berghuis et al. 2007, Harkany et al. 2008, Ohno-Shosaku et al. 2001). ECs are removed from the synaptic junction after CB1R activation by process of cellular transport followed by hydrolysis. Anandamide is hydrolyzed in postsynaptic neurons by fatty acid amide hydrolase (FAAH), thus terminating its action (Varvel et al. 2006, Varvel et al. 2007). After CB1R activation, 2-AG is hydrolyzed in presynaptic neurons by monoacylglycerol lipase (MAGL) [see recent reviews (Basavarajappa 2015, Lu & Anderson 2017)] (Fig. 4A and B). This retrograde signaling provides a mechanism for inhibitory feedback to regulate neurotransmitter release in the brain. This unique function of ECs has provided a strong rationale for investigating them as therapeutic targets for autoimmune disease, stroke and some other severe neurodegenerative diseases, including Alzheimer’s disease (AD), Huntington’s disease (HD) and Parkinson’s disease (PD). We have recently reviewed the functions of EC system in the normal brain (Basavarajappa 2015, Lu & Anderson 2017); therefore, the current study aims to present the new understanding of the role and involvement of the EC system in neurodegenerative disorders.
Figure 1. CB1R signaling pathway.
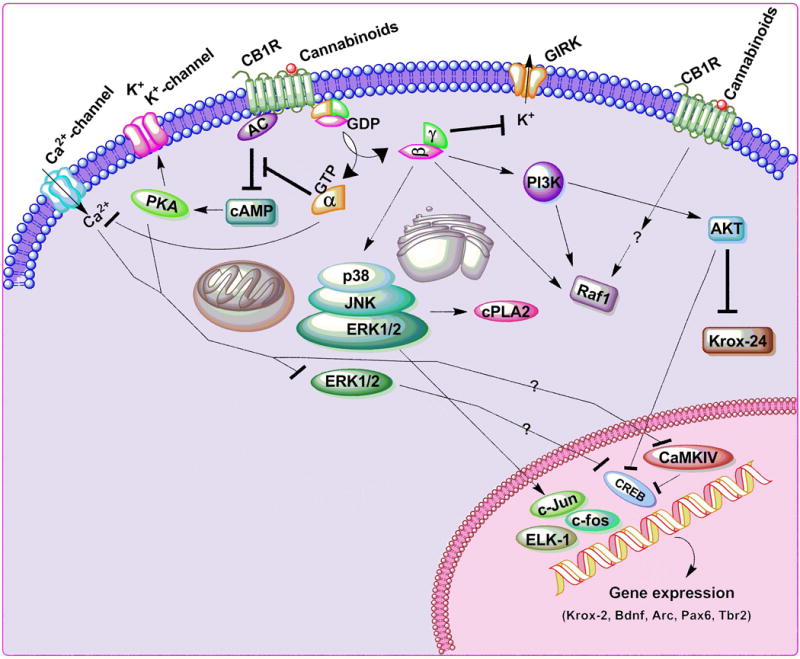
Both endogenous and synthetic cannabinoids elicit their effects by binding to CB1Rs (Breivogel & Childers 2000, Derkinderen et al. 2003, Mackie 2006, Mechoulam & Parker 2013). CB1Rs are seven-transmembrane-domain, G protein-coupled receptors located in the cell membrane (Howlett 1995). CB1R signaling leads to inhibition of adenylate cyclase (AC) activity (Childers et al. 1994, Pinto et al. 1994, Howlett & Mukhopadhyay 2000), N-type voltage-gated calcium channels (Caulfield & Brown 1992, Mackie & Hille 1992, Nogueron et al. 2001, Pan et al. 1996), N-type and P/Q-type calcium channels and D-type potassium channels (Howlett & Mukhopadhyay 2000, Howlett et al. 2002) and activate A-type and inwardly rectifying potassium channels (GIRKs) (Mu et al. 1999). CB1Rs also participate in the regulation of neurotransmitter release (Freund et al. 2003, Howlett et al. 2002) and inhibit synaptic transmission (Freund et al. 2003, Howlett et al. 2002). The actions on Ca2+ channels and AC are thought to be mediated by the G protein α subunits, while the βγ subunits activate GIRK and PI3K. The βγ complex further activates the p38/JNK/ERK1/2 pathways, followed by phosphorylation of several downstream targets, such as cPLA2, ELK-1, c-fos, c-Jun and CREB, leading to the expression of target genes such as krox-24 and BDNF (Derkinderen et al. 2003, Graham et al. 2006). PI3K mediates the AKT-induced inhibition of CREB activation (Graham et al. 2006). Inhibition of AC and the subsequent decrease in cAMP reduces the activation of cAMP-dependent protein kinase (PKA), resulting in reduced phosphorylation of K+ channels (Basavarajappa & Arancio 2008, Mechoulam & Parker 2013, Ozaita et al. 2007). Inhibition of ERK1/2 activation followed by inhibition of CaMKIV and CREB phosphorylation has also been found under certain conditions in vivo, leading to inhibition of Arc expression (Basavarajappa 2015, Basavarajappa & Subbanna 2014). Stimulatory effects are shown by (→) arrows and inhibitory effects by (⊥) arrows.
Figure 2. CB2R signaling pathway.
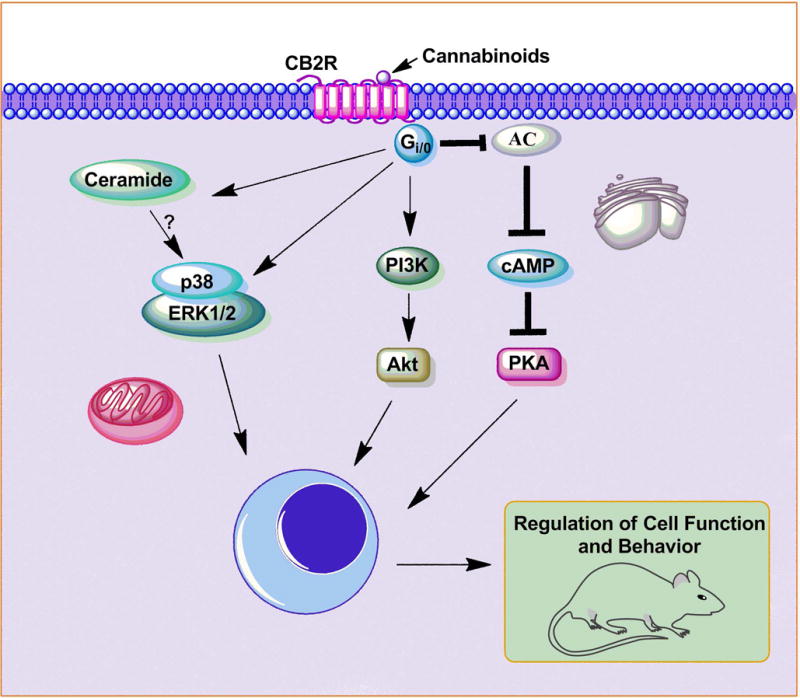
Both endogenous and synthetic cannabinoids bind to CB2Rs. CB2Rs are also seven-transmembrane-domain, G protein-coupled receptors located in the cell membrane (Bouaboula et al. 1996, Pertwee 1997, Buckley et al. 1998, Onaivi et al. 2006). Activation of CB2R is coupled to several different cellular pathways, including AC, cAMP, PKA, ERK1/2, p38 MAPK, and AKT and a pathway for de novo synthesis of ceramide (Bouaboula et al. 1996, Carracedo et al. 2006a, Carracedo et al. 2006b, Carrier et al. 2004, Choi et al. 2013, Gertsch et al. 2004, Herrera et al. 2005, Molina-Holgado et al. 2002a, Palazuelos et al. 2006, Samson et al. 2003). These signaling cascades may participate in the regulation of cell function and behavior. Stimulatory effects are shown by (→) arrows and inhibitory effects by (⊥) arrows.
Figure 3.
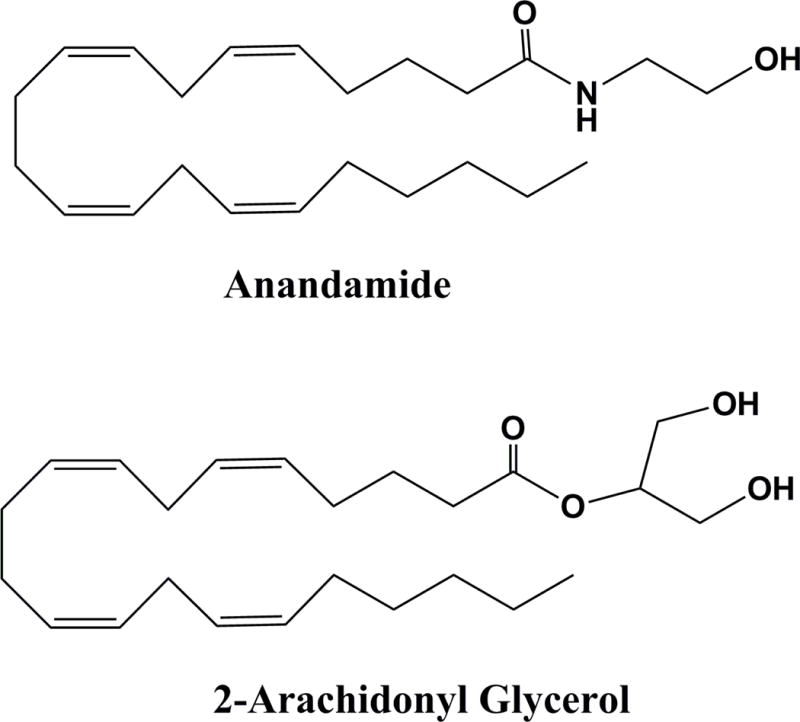
The AEA and 2-AG chemical structures.
Figure 4. The schematic enzymatic pathway that regulates catabolism of AEA (A) and 2-AG (B).
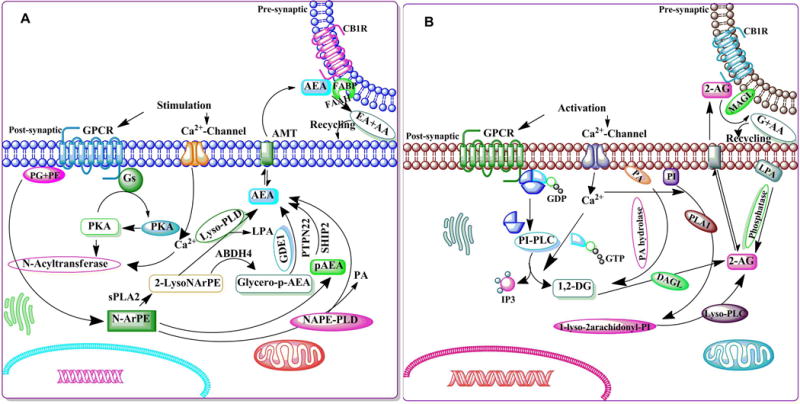
A. Stimulation of AC and PKA potentiates the N-acyltransferase (Ca2+-dependent transacylase, CDTA). An arachidonic acid chain is transferred by CDTA from the sn-1 position of a phospholipid to the primary amine of phosphatidylethanolamine, in a Ca2+-dependent manner, forming N-arachidonoyl phosphatidylethanolamine (N-ArPE), an intermediate. This N-ArPE is then hydrolyzed by a phospholipase D (PLD)-like enzyme to yield anandamide (AEA) (Natarajan et al. 1981, Schmid et al. 1983, Di Marzo et al. 1994). It is not clear whether the N-acyltransferase (NAT) or the N-acylphosphatidylethanolamine-specific phospholipase D (NAPE-PLD) controls the rate-limiting step of AEA synthesis (Di Marzo 1998, Sugiura et al. 2002, Hansen et al. 2000). NAPE-PLD knockout mice exhibit normal AEA biosynthesis, suggesting the involvement of other enzymes (Leung et al. 2006). Another pathway that regulates the conversion of NAPE into 2-lysol-NAPEs by the activity of secretory PLA2 (sPLA2) has also been proposed. 2-Lysol-NAPEs by the action of selective lysophospholipase D (lyso-PLD) (Sun et al. 2004) is then converted into N-acyl-ethanolamides, including AEA. 2-Lysol-NAPEs, through the action of abhydrolase domain 4 (ABHD4) (Liu et al. 2008), are turned into glycero-p-AEA, which is then converted by glycerol phosphodiesterase (GDE1) (Simon & Cravatt 2008) into AEA. A recent study using mouse brain and RAW264.7 macrophages proposed the existence of an analogous pathway where NAPE converted into pAEA by the action of PLC. The pAEA is subsequently dephosphorylated by a protein tyrosine phosphatase (PTPN22) (Liu et al. 2006). As a putative neuromodulator, AEA that is released into the synaptic cleft is expected to be rapidly inactivated. In general, there are two known mechanisms for removing endocannabinoids from the synaptic cleft to ensure rapid signal inactivation: re-uptake or enzymatic degradation. AEA is inactivated by reuptake (Beltramo & Piomelli 2000, Bisogno et al. 2001) via an uncharacterized membrane transport molecule, the ‘AEA membrane transporter’ (AMT) (Hillard et al. 1997, Beltramo et al. 1997, Beltramo & Piomelli 2000, Hillard & Jarrahian 2000, Maccarrone et al. 1998, Giuffrida et al. 2001, Basavarajappa et al. 2003), and subsequently, undergoes intracellular enzymatic degradation. FAAH metabolizes AEA to arachidonic acid, and ethanolamine leading to rapid clearance of AEA from extracellular compartments (Deutsch et al. 2001, Glaser et al. 2003). B. Intracellular Ca2+ initiates 2-AG biosynthesis by activating the process of formation of diacylglycerol (DAG) (Prescott & Majerus 1983, Sugiura et al. 1995) in the membrane by stimulating the phosphatidyl-inositol-phospholipase C (PI-PLC) pathway. 2-AG is the product of DAG-lipase (DAGL) acting on DAG (Bisogno et al. 1999b, Carrier et al. 2004). The second route involves hydrolysis of phosphatidylinositol (PI) by phospholipase A1 (PLA1) and hydrolysis of the resultant lyso-PI by a specific lyso-PLC (Sugiura et al. 1995). 2-AG is also synthesized through the conversion of 2-arachidonyl lysophosphatidic acid (LPA) by phosphatase (Nakane et al. 2002). 2-AG activates CB1Rs with greater efficacy than does AEA. Like AEA, 2-AG is inactivated by reuptake (Beltramo & Piomelli 2000, Bisogno et al. 2001) via the AMT (Hillard et al. 1997, Beltramo et al. 1997, Beltramo & Piomelli 2000, Hillard & Jarrahian 2000, Maccarrone et al. 1998, Giuffrida et al. 2001, Basavarajappa et al. 2003) and subsequently undergoes intracellular enzymatic degradation (Di Marzo et al. 1994, Day et al. 2001, Deutsch et al. 2001) by monoacylglycerol lipase (MAGL).
EC system and AD
The distribution of CB1Rs in the rodent adult brain is highly heterogeneous, with the highest densities of receptors present in the basal ganglia, including the substantia nigra pars reticulata (SNr) and the globus pallidus, in the hippocampus, particularly within the dentate gyrus, and in the molecular layer of the cerebellum. Low levels of CB1Rs are also found in the brainstem (Howlett 2002). There is a similar distribution of CB1Rs in humans (Glass et al. 1997, Biegon & Kerman 2001). The highest densities are found in limbic cortices, with much lower levels observed within primary sensory and motor regions, pointing an important role in motivational (limbic) and cognitive (Association) information processing. CB1Rs have been shown to be localized presynaptically on GABAergic interneurons and glutamatergic neurons (Katona et al. 2001, Katona et al. 1999, Katona et al. 2006), and this localization of CB1Rs is consistent with the proposed role of ECs in modulating GABA and glutamate neurotransmission (Ohno-Shosaku et al. 2001, Ohno-Shosaku et al. 2000, Ohno-Shosaku et al. 1998, Ohno-Shosaku et al. 2002, Wilson et al. 2001, Wilson & Nicoll 2001, Wilson & Nicoll 2002). The discovery of ECs such as AEA and 2-AG and the widespread localization of CB1Rs in the brain have stimulated considerable excitement regarding the role of the EC system in Alzheimer’s disease (AD). AD is the most devastating neurodegenerative disorder in humans, and the most common symptom of AD is difficulty in retaining and recalling recently learned information. Cognitive deficits of AD patients correlate with cerebral disturbances in sensitive brain areas, largely in the frontal cortex and the hippocampal region, areas that are rich in CB1Rs (Biegon & Kerman 2001, Glass et al. 1997). AD pathology is characterized by the accumulation of β-amyloid (Aβ), neurofibrillary tangles consisting of hypophosphorylated tau, the loss of particular subsets of neurons, and neuroinflammation resulting from glial activation. Although the existence of familial AD, which shows early onset, has been characterized, this form only accounts for a small proportion of cases (Campion et al. 1999, Hardy 1996b, Hardy 1996a, Hardy & Selkoe 2002). The real cause of sporadic AD is unknown, but numerous risk factors have been identified, including hypercholesterolemia, ischemic stroke, hypertension, the ApoE4 allele and diabetes (Carnevale et al. 2016, Grammas 2011, Hamel et al. 2016).
Limited numbers of studies have reported reduced densities of CB1Rs in aged animals (Mailleux & Vanderhaeghen 1992a, Romero et al. 1998). ECs were shown to decrease significantly with age in the mouse hippocampus and frontal cortex (Maroof et al. 2014). An autoradiographic examination of sections of normal post-mortem brain suggested a no significant reduction in CB1R binding with age (Ahmad et al. 2014). Several investigators have studied the changes associated with the EC system during the progression of AD disease. Correlation analyses based on post-mortem cortical brain tissues (Brodmann area 10) from a cohort of neuropathologically confirmed AD patients indicated lower CB1R levels than in age-matched controls (Solas et al. 2013). These lower CB1R levels correlated with hypophagia but not with any AD molecular markers or cognitive status (Solas et al. 2013). The levels of AEA were reduced in the hippocampus of a mouse genetic model of AD (TgAPP-2576) (Kofalvi et al. 2016). The ECs acting through transient receptor potential cation channel subfamily V member 1 (TRPV1) were suggested to play a significant role in Aβ-induced cognitive impairment in D3 receptor KO mice (Micale et al. 2010).
Cannabinoid receptor agonists such as AEA and noladin ether have been shown to provide protection against the Aβ-peptide induced neurotoxicity in neurons differentiated from the human teratocarcinoma cell line NTERA-2/cl-D1 in a CB1R- and mitogen-activated protein kinase (MAPK) pathway-dependent manner (Milton 2002). In another study, the injection of the CB1R antagonist SR141716A failed to provide protection against Aβ-induced amnesia (Mazzola et al. 2003). However, in a triple transgenic mouse model of AD (3xTg-AD), the activity of CB1R was found to be up-regulated in the anterior thalamus at the age of 4 months while the activity decreased in nucleus basalis of Meynert at 15 months of age (Manuel et al. 2016).
It has been found that senile plaques in AD patients express CB1R and CB2R and are associated with markers of microglial activation. CB1R-enriched neurons were significantly reduced in areas of microglial activation (Ramirez et al. 2005). Also, G-protein coupling and CB1R protein expression are significantly reduced in AD brains (Ramirez et al. 2005). However, CB2R levels were found enhanced in AD patients and well correlated with two relevant AD molecular markers (Aβ-42 levels and senile plaque score) but not with cognitive status (Solas et al. 2013). Additionally, in AD brains, the nitration of CB1R and CB2R proteins was elevated (Ramirez et al. 2005). It has been reported that AD-like pathology and learning and memory impairments induced by Aβ administration in rats was rescued by WIN-55,212-2 (CB1R agonist) (Ramirez et al. 2005). Additionally, cannabinoids (HU-210, WIN-55,212-2, and JWH-133) prevented Aβ-induced microglial activation in cultured glial cells (Ramirez et al. 2005). Further, Aβ-induced CD40-mediated microglial phagocytosis was inhibited by the CB2R agonist JWH-015 (Ehrhart et al. 2005). Similarly, treatment with WIN, 2-AG, or methanandamide prevented the hemichannel activity and inflammatory profile induced by Aβ in astrocytes; excitotoxic glutamate release and neuronal damage in hippocampal slices treated with Aβ (Gajardo-Gomez et al. 2017). Notably, CB2R and the AEA-hydrolyzing enzyme FAAH have been shown to be selectively expressed in astrocytes and microglia associated with neuritic plaques (Benito et al. 2003); however, these AD patients showed no difference in CB1R expression in the neuritic plaques (Benito et al. 2003). Other studies have reported increased FAAH gene expression and decreased DNA methylation at the FAAH gene promoter in peripheral blood mononuclear cells from late-onset AD patients (D’Addario et al. 2012) and decreased FAAH activity in cortical membrane tissues of AD patients (Pascual et al. 2014). Other studies have reported increased DAG lipase alpha (DAGLα) in the hippocampus of AD patients (Farooqui et al. 1988) and increased DAGLα followed by enhanced 2-AG levels in the hippocampus of Aβ peptide-treated rodents. Eight-month-old AβPPswe/PS1ΔE9 mice displayed significantly lower levels of striatal 2-AG than wild-type mice but exhibited enhanced cannabinoid receptor/effector coupling (Maroof et al. 2014). Superfusion of mouse hippocampi with Aβ was reported to significantly prolong depolarization-induced suppression of inhibition leading to synapse silencing in AD (Mulder et al. 2011). Also, early administration of an EC cellular reuptake inhibitor (VDM11) rescued both hippocampal damage and loss of memory retention in rats (van der Stelt et al. 2006). Thus, increased 2-AG signaling, particularly near the senile plaques, can exacerbate synaptic failure in AD (Mulder et al. 2011). In another study (Tanveer et al. 2012), it was reported that AEA upregulates Notch-1 signaling in cultured neurons. Exposure of cultured neurons to Aβ (1–42) increases expression of the endogenous inhibitor of Notch-1, numb (Nb), leading to impaired Notch-1 signaling. The addition of AEA and 2-AG prevented Nb expression and enhanced Notch-1 signaling. The stimulatory effects of AEA on Notch-1 signaling persisted in the presence of Aβ (1–42). Through Notch-1 signaling, AEA may be able to help restore neurogenesis and cognition in AD (Tanveer et al. 2012). Interestingly, elevation of 2-AG through pharmacological inhibition of MAGL significantly reduced Aβ-induced neurodegeneration via CB1R-dependent suppression of ERK1/2, NF-κB phosphorylation and cyclooxygenase (COX-2) expression in cultured hippocampal neurons (Chen et al. 2011). In another study (Yan et al. 2016), increasing 2-AG via inhibiting MAGL prevented prostaglandin (PGE2) production, neuroinflammation-associated Aβ42 accumulation, and neurodegeneration in C57BL/6J and APP/PS1 mice [Double transgenic mice over expressing human amyloid precursor protein (APP) and a mutant human presenilin 1 (PS1)] exposed to NO2 inhalation, which promotes pathological abnormalities and cognitive defects related to AD (Yan et al. 2016). Further, inhibition of MAGL significantly suppressed the production and accumulation of Aβ by reducing the expression of β-site amyloid precursor protein cleaving enzyme 1 (BACE1) in a mouse model of AD (Chen et al. 2012). MAGL inhibition also prevented neuroinflammation and decreased neurodegeneration by suppressing microglial and astrocytic activation. Inhibition of MAGL also maintained the integrity of hippocampal synaptic structure and function, improved long-term synaptic plasticity, spatial learning, and memory (Chen et al. 2012). Expression of the non-coding small RNA miR-188-3p, which targets BACE1, was demonstrated to be significantly reduced in the AD postmortem brains and APP transgenic (TG) mice (Zhang et al. 2014), a mouse model of AD, and MAGL inhibition restored the decreased miR-188-3p expression. Overexpression of miR-188-3p in the hippocampus reduced the levels of BACE1, Aβ, and neuroinflammation and prevented impairments in hippocampal basal synaptic transmission, long-term potentiation (LTP), learning, and memory in TG mice. MiR-188-3p loss of function prevented 2-AG-induced suppression of BACE1. Moreover, 2-AG or peroxisome proliferator-activated receptor-γ (PPARγ) agonists and PPARγ antagonism or NF-κB activation upregulated miR-188-3p expression. Reducing Aβ and neuroinflammation by inhibiting MAGL was blocked by PPARγ antagonism. Also, BACE1 suppression by 2-AG and PPARγ activation was eliminated by knockdown of NF-κB, and improved synaptic and cognitive function in TG mice was prevented by 2-AG signaling (Zhang et al. 2014). APP/PS1 mice lacking CB2R exhibit increased Aβ deposits in cerebral cortex, hippocampus, and cortical Aβ40 soluble levels in the early symptomatic phase (6 months) but not in the pre-symptomatic phase (3 months) (Aso et al. 2016, Koppel et al. 2014). However, lack of CB2R (CB2RKO) does not accelerate the memory impairment in APP/PS1 mice, even though CB2RKO mice themselves exhibit memory impairments (Li & Kim 2016b, Li & Kim 2016a, Garcia-Gutierrez et al. 2013). These findings together demonstrate that CB2Rs may assist in Aβ processing and thereby prevent neuroinflammation in AD (Koppel et al. 2014, Schmole et al. 2015). The treatment of APP/PS1/CB2R KO and APP/PS1/CB2R WT mice with Δ9-THC and cannabidiol (CBD) at a therapeutic dose are effective in rescuing the memory and learning impairments, Aβ42 contents in plaques, and astroglial reactivity (Aso et al. 2015), irrespective of the presence or lack of CB2Rs. Further, pre-treatment with CBD was found to suppress the expression of proteins involved in tau phosphorylation and Aβ production in Mesenchymal stem cells derived from gingiva (GMSCs) (Libro et al. 2016). In contrast, administration of Δ9-THC + CBD in APP/PS1/CB2 KO mice has no effect on the microglial reactivity or tau phosphorylation around Aβ plaques. These findings indicate that signaling pathways other than those associated with CB2R are responsible for the beneficial effects of Δ9-THC and CBD in APP/PS1 mice (Aso et al. 2016, Aso et al. 2015). Activation of CB1Rs or inhibition of endocannabinoid-degrading enzymes [FAAH, MAGL and alpha/beta-Hydrolase domain containing 6 (ABHD6)] was shown to enhance Aβ clearance across the blood-brain barrier through increased expression of the low-density lipoprotein receptor-related protein 1 (LRP1) (Shibata et al. 2000). LRP1 has been demonstrated to function in the brain-to-blood transport of Aβ (Shibata et al. 2000). Infact, LRP1 levels in the brain and plasma were elevated following activation of CB1Rs (Bachmeier et al. 2013). Together, these findings provide insight into the additional mechanisms by which cannabinoid-based strategies help to reduce Aβ deposition in the AD brain. Exposure to a low concentration of Δ9-THC reduced Aβ levels in N2a-variant Aβ protein precursor (AβPP) cells and inhibited Aβ aggregation by directly interacting with Aβ peptide (Cao et al. 2014). Δ9-THC competitively binds to the peripheral anionic site of acetylcholine esterase (AChE) and prevents AChE-induced Aβ-peptide aggregation (Eubanks et al. 2006). Δ9-THC attenuates Aβ accumulation in a human CNS cell line (MC65 cells) that is induced to express Aβ proteotoxicity that initiates inflammatory responses (Currais et al. 2016). It has been shown that prolonged oral administration of two different cannabinoid agonists (WIN 55,212-2 and JWH-133) rescues neuroinflammation, reduces Aβ levels and improves cognitive performance in Tg APP 2576 mice (Martin-Moreno et al. 2012).
A previous report showed that the activation of CB1R and CB2R upregulates PPARγ signaling and attenuates Aβ-induced neuroinflammation, neurodegeneration and spatial memory impairment in animals (Fakhfouri et al. 2012). In another study (Janefjord et al. 2014), 2-AG and CBD provided protection against Aβ-evoked loss of SH-SY5Y cell viability, whereas JWH-015, Δ9-THC, CBD, abnormal-cannabidiol (Abn-CBD) and O-1602 (GPR18/GPR55 ligands) all protected SH-SY5Y cells from BV-2 conditioned media activated via lipopolysaccharide (LPS, microglial activation). While cannabinoid ligands variably altered the morphology of Aβ fibrils and aggregates, there was no clear correlation between effects on Aβ morphology and neuroprotective actions. These findings indicate a neuroprotective activity of cannabinoid ligands via actions at microglial and neuronal cells (Janefjord et al. 2014). In another animal model of AD (APP/PS1) mice, deletion of CB2R reduced microglial activation and infiltration of macrophages. Furthermore, these mice expressed low levels of pro-inflammatory chemokines and cytokines in the brain, as well as decreased concentrations of soluble Aβ 40/42 (Schmole et al. 2015). Activation of CB2R with a lower dose of JWH-015 was shown to remove native Aβ from human tissue sections as well as to clear synthetic pathogenic Aβ peptide from a human macrophage cell line (THP-1) but not from U373MG astrocytoma cells. This effect was reversed by the selective CB2R antagonist SR144528 (Tolon et al. 2009). Further, a novel CB2R agonist (MDA7) provided protection against Aβ fibril-induced microglial and astrocyte activation, normalized CB2R expression, promoted Aβ clearance, attenuated synaptic plasticity deficits, learning and memory impairments (Wu et al. 2013).
CB2R activation has been reported to transform microglial cells from the M1 to the M2 phenotype (Orihuela et al. 2016) and to favor phagocytosis (Mecha et al. 2015). In an in vitro experiment, selective CB2 agonists prevented the Aβ-induced release of proinflammatory cytokines by decreasing the intracellular calcium concentration and enhanced phagocytosis in microglial cells (Martin-Moreno et al. 2011). CB2R activation by JWH-015 increased Aβ-induced astrocytic proliferation in cell culture (Esposito et al. 2007). Administration of the CB2R agonist JWH-133 has been shown to prevent the activation of microglial cells and the release of proinflammatory cytokines in the vicinity of Aβ deposits in APP transgenic mice (Aso et al. 2013). Treatment with 1-phenylisatin, selective modulator of CB2R, prevented streptozotocin and aluminum trichloride + d-galactose induced impairment of learning-memory and brain damage in mouse model of AD (Jayant et al. 2016). Inhibition of MAGL decreased the astroglial reaction to Aβ plaques in 5xFAD mice lacking CB2R [54]. In contrast, natural cannabinoids such as THC+CBD failed to reduce the Aβ plaques and rescue memory deficits in APP/PS1 mice lacking CB2R (Aso et al. 2016). The non-selective CBR agonist WIN-55,212-2 inhibited Aβ-induced tau hyperphosphorylation via CB1R but not CB2R in PC12 neuronal cells (Esposito et al. 2006). In another study, Δ9-THC reduced phosphorylated tau in N2a/APPswe cells (Cao et al. 2014). The CB2R agonist JWH-133 decreased tau phosphorylation at Thr181 and also decreased the expression of the active forms of GSK3β, p38 and SAPK/JNK in APP/PS1 mice (Aso et al. 2013). Further, the lack of CB2R failed to alter tau phosphorylation in mice (Aso et al. 2016). The lack of CB2R reduced total tau without any effect on tau phosphorylation in J20APP mice (Koppel et al. 2014). In other studies, JWH-133 decreased the levels of superoxide dismutase (SOD) 1 and 2 around plaques in APP/PS1 mice (Aso et al. 2013). In line with these findings, CBD prevented Aβ-induced ROS production, lipid peroxidation, caspase-3-mediated apoptosis and elevation of the intracellular calcium concentration in PC12 neuronal cells (Iuvone et al. 2004). In a mouse model of tauopathy exposed to Sativex, a mixture of Δ9-THC and CBD, both ROS levels and mitochondrial activity were decreased (Casarejos et al. 2013). In a published case report, treatment with an analog of Δ9-THC (nabilone or dronabinol) improved the behavior and food intake in AD patients after 6 weeks of treatment (Volicer et al. 1997). Similarly, the use of nabilone improved reduced agitation and aggressiveness exhibited by advanced AD patients (Passmore 2008). Upregulation of CB2R and its preferential distribution near Aβ plaques has been reported in several animal models of AD and in post mortem patients. However, in a study conducted by Ahmad R et al., 2016 it was found that in vivo availability of CB2R is decreased in AD patients compared to healthy individuals (Ahmad et al. 2016).
EC system in Huntington’s disease
Huntington’s disease (HD) is life-threatening and inherited progressive and neurodegenerative disorder caused by the extension of an unstable trinucleotide (cytosine-adenine-guanine, CAG) repeat in the Huntingtin gene (Htt) to more than 36 repeats, resulting in a polyglutamine (polyQ) expansion in the N-terminus of Huntingtin. HD is characterized by progressive motor abnormalities (chorea) and cognitive deterioration (dementia) accompanied by neuronal death in some basal ganglia structures, such as the striatum, globus pallidus, and SN, and to a lesser extent, the cerebral cortex. The mechanism of disease progression remains unknown, and there is currently no successful treatment available to prevent or slow the disease. Although considerable research suggests that there is apoptotic neuron loss in HD (Hickey & Chesselet 2003), the molecular mechanisms responsible for such a loss of neurons is unknown. Changes in protein aggregation (Arrasate & Finkbeiner 2012), mitochondrial function (Quintanilla & Johnson 2009), oxidative stress (Browne et al. 1999), and glial activation leading to inflammatory and cytotoxic events (Tai et al. 2007) have been suggested to play important roles in causing neuronal loss and disease onset. ECs, their synthesizing and degrading enzymes, and their receptors are particularly abundant in basal ganglia structures (Bisogno et al. 1999a, Herkenham et al. 1991, Mailleux & Vanderhaeghen 1992b). The abundance of the EC system in the basal ganglia suggests that the activation or inhibition of EC signaling system might have a significant influence on motor responses. Interestingly, most post-mortem studies of HD patients published to date have reported a massive loss of CB1R in various basal ganglia structures. A dramatic loss of CB1R immunoreactivity in the globus pallidus was observed in human HD brain (Richfield & Herkenham 1994).
Quantitative autoradiography of tissue sections from post-mortem HD brain showed a greater loss of CB1Rs in the lateral pallidum and SNr (Glass et al. 1993). Studies using various HD animal models have also shown similar patterns of decrease in CB1R expression (Bisogno et al. 2008, Dowie et al. 2009, Glass et al. 2000). For example, HD R6/1 mice showed reduced CB1R mRNA and CB1R ligand binding in the striatum (Dowie et al. 2009). In another study, a progressive decline in CB1R with significant decreases in AEA and 2-AG levels was observed in the striatum of transgenic HD R6/2 mice with 150 CAG repeats (Bisogno et al. 2008). Interestingly, HD mice showed loss of CB1R mRNA in the lateral striatum, cortex, and hippocampus in the early onset of disease suggesting that decrease in the CB1R mRNA is a hallmark in the initial phase of the HD disease (Denovan-Wright & Robertson 2000, McCaw et al. 2004). Also, it was found that CB1R protein and its mRNA levels decreased with the motor hyperkinesia in the caudate putamen of rats injected with 3-nitropropionic acid (3-NP) (Lastres-Becker et al. 2002). Also, 3-NP injected HD rats showed reduced CB1R binding, GTPγS binding, AEA and 2-AG in the SN but not in the cerebral cortex (Lastres-Becker et al. 2001b). However, investigations have shown significant decreases in the AEA and 2-AG levels in the striatum (Bisogno et al. 2008), a significant reduction in 2-AG levels and increase in AEA levels in the cortex with no significant difference in the hippocampus in the late phase of HD (Bisogno et al. 2008). These observations suggest that impairment of the EC system in HD is region specific as well as stage-specific. In contrast, another study showed that 2-AG was increased and AEA was decreased significantly in R6/1 mice (Dowie et al. 2009). Also, reductions in the activity of NAPE-PLD and DAGL as well as in cannabinoid binding with increased 2-AG levels were observed in the striatum of the R6/2 mouse model of HD (Bari et al. 2013). FAAH activity was dramatically decreased along with a 6-fold increase in AEA levels in the lymphocytes of HD patients (Battista et al. 2007). These findings clearly suggest that endocannabinoids are dysregulated in different brain regions of HD animal models.
It is very clear that cannabinoid signal becomes hypofunctional in the basal ganglia in HD. However, it has also been suggested that loss of CB1R may be initially related to the progressive and selective loss of medium spiny GABAergic neurons in the striatum, where these receptors are abundant (Rikani et al. 2014). Studies using R6/2 mice at presymptomatic ages showed impairment of the sensitivity of striatal GABA synapses to cannabinoid stimulation (Centonze et al. 2005). It has been shown that loss of CB1Rs function contributes to the progression of HD, the aggravation of HD symptoms and neuropathology in a mouse model that expresses human mutant Huntingtin exon 1 and lacks CB1R (Blazquez et al. 2011). It has also been shown that lack of CB1R in N171-82Q transgenic mice worsens motor function, increases mouse susceptibility to 3-NP, and contributes to the physiological development of HD (Mievis et al. 2011).A marked loss of CB1R immunoreactivity in the globus pallidus has been associated with the upregulation of GABA (A and B) receptor in human HD brain (Allen et al. 2009). It has also been shown that the severity of HD pathology was associated with increased numbers of progenitor cells with functional CB1Rs (Curtis et al. 2006). These findings suggest that CB1R agonists or inhibitors of EC inactivation have the potential therapeutic value in treating various forms of HD.
In late-onset HD, the loss of CB1R is accompanied by increased expression of CB2R, particularly in glial cells, during striatal degeneration (Fernandez-Ruiz et al. 2008, Fernandez-Ruiz et al. 2007). In a rat model of HD (intrastriatal injection of the mitochondrial complex II inhibitor malonate), the selective CB2R antagonist SR144528 protected striatal projection neurons from malonate-induced death through a mechanism involving glial cells (Sagredo et al. 2009). The CB2R expression has been shown to increase in striatal microglia in a transgenic mouse model of HD and also in patients (Palazuelos et al. 2009). Further, another study demonstrated that the genetic ablation of CB2R exacerbated HD and that administration of CB2R-selective agonists reduced striatal neurodegeneration through microglial activation (Palazuelos et al. 2009). Along with CB1R, adenosine A2A receptor (A2AR) plays an important role in the control of neuronal excitability. The A2AR-CB1R form heteromeric complexes and recent studies revealed that the complex becomes dysfunctional in caudate putamen of advanced HD patients (Moreno et al. 2017).
One study in HD rats demonstrated that Δ9-THC treatment provided neuroprotection against 3-NP-induced toxicity (Lastres-Becker et al. 2004). However, in the malonate-injected rat model of HD, Δ9-THC exacerbated malonate-induced striatal pathology (Lastres-Becker et al. 2004). Also, SR141716 has also been reported to exacerbate malonate-induced striatal pathology (Lastres-Becker et al. 2003). Consistent with this observation, another study suggests that Δ9-THC inhibited CB1R signaling by increasing β-arrestin expression and exacerbating neuronal loss in a cell culture model of HD. However, CBD treatment prevented the Δ9-THC-induced loss of CB1R and rescued neuronal loss (Laprairie et al. 2016). In another study, CBD completely reversed 3-NP-induced toxicity in HD rats, and this effect was not reversed by SR141716, suggesting that CBD effects are independent of CB1Rs (Sagredo et al. 2007). Similarly, WIN55,212-2 administration has a protective effect on the 3-NP-induced striatal neurotoxicity in rat model of HD which could be related to endocannabinoid signaling system (ECS) stimulation and induction of NMDA receptor hypofunction (Maya-Lopez et al. 2017). VCE-003.2, a cannabigerol quinone derivative, inhibited the upregulation of proinflammatory markers and improved antioxidant defenses in 3NP-injected mice brain (Diaz-Alonso et al. 2016). Administration of WIN-55,212-2 dose-dependently and in a CB1R-dependent manner decreased the extracellular glutamate and attenuated the striatal damage induced by quinolinic acid in rat experimental models of HD (Pintor et al. 2006). Administration of AEA to HD rats caused hypokinesia and decreased nigrostriatal dopaminergic activity, but this effect was mediated through activation of vanilloid-like receptor rather than CB1R (de Lago et al. 2004). Cannabis sativa and its components have been shown to provide protection against many movement disorders, such as tremor (Clifford 1983, Consroe et al. 1997, Gapen 1982), dystonia (Consroe et al. 1986, Sandyk et al. 1986), L-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesia (LID) in Parkinson’s disease (PD) (Sieradzan et al. 1998), and Gilles de la Tourette syndrome (TS) (Muller-Vahl et al. 1999a, Muller-Vahl et al. 1999b). However, in several clinical trials, cannabinoids failed to provide protection against HD. In a case report of a female HD patient with irritability, cannabis improved motor skills and cognitive behavior, and these improvements were maintained by treatment with nabilone (Curtis & Rickards 2006). In another pilot study with HD patients, treatment with nabilone improved motor skills, chorea, and cognition to moderate levels (Curtis et al. 2009). In a double-blind, randomized, placebo-controlled, cross-over pilot clinical trial, no significant differences on motor, cognitive, behavioral and functional scores were detected among HD patients treated with Sativex and placebo (Lopez-Sendon Moreno et al. 2016). Furthermore, in a controlled clinical trial, CBD failed to provide any symptomatic protection against chorea in 15 neuroleptic-free HD patients (Consroe et al. 1991). In contrast, in an uncontrolled clinical trial, administration of a single dose of nabilone in one patient with HD resulted in a marked increase in choreatic movements (Müller-Vahl et al. 1999). Taken these findings, it can be speculated that CBR antagonists might have potential therapeutic value in treating HD. Further mechanistic studies are warranted to investigate the function of the cannabinoid system in HD because there is substantial evidence that CB1Rs regulate motor function.
EC system in Parkinson’s disease
Parkinson’s disease (PD) is the second most common neurodegenerative disease and is characterized by the progressive loss of dopamine (DA)-containing neurons of the SN, leading to basal ganglia circuit deficits. The irreversible loss of the DA-mediated control of striatal function leads to bradykinesia, tremor, and rigidity in PD (Bartels & Leenders 2009, Branchi et al. 2010, Branchi et al. 2008, de Lau & Breteler 2006). The striatum is a major forebrain nucleus that interconnects cortical and thalamic circuits and acts as the input nucleus of the basal ganglia. Striatal projection neurons send information to the SN pars reticulata (direct pathway) or the lateral globus pallidus (indirect pathway). Deficits in the neural activity of these two pathways have been suggested to cause the motor deficits observed in PD. However, the differences in the cellular and synaptic mechanisms of these circuits are not well understood. PD progression has been shown to be associated with multiple molecular changes in the brain. For instance, inflammation through activation of microglia in the SN (McGeer et al. 1988) and an increase in TNF-α, IL-1β, IL-2, IL-4, and IL-6 (Gerhard et al. 2006, Ouchi et al. 2005, Taylor et al. 2013), have been suggested to be significant pathogenic factors in sporadic PD, in which they lead to loss of dopaminergic (DA) neurons in the SN and thus to the DA denervation in the striatum. Missense mutations in the α-synuclein (α-syn) gene have been shown to cause autosomal dominant familial PD (Kruger et al. 1998, Polymeropoulos et al. 1997, Zarranz et al. 2004), and it has been suggested that α-syn causes the microglia-mediated inflammatory response in PD (Aureli et al. 2014, Austin et al. 2006, Gao et al. 2008, Klegeris et al. 2008, Reynolds et al. 2008a, Reynolds et al. 2008b, Thomas et al. 2007, Zhang et al. 2005). Current treatments such as dopaminergic replacement therapy have been proposed to alleviate some of the symptoms of PD, but there are no existing therapies that can rescue any of the underlying pathological conditions of PD (Calne et al. 2005, Denora et al. 2012, Di Gioia et al. 2015, Trapani et al. 2011). Together, these findings suggest that both environmental and genetic factors may contribute to the initiation of neurodegeneration followed by neuroinflammation leading to PD pathology.
Several mechanisms have been shown to play significant roles in the selective DA neuron degeneration observed in PD, such as mitochondrial dysfunction, oxidative stress, and excitotoxicity. Enhanced EC tone in the globus pallidus has been shown to cause Parkinsonian symptomology (Maneuf et al. 1996). Although CB1Rs are not abundant in DA neurons of the SN, the alleged involvement of the ECs in DA neuron degeneration is apparent in many studies. A study demonstrated increased 2-AG in the globus pallidus of rats treated with reserpine (Di Marzo et al. 2000). EC signaling was also shown to contribute to the pathophysiology of parkinsonism and LID in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned, non-human primate models of PD (van der Stelt et al. 2005). The impairments in EC transmission may contribute to LID which may be alleviated by the activation of CB1Rs (Ferrer et al. 2003). However, enhanced AEA levels have also been shown in rat models of PD (Di Marzo et al. 2000, Gubellini et al. 2002). In the reserpine-treated rodent model of PD, impaired locomotion is associated with a sevenfold increase in the levels of the 2-AG in the globus pallidus, but no such change was observed in the other five brain regions analyzed (Di Marzo et al. 2000). In this model, stimulation of locomotion in the reserpine-treated rat with quinpirole (D2 dopamine receptor agonist) and R-(+/−)-3-allyl-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide (Cl-APB) (D1 dopamine receptor agonist) results in decreased levels of both AEA and 2-AG in the globus pallidus (Di Marzo et al. 2000). Co-administration of quinpirole and SR141716A (CB1R antagonist) rescued deficits in locomotion in the reserpine-treated rat (Di Marzo et al. 2000). However, reserpine treatment caused a decrease in CB1R mRNA expression in the rat striatum (Silverdale et al. 2001). Consistent with these data from animal models, higher levels of ECs were also found in the cerebrospinal fluid of PD patients (Pisani et al. 2005). Increased 2-AG levels were observed in a time- and region-specific manner following MPTP administration in mice (Mounsey et al. 2015). URB597, an inhibitor of FAAH, exhibited protective effects by inhibiting dopaminergic neuronal death, decreased microglial immunoreactivity and improving motor alterations in MPTP-lesioned mice (Celorrio et al. 2016, Viveros-Paredes et al. 2017). It has also been reported that CB1R binding and the activation of G proteins by cannabinoid agonists were significantly increased in the PD post-mortem basal ganglia brain region and that this increase could be rescued by chronic L-DOPA therapy (Lastres-Becker et al. 2001a). Enhanced levels of CB1Rs were also found in MPTP-treated marmosets, a primate PD model (Lastres-Becker et al. 2001a). Blockade of CB1Rs with lower doses of SR141716A partially prevented the hyperkinesia in a rat model of PD (Gonzalez et al. 2006). However, in post-mortem brain tissues derived from healthy subjects and PD patients, the level of CB1R mRNA was reduced in the caudate nucleus, anterior dorsal putamen and an outer segment of the globus pallidus but no changes were observed in the other brain areas examined (Hurley et al. 2003). Chronic L-DOPA treatment of 6-hydroxydopamine (6-OHDA)-lesioned rats significantly increased CB1R mRNA expression in the denervated striatum (Zeng et al. 1999). Also, enhanced levels of AEA in the striatum and decreased the activity of FAAH was reported in a rat model of PD (induced by unilateral nigral lesion with 6-OHDA). The frequency and amplitude of glutamate-induced spontaneous excitatory post-synaptic currents were augmented in striatal spiny neurons recorded from PD rats (Maccarrone et al. 2003). Chronic treatment of PD rats with L-DOPA rescued both deficits in the EC system and glutamatergic activity (Maccarrone et al. 2003). Blockade of CB1R by injections of SR141716A into the striatum, globus pallidus, and to a lesser extent, the subthalamic nucleus, reduced motor asymmetry in PD rats (El-Banoua et al. 2004). Administration of SR141716A in PD rats with very severe nigral degeneration was shown to exert antiparkinsonian effects (Fernandez-Espejo et al. 2005). Daily administrations of Δ9-THC or cannabidiol (CBD) for two weeks rescued the 6-OHDA-induced (PD model) reduction of dopamine contents and the decrease of tyrosine hydroxylase (TH) activity in the lesioned striatum, and those effects were accompanied by a decrease in TH-mRNA levels in the SN (Lastres-Becker et al. 2005). Increased density of CB1R in the SN was observed in a rat model of PD induced by an intracerebroventricular injection of 6-OHDA (Gonzalez et al. 2006). Also, a low dose of SR141716A attenuated hypokinesia associated with nigral cell death in this model (Gonzalez et al. 2006). Administration of the CB1R selective antagonist, CE (1-[7-(2-chlorophenyl)-8-(4-chlorophenyl)-2-methylpyrazolo[1,5-a]-[1,3,5]triazin-4-yl]-3-ethylaminoazetidine-3-carboxylic acid amide benzenesulfonate) to L-DOPA-primed MPTP-treated rhesus monkeys with moderate and severe parkinsonism increased responses to L-DOPA (Cao et al. 2007). In models of PD, EC-mediated long-term depression (LTD) is absent from the indirect pathway, but this could be rescued by a D2 receptor agonist or inhibitors of endocannabinoid degradation. Co-administration of these drugs in vivo reduces parkinsonian motor deficits. These observations suggest that EC-mediated depression of synapses in the indirect pathway plays a significant role in the control of movement (Kreitzer & Malenka 2007). In genetic models of PD (various mouse mutants generated by deletion of specific genes associated with the development of PD in humans [PARK1 (alpha-synuclein), PARK2 (parkin) or PARK6 (PINK1)], CB1Rs have been reported to exhibit a biphasic response, with losses at early stages, during which the dopaminergic dysfunction is possibly the major event, followed by upregulation at advanced stages, which are characterized by the presence of nigrostriatal pathology including neuronal death (Garcia-Arencibia et al. 2009). Acute injections of L-DOPA or rimonabant produced similar improvements in contralateral forepaw stepping in rats with unilateral 6-OHDA lesions, and their co-administration enhanced stepping more than either drug alone (Kelsey et al. 2009). Rats with 6-OHDA injected into the striatum exhibit emotional and cognitive alterations that were rescued by acute injection of SR141716A (Tadaiesky et al. 2010). Blockade of CB1R was shown to protect nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation (Chung et al. 2011). Rats with unilateral intrastriatal 6-OHDA lesions treated chronically with MSX-3/or rimonabant showed rescue of dopaminergic cell death and neuroinflammation in the SN pars compacta (SNc) (Cerri et al. 2014). In a primate model of PD, SR141716A failed to ameliorate the motor deficits of parkinsonism (Meschler et al. 2001). Together, these findings suggest a direct link between EC signaling and symptoms of Parkinson’s disease and suggest that modulation of the EC signaling system might prove useful in treating PD.
Several lines of research imply that inflammation plays a significant role in the neurodegenerative process, and because cannabinoids possess anti-inflammatory and neuroprotective actions, recent preclinical research provides much evidence suggesting that CB2Rs play a major role in limiting the inflammatory responses secondary to microglial activation. Pharmacological activation of microglial CB2Rs in the MPTP mouse model of PD led to reduced microglial activation and functional deficits (Price et al. 2009), reduced the release of pro-inflammatory cytokines (Ehrhart et al. 2005, Klegeris et al. 2003, Molina-Holgado et al. 2002b), and increased the levels of anti-inflammatory cytokines (Molina-Holgado et al. 2003). Consistent with these observations, CB2R knockout (KO) mice exhibited enhanced microglial activation and exacerbation of the PD pathology with neural alterations and functional deficits (Molina-Holgado et al. 2003). Similar changes have also been reported in other PD models, such as MPTP-lesioned and LPS-injected mice (Garcia et al. 2011, Gomez-Galvez et al. 2016, Price et al. 2009). Also, overexpression of CB2R provides protection against 6-OHDA-induced nigrostriatal lesions in mice (Ternianov et al. 2012). Activation of CB2R with selective agonists improved function in MPTP-lesioned mice (Price et al. 2009) and LPS-injected mice (Garcia et al. 2011) but not in 6-OHDA-lesioned rats (Garcia-Arencibia et al. 2007). For example, the selective CB2R agonist HU-308 rescued the LPS-induced decrease in TH-positive neurons. Increased CD68 immunostaining in the striatum (activated microglia and infiltrated peripheral macrophages) was also observed (Gomez-Galvez et al. 2016). Also, HU-308 significantly rescued striatal iNOS gene expression after LPS injection (Gomez-Galvez et al. 2016). Consistent with these observations, HU-308 rescued the loss of TH+ neurons in the SN of LPS-injected mice (Garcia et al. 2011). JWH-015, an agonist of CB2R, also reduced MPTP-induced microglial infiltration, and this effect was due specifically to CB2R activation, as it was rescued by the CB2R antagonist N-(1,3-Benzodioxol-5-ylmethyl)-1,2-dihydro-7-methoxy-2-oxo-8-(pentyloxy)-3-quinolinecarboxamide, JTE-907 (Price et al. 2009). Similarly, the CB2R agonist β-caryophyllene demonstrated neuroprotection by virtue of its anti-inflammatory and antioxidant properties (Javed et al. 2016). Studies using leucine-rich repeat kinase 2 (LRRK2)-transgenic mice also demonstrated CB2 receptor as a potential pharmacological target to treat PD (Palomo-Garo et al. 2016). In recent study, upregulation of the CB2R was shown in inflammation-driven PD rat model (Concannon et al. 2016). Although more studies are necessary, these findings suggest that CB2Rs function in the pathophysiology of PD. Use of CB2R selective agonists without the psychoactive effects may provide neuroprotection against the neurodegenerative processes of PD.
Conclusion
Several lines of evidence suggest a primary function of endocannabinoids, CBRs and other components of EC system in the degenerative process. Various investigators using a variety of preclinical models have discovered the therapeutic intervention strategies modulating EC system. Furthermore, current approaches to the development of novel therapeutic strategies for neurodegenerative diseases have focused not only on their neuroprotective properties but also on alleviating symptoms of the neurodegenerative diseases. These approaches are based on the well-characterized role of cannabinoids that have the ability to control both anti-inflammatory and neuroprotective functions. Therefore, the use of CB2R agonists and CB1R antagonists endeavor an interesting, unique and potential therapeutic advance for neurodegenerative disorders.
Table 1.
Changes in the endocannabinoid system in neurodegenerative disease.
| Changes in the EC system components in Alzheimer diseases | ||
|---|---|---|
|
| ||
| Study model | Changes in EC system | References |
| Preclinical Studies | ||
| AβPPswe/PS1ΔE9 model of AD | < Striatal AG level >CBR/effector coupling |
(Maroof et al. 2014) |
| TgAPP-2576 mice | <AEA in the hippocampus | (Kofalvi et al. 2016) |
| APPSwe/PS1dE9 Tg mice | >DAGLα, DAGLβ and 2-AG in hippocampus | (Mulder et al. 2011) |
| 3xTg-AD mice |
> CB1R activity in anterior thalamus at 4 months < CB1R activity in nucleus basalis of Meynert at 15 month |
(Manuel et al. 2016) |
| Clinical Studies | ||
| AD Patients | No difference in CB1R.in overall brain region | (Ahmad et al. 2014) |
| < CB1R expression in frontal cortex > CB2R expression in frontal cortex |
(Solas et al. 2013) | |
| < G-protein coupling and CB1R protein expression. >Nitrated CB1R and CB2R protein levels in whole brain |
(Ramirez et al. 2005) | |
| >FAAH protein, >CB2R in neuritic plaque-associated astrocytes and microglia, CB1R unchanged in whole brain. |
(Benito et al. 2003) | |
| Peripheral blood mononuclear cells (PBMCs) with late-onset AD Patients | >FAAH gene and protein and its activity | (D’Addario et al. 2012) |
| AD Patients. | <FAAH activity in cortex | (Pascual et al. 2014) |
| < DAGL α levels in hippocampus | (Farooqui et al. 1988) | |
| AD patients | CB1R unchanged | (Mulder et al. 2011) |
| AD patients | < in vivo availability of CB2R | (Ahmad et al. 2016) |
|
| ||
| Changes in the EC system components in Huntington diseases | ||
|
| ||
| Preclinical Studies | ||
| R6/1Tg mice | <CB1 mRNA in striatum <CB1R protein binding in basal ganglia >2AG in cortex <AEA in hippocampus |
(Dowie et al. 2009) |
| <2AG, AEA,PEA in striatum <2AG in hippocampus >AEA in cortex |
(Bisogno et al. 2008) | |
| HD mouse model | < CB1R mRNA in lateral striatum, cortex and hippocampus | (Denovan-Wright & Robertson 2000, McCaw et al. 2004) |
| R6/2 HD mice | >2AG < NAPE-PLD activity, < DAGL activity, < CBR binding in striatum | (Bari et al. 2013) |
| 3-NP injected HD rat model | < CB1R mRNA overall brain <CB1R binding in caudate putamen <CB1R in basal ganglia |
(Lastres-Becker et al. 2002) |
| <CB1R binding in basal ganglia <AEA,2AG in striatum >AEA in substantia nigra |
(Lastres-Becker et al. 2001b) | |
| Post mortem samples from high grade HD patients | < A2AR-CB1R heteromeric complexes in caudate putamen | (Moreno et al. 2017) |
| Clinical Studies | ||
| Peripheral lymphocytes from HD patients | < FAAH activity >AEA levels |
(Battista et al. 2007) |
| HD Patients | <CB1R immunoreactivity in globus pallidus | (Allen et al. 2009) |
| HD Patients | <CB1R in striatal nerve in lateral pallidum | (Richfield & Herkenham 1994) |
| <CB1R binding in substantia nigra and pars reticulate<CB1R in basal ganglia | (Glass et al. 1993) | |
|
| ||
| Changes in the EC system components in Parkinson’s diseases | ||
|
| ||
| Preclinical Studies | ||
| Reserpine treated rats | > 2-AG in globus pallidus Impaired locomotion > AEA in globus pallidus & substantia nigra |
(Di Marzo et al. 2000) |
| < CB1R mRNA expression in striatum | (Silverdale et al. 2001) | |
| MPTP-lesioned cynomolgus monkeys | > 2-AG in striatum & substantia nigra > AEA in striatum & globus pallidus |
(van der Stelt et al. 2005) |
| Rats treated with L-DOPA | > AEA in basal ganglia | (Ferrer et al. 2003) |
| 6-OHDA-lesioned rats | < AEA in caudate-putamen | (Ferrer et al. 2003) |
| > CB1Rs in substantia nigra | (Gonzalez et al. 2006) | |
| MPTP-lesioned mice | > 2-AG in substantia nigra > TH+ neurons |
(Mounsey et al. 2015) |
| MPTP-lesioned marmosets treated with L-DOPA | > CB1R binding in Caudate nucleus & putamen | (Lastres-Becker et al. 2001a) |
| 6-OHDA-lesioned rats treated with L-DOPA | > CB1R mRNA expression in Denervated striatum | (Zeng et al. 1999) |
| > AEA in striatum < FAAH activity |
(Maccarrone et al. 2003) | |
| Mouse astrocytes, co-incubated with LPS | < NO release < iNOS expression |
(Ehrhart et al. 2005, Klegeris et al. 2003, Molina-Holgado et al. 2002b) |
| PARK1, PARK2 and PARK6 mutant mice | < CB1R-mRNA in caudate-putamen, substantia nigra & globus pallidus < CB1R binding in early stages > CB1R-mRNA & CB1R binding in older age |
(Garcia-Arencibia et al. 2009) |
| MPTP-lesioned mice treated with WIN 55,212-2/JWH015 | < microglial activation & functional deficits < pro-inflammatory cytokines > anti-inflammatory cytokines |
(Ehrhart et al. 2005, Klegeris et al. 2003, Molina-Holgado et al. 2002b, Price et al. 2009, Molina-Holgado et al. 2003) |
| MPTP-lesioned CB2R KO mice | > microglial activation Exacerbation of PD pathology |
(Price et al. 2009) |
| LPS-lesioned mice | > CB2R in striatum & substantia nigra | (Garcia et al. 2011, Gomez-Galvez et al. 2016, Price et al. 2009) |
| LPS-lesioned CB2R KO mice | > CD68 immunostaining in striatum | (Garcia et al. 2011, Gomez-Galvez et al. 2016, Price et al. 2009) |
| Post-mortem brain tissue from PD patients receiving L-DOPA and direct acting dopamine agonists treatment | < CB1R mRNA in caudate nucleus, anterior dorsal putamen, external segment of globus pallidus | (Hurley et al. 2003) |
| LRRK2-transgenic mice | > motor deficits | (Palomo-Garo et al. 2016) |
| Inflammation-driven PD rat model | > CB2R expression | (Concannon et al. 2016) |
| Clinical Studies | ||
| Cerebrospinal fluid of PD patients | > ECs | (Pisani et al. 2005) |
| Brain tissues from PD patients | > CB2R in microglial cells | (Garcia et al. 2011, Gomez-Galvez et al. 2016, Price et al. 2009) |
Symbols used: <: decreased, >: increased
AD: Alzheimer diseases, HD: Huntington diseases, PD: Parkinson’s disease, Tg mice: Transgenic mice, CB1R: Cannabinoid receptor type 1, CB2R: Cannabinoid receptor type 2, 2-AG: 2-Arachidonoylglycerol, AEA: N-arachidonoyl ethanolamine, FAAH: Fatty acid amide hydrolase, NAPE-PLD: N-acyl phosphatidylethanolamine phospholipase D, DAGL: Diacylglycerol lipase, 3-NP: 3-nitropropionic acid, MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, L-DOPA: 3,4-Dihydroxy-L-phenylalanine, 6-OHDA: 6-hydroxydopamine, LPS: lipopolysaccharide, TH: tyrosine hydroxylase, PARK: genes associated with the development of Parkinson’s disease in humans [PARK1 (alpha-synuclein), PARK2 (parkin) or PARK6 (PINK1)], ECs: Endocannabinoids, NO: nitric oxide, iNOS: inducible NO synthase, ROS: reactive oxygen species, KO: knock out, A2AR: adenosine A2A receptor, LRRK2: leucine-rich repeat kinase 2.
Table 2.
EC system targeted pharmacological compounds treating neurodegenerative diseases.
| EC system targeted pharmacological compounds treating Alzheimer diseases | |||
|---|---|---|---|
|
| |||
| Study model | Drugs | Effects | References |
| Preclinical studies | |||
| Aβ injected rats | WIN-55, 212 22 | <Aβ induced microglial activation, <cognitive impairment. | (Ramirez et al. 2005) |
| Aβ treated microglial cells | HU-210 | ||
| Rat cortical co cultures | WIN-55, 212 22 JWH-133 |
Block Aβ induced activation, Rescued microglia-mediated neurotoxicity |
|
|
Astrocytes & hippocampal Slices treated with Aβ |
WIN, 2-AG, methanandamide |
< hemichannel activity & inflammatory Profile in astrocytes < excitotoxic glutamate release & neuronal damage in hippocampal slices |
(Gajardo-Gomez et al. 2017) |
| Tetra carcinoma cell lines, Ntera 2/cl-D1 neurons |
AEA, noladin ether | Protection against Aβ peptide-induced neurotoxicity | (Milton 2002) |
| Rodents treated with the Aβ- peptide (1–42) (BAP). | VDM-11, | > endocannabinoid levels reversed hippocampal damage in rats |
(van der Stelt et al. 2006) |
| Aβ- peptide (1–42) treated microglial cells | JWH-015 | Suppressed IFN-γ-induced CD40 expression, attenuated CD40-mediated inhibition of microglial phagocytosis of Aβ1–42 peptide | (Ehrhart et al. 2005) |
| Cultured hippocampal neurons | Exogenous 2-AG | Protection against Aβ peptide-induced neurodegeneration >2-AG, < ERK1/2 and NF-kB phosphorylation. |
(Chen et al. 2011) |
| URB602, JZL184 | < cox-2 expression thereby rescuing from neurodegeneration and apoptosis | ||
| C57BL/6J APP/PS1 mice exposed to NO2 |
JZL184 | Prevented prostaglandin production, <Aβ42 accumulation |
(Yan et al. 2016) |
| 5XFAD APP TG mice, | JZL184 | < BACE1, < Aβ production | (Chen et al. 2012) |
| 5XFAD TG mice Brains of AD humans |
JZL184, GW9662 | > MiR188-3 expression, < BACE1 and Aβ formation. Improved synaptic and cognition functions, LTP |
(Zhang et al. 2014) |
| AβPP/PS1 mice | Δ9 THC + CBD botanical extract | < Aβ42 content, Improved learning and memory |
(Aso et al. 2015) |
| Mesenchymal stem cells | CBD | < expression of AD-related genes | (Libro et al. 2016) |
| N2a/AβPPswe cells | Δ9 THC | <Aβ levels, >mitochondrial function | (Cao et al. 2014) |
| Human neuron cells, MC65 cell line |
Δ9 THC | Attenuates Aβ accumulation | (Currais et al. 2016) |
| TG APP2576 mice | WIN 55, 212 22, JWH-133 | Rescued neuroinflammation Reduced Aβ levels Improves cognitive performance |
(Martin-Moreno et al. 2012) |
| Aβ injected rats | WIN55212-2 | Upregulated PARP signaling Attenuate Aβ-induced neuroinflammation, neurodegeneration Improves spatial memory |
(Fakhfouri et al. 2012) |
| Aβ evoked Neuroblastoma (SH-SY5Y)cells | 2-AG, CBD | Direct neuroprotective against Aβ toxicity | (Janefjord et al. 2014) |
| BV-2 cells in conditioned media | JWH015, THC, CBD, O-1602 | Reduced Aβ fibrils and their aggregation | |
| Human macrophage (THP-1) cell line | JWH-015 | Cleared native Aβ peptide | (Tolon et al. 2009) |
| Aβ injected rats | Piperidine (MDA7) | Ameliorated CD11b and GFAP, < inter leukin-1β, <Aβ. Restored synaptic plasticity, cognition, and memory. |
(Wu et al. 2013) |
| Aβ(1-40) treated Rat cultured cells, N13 microglial cells, BV-2 cells |
CBD, WIN 55,212-2 | < cytokines gene expression, Modulated microglial cell function |
(Martin-Moreno et al. 2011) |
| Aβ(1-42) induced C6 rat glioma cells |
JWH-015 | >Aβ-induced astrocytic proliferation | (Esposito et al. 2007) |
| APP/PS1 mice | JWH133 | < GSK3β, p38 and SAPK/JNK, < phosphorylated tau at Thr181 site. < SOD (oxidative damage) around plaques >Cognitive performance |
(Aso et al. 2013) |
| Streptozotocin & aluminum trichloride + d-galactose induced mice | 1-phenylisatin |
Improved learning & memory < brain damage |
(Jayant et al. 2016) |
| PC12 neuronal cells | WIN55,212-2 | Inhibited Aβ-induce Tau hyperphosphorylation | (Esposito et al. 2006) |
| CBD | <ROS production <Lipid peroxidation <CC3 mediated apoptosis |
(Iuvone et al. 2004) | |
| Clinical Studies | |||
| AD patients | Δ9 THC | Prevents AChE-induced Aβ peptide aggregation | (Eubanks et al. 2006) |
| AD patients | Analog of THC (nabilone or dronabinol) | Improved behavior and food intake | (Volicer et al. 1997) |
| Nabilone | < Agitation and aggressiveness | (Passmore 2008) | |
|
| |||
| EC system targeted pharmacological compounds treating Huntington diseases | |||
|
| |||
| Preclinical studies | |||
| Malonate injected HD rats models |
SR144528 | < Neurodegeneration of striate projection neurons |
(Sagredo et al. 2009) |
| Tg HD mice | HU-308, Cannabinor, PRS-639, PRS-486 | < Striatal neurodegeneration | (Palazuelos et al. 2009) |
| 3-NP injected HD rats | Δ9 THC | Induced Neuroprotection | (Lastres-Becker et al. 2004) |
| 3-NP injected rats | WIN55,212-2 | Neuro-protection | (Maya-Lopez et al. 2017) |
| 3-NP injected mice | VCE-003.2 | Neuro-protection | (Diaz-Alonso et al. 2016) |
| Huntington expressing medium spiny mutant projection neurons (STHdh(Q111/Q111) | 2-AG AEA CP55,940 CBD |
Enhanced cell viability | (Laprairie et al. 2016) |
| Striatal neurons, 3NP injected HD rats |
CBD | Neuro-protection Reversed 3NP induced toxicity |
(Sagredo et al. 2007) |
| Quinolic acid exposed HD rats | WIN55,212-2 | < Extracellular glutamate Attenuated striatal damage |
(Pintor et al. 2006) |
| HD rats | AEA | > Hypokinesia < Nigrostriatal dopaminergic activity |
(de Lago et al. 2004) |
| Clinical Studies | |||
| Female HD patient | Cannabis Nabilone |
improved motor skills and cognitive behavior | (Curtis & Rickards 2006) |
| HD patients | Nabilone | Little changes in motor skill Chorea and cognition |
(Consroe et al. 1991) |
| HD patients | Nabilone | < Choreatic movements | (Müller-Vahl et al. 1999) |
|
| |||
| EC system targeted pharmacological compounds treating Parkinson’s diseases | |||
|
| |||
| Preclinical studies | |||
| Reserpine treated rats | Quinpirole | < AEA, < 2-AG in globus pallidus | (Di Marzo et al. 2000) |
| Cl-APB | < AEA, < 2-AG in globus pallidus | (Di Marzo et al. 2000) | |
| 6-OHDA-lesioned rats | SR141716A | Prevented hypokinesia at low dose | (Gonzalez et al. 2006) |
| SR141716A | < motor asymmetry | (El-Banoua et al. 2004, Fernandez-Espejo et al. 2005) | |
| SR141716A | Improved emotional & cognitive alterations | (Tadaiesky et al. 2010) | |
| SR141716A | > DA neuron survival in substantia nigra > astrocyte cell density |
(Cerri et al. 2014) | |
| AM251 | Antiparkinsonian effects | (Fernandez-Espejo et al. 2005) | |
| Δ9-THC | > dopamine content, > TH activity & > TH mRNA in striatum & substantia nigra | (Lastres-Becker et al. 2005) | |
| CBD | > dopamine content, > TH activity & > TH mRNA in striatum & substantia nigra | (Lastres-Becker et al. 2005) | |
| CBD | Neuroprotection when administered immediately after the lesion | (Garcia-Arencibia et al. 2007) | |
| MSX-3 | < dopaminergic cell death & neuro-inflammation in substantia nigra | (Cerri et al. 2014) | |
| Δ9-THCV | Improved motor function, > TH+ neurons | (Garcia et al. 2011) | |
| AM404 | Prevented DA depletion, > TH+ neurons, <parkinsonian motor asymmetries | (Garcia-Arencibia et al. 2007) | |
| L-DOPA primed MPTP-treated rhesus monkeys | CE | > antiparkinsonian effect | (Cao et al. 2007) |
| 6-OHDA-lesioned rats treated with L-DOPA | WIN 55,212-2 | < oro-lingual involuntary movements | (Ferrer et al. 2003) |
| SR141716A | Improved contralateral forepaw stepping | (Kelsey et al. 2009) | |
| MPTP-lesioned mice | WIN 55,212-2 | > survival of DA neurons in substantia nigra & striatum > dopamine levels < ROS production < pro-inflammatory cytokines expression Improved motor function |
(Chung et al. 2011) |
| WIN 55,212-2 | > TH+ neurons in substantia nigra & dorsal striatum > dopamine & 3,4-dihydroxyphenylacetic acid < microglial activation Improved motor function Neuroprotection is independent of CB1R |
(Price et al. 2009) | |
| HU210 | > survival of DA neurons in substantia nigra & striatum > dopamine levels < ROS production < pro-inflammatory cytokines expression Improved motor function |
(Chung et al. 2011) | |
| JWH015 | < microglial activation | (Price et al. 2009) | |
| MPTP-lesioned mice | URB597 |
< dopaminergic neuronal death < microglial immunoreactivity Improved motor functions |
(Celorrio et al. 2016, Viveros-Paredes et al. 2017) |
| MPTP-lesioned monkeys | SR141716A | Failed to relieve the motor deficits of parkinsonism | (Meschler et al. 2001) |
| MPTP-lesioned marmosets treated with L-DOPA | SR141716A | < LID | (van der Stelt et al. 2005) |
| Human microglia and THP-1 cells treated with LPS/IFN-γ | JWH015 | < neurotoxicity | (Ehrhart et al. 2005, Klegeris et al. 2003, Molina-Holgado et al. 2002b) |
| Mouse astrocytes co-incubated with LPS | SR141716A | > NO release | (Ehrhart et al. 2005, Klegeris et al. 2003, Molina-Holgado et al. 2002b) |
| HU210 | < NO release < iNOS expression |
(Ehrhart et al. 2005, Klegeris et al. 2003, Molina-Holgado et al. 2002b) | |
| CP-55940 | < NO release < iNOS expression |
(Ehrhart et al. 2005, Klegeris et al. 2003, Molina-Holgado et al. 2002b) | |
| Mouse glial cells co-incubated with LPS | SR141716A | < IL-1ra release | (Molina-Holgado et al. 2003) |
| SR144528 | < IL-1ra release | (Molina-Holgado et al. 2003) | |
| HU210 | > IL-1ra release | (Molina-Holgado et al. 2003) | |
| LPS-lesioned mice | HU-308 | < neurodegeneration > TH+ neurons < CD68 immunostaining < iNOS expression |
(Gomez-Galvez et al. 2016, Garcia et al. 2011) |
| Δ9-THCV | > TH+ neurons Prevented neurodegeneration |
(Garcia et al. 2011) | |
| Rotenone-lesioned rats | β-caryophyllene |
< IL-1β, IL-6, TNF-α, NF-κB, COX-2 & iNOS expression < lipid peroxidation > antioxidant enzyme activity > survival of DA neurons |
(Javed et al. 2016) |
| Post-mortem brain of humans affected by PD | WIN 55,212-2 | > CB1R binding | (Lastres-Becker et al. 2001a) |
Symbols used: <: decreased, >: increased
AD: Alzheimer diseases, Aβ: Beta amyloid, HD: Huntington diseases, Tg mice: Transgenic mice, 3-NP: 3-nitropropionic acid, SOD: Superoxide dismutase, ROS: Reactive oxygen species, GFAP: Glial fibrillary acidic protein, CC3: Cleaved Caspase-3, AChE: Acetylcholinesterase, CB1R: cannabinoid receptor type 1, CB2R: cannabinoid receptor type 2, MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, L-DOPA: 3,4-Dihydroxy-L-phenylalanine, 6-OHDA: 6-hydroxydopamine, LPS: lipopolysaccharide, WIN-55, 212 2: CB1R and CB2R agonist, HU-210: CB1R and CB2R agonist, JWH-133: selective CB2R agonist, AEA: N-arachidonoylethanolamine, VDM-11: potent cannabinoid reuptake inhibitor, JWH015: selective CB2R agonist, 2-AG: 2-Arachidonoylglycerol, MAGL: monoacylglycerol lipase, URB602: selective MAGL inhibitor, JZL184: irreversible MAGL inhibitor, GW9662: selective PPAR antagonist, O-1602: synthetic compound most closely related to abnormal cannabidiol, MDA7: selective CB2R agonist, SR144528: selective CB2R antagonist, HU308: Selective CB2R agonist, CP 55 940: synthetic cannabinoid, Quinpirole: D2 dopamine receptor agonist, Cl-APB: R-(+/−)-3-allyl-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide, D1 dopamine receptor agonist, SR141716A: CB1R antagonist, LID: levodopa-induced dyskinesia, NO: nitric oxide, IL-1ra: IL-1 receptor antagonist, DA: dopaminergic, TH: tyrosine hydroxylase, Δ9-THC: (-)-trans-Δ9-tetrahydrocannabinol, CB1R and CB2R agonist, CBD: Cannabidiol, CB1R and CB2R antagonist, CE: 1-[7-(2-chlorophenyl)-8-(4-chlorophenyl)-2-methylpyrazolo[1,5-a]-[1,3,5]triazin-4-yl]-3-ethylaminoazetidine-3-carboxylic acid amide benzenesulfonate, CB1R antagonist, MSX-3: 3,7-Dihydro-8-[(1E)-2-(3-Methoxyphenyl)ethenyl]-7-methyl-3-[3-(phosphonooxy)propyl-1-(2-propynyl)-1H-purine-2,6-dione disodium salt, selective adenosine/P2 nucleotide receptor antagonist, THP-1: human monocytic cell line, IFN-γ: interferon-γ, Δ9-THCV: Δ9-tetrahydrocannabivarin, CB1R and CB2R antagonist, AM251: CB1R antagonist, AM404: N-arachidonoylaminophenol, AEA transport inhibitor, URB597: fatty acid amide hydrolase inhibitor, β-caryophyllene: CB2R agonist, VCE-003.2: cannabigerol quinone derivative.
Acknowledgments
This study was partially supported by a grant from the National Institute of Alcohol and Alcoholism (RO1-AA019443) to B.S.B.
Abbreviations
- CB1R
cannabinoid receptor type 1
- CB2R
cannabinoid receptor type 2
- EC
endocannabinoid
- miRNAs
microRNAs
- GABAA
gamma-amino butyric acid-A receptor
- MAPK
mitogen-activated protein kinase
- Δ9-THC
delta-9-tetrahydrocannabinol
- AEA
arachidonylethanolamide (anandamide)
- 2-AG
2-arachidonylglycerol
- VR1
vanilloid receptor type 1
- NAPE-PLD
N-acylphosphatidylethanolamine-specific phospholipase D
- GDE1
glycerophosphodiesterase
- ABHD4
abhydrolase domain 4
- PTPN22
phosphatase
- AMT
AEA membrane transporter
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- PGH2
prostaglandin H2
- BDNF
brain-derived neurotrophic factor
- DAGL
diacylglycerollipase
- FAK
focal adhesion kinase
- AMPAR
a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- GPCR
G protein coupled receptor
- NAT
N-acyltransferase
- N-ArPE
N-arachidonyl phosphatidylethanolamine
- NMDA
N-methyl-D-aspartate
Footnotes
Involves human subjects: No
If yes: Informed consent & ethics approval achieved: yes => if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if No or if it is a Review or Editorial, skip complete sentence => if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
Conflicts of interest: None
=> if ‘none’, insert “The authors have no conflict of interest to declare.”
=> otherwise insert info unless it is already included
Conflict of interest disclosure
The authors declare no competing financial interests. All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Ahmad R, Goffin K, Van den Stock J, et al. In vivo type 1 cannabinoid receptor availability in Alzheimer’s disease. Eur Neuropsychopharmacol. 2014;24:242–250. doi: 10.1016/j.euroneuro.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Ahmad R, Postnov A, Bormans G, Versijpt J, Vandenbulcke M, Van Laere K. Decreased in vivo availability of the cannabinoid type 2 receptor in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2016;43:2219–2227. doi: 10.1007/s00259-016-3457-7. [DOI] [PubMed] [Google Scholar]
- Allen KL, Waldvogel HJ, Glass M, Faull RL. Cannabinoid (CB(1)), GABA(A) and GABA(B) receptor subunit changes in the globus pallidus in Huntington’s disease. J Chem Neuroanat. 2009;37:266–281. doi: 10.1016/j.jchemneu.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Finkbeiner S. Protein aggregates in Huntington’s disease. Exp Neurol. 2012;238:1–11. doi: 10.1016/j.expneurol.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso E, Andres-Benito P, Carmona M, Maldonado R, Ferrer I. Cannabinoid Receptor 2 Participates in Amyloid-beta Processing in a Mouse Model of Alzheimer’s Disease but Plays a Minor Role in the Therapeutic Properties of a Cannabis-Based Medicine. Journal of Alzheimer’s disease: JAD. 2016;51:489–500. doi: 10.3233/JAD-150913. [DOI] [PubMed] [Google Scholar]
- Aso E, Juves S, Maldonado R, Ferrer I. CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AbetaPP/PS1 mice. Journal of Alzheimer’s disease: JAD. 2013;35:847–858. doi: 10.3233/JAD-130137. [DOI] [PubMed] [Google Scholar]
- Aso E, Sanchez-Pla A, Vegas-Lozano E, Maldonado R, Ferrer I. Cannabis-based medicine reduces multiple pathological processes in AbetaPP/PS1 mice. Journal of Alzheimer’s disease: JAD. 2015;43:977–991. doi: 10.3233/JAD-141014. [DOI] [PubMed] [Google Scholar]
- Aureli C, Cassano T, Masci A, et al. 5-S-cysteinyldopamine neurotoxicity: Influence on the expression of alpha-synuclein and ERp57 in cellular and animal models of Parkinson’s disease. J Neurosci Res. 2014;92:347–358. doi: 10.1002/jnr.23318. [DOI] [PubMed] [Google Scholar]
- Austin SA, Floden AM, Murphy EJ, Combs CK. Alpha-synuclein expression modulates microglial activation phenotype. J Neurosci. 2006;26:10558–10563. doi: 10.1523/JNEUROSCI.1799-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmeier C, Beaulieu-Abdelahad D, Mullan M, Paris D. Role of the cannabinoid system in the transit of beta-amyloid across the blood-brain barrier. Mol Cell Neurosci. 2013;56:255–262. doi: 10.1016/j.mcn.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Bari M, Battista N, Valenza M, et al. In vitro and in vivo models of Huntington’s disease show alterations in the endocannabinoid system. The FEBS journal. 2013;280:3376–3388. doi: 10.1111/febs.12329. [DOI] [PubMed] [Google Scholar]
- Bartels AL, Leenders KL. Parkinson’s disease: the syndrome, the pathogenesis and pathophysiology. Cortex. 2009;45:915–921. doi: 10.1016/j.cortex.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS. Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling. Brain sciences. 2015;5:456–493. doi: 10.3390/brainsci5040456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS, Arancio O. Synaptic Plasticity: Emerging Role for Endocannabinoid system. In: Kaiser TF, Peters FJ, editors. Synaptic Plasticity: New Research. Nova Science Publishers, Inc; NY, USA: 2008. pp. 77–112. [Google Scholar]
- Basavarajappa BS, Saito M, Cooper TB, Hungund BL. Chronic ethanol inhibits the anandamide transport and increases extracellular anandamide levels in cerebellar granule neurons. Eur J Pharmacol. 2003;466:73–83. doi: 10.1016/s0014-2999(03)01557-7. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS, Subbanna S. CB1 Receptor-Mediated Signaling Underlies the Hippocampal Synaptic, Learning and Memory Deficits Following Treatment with JWH-081, a New Component of Spice/K2 Preparations. Hippocampus. 2014;24:178–188. doi: 10.1002/hipo.22213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battista N, Bari M, Tarditi A, et al. Severe deficiency of the fatty acid amide hydrolase (FAAH) activity segregates with the Huntington’s disease mutation in peripheral lymphocytes. Neurobiol Dis. 2007;27:108–116. doi: 10.1016/j.nbd.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Piomelli D. Carrier-mediated transport and enzymatic hydrolysis of the endogenous cannabinoid 2-arachidonylglycerol. Neuroreport. 2000;11:1231–1235. doi: 10.1097/00001756-200004270-00018. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- Benito C, Nunez E, Tolon RM, Carrier EJ, Rabano A, Hillard CJ, Romero J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J Neurosci. 2003;23:11136–11141. doi: 10.1523/JNEUROSCI.23-35-11136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, et al. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- Biegon A, Kerman IA. Autoradiographic study of pre- and postnatal distribution of cannabinoid receptors in human brain. NeuroImage. 2001;14:1463–1468. doi: 10.1006/nimg.2001.0939. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Berrendero F, Ambrosino G, Cebeira M, Ramos JA, Fernandez-Ruiz JJ, Di Marzo V. Brain regional distribution of endocannabinoids: implications for their biosynthesis and biological function. Biochem Biophys Res Commun. 1999a;256:377–380. doi: 10.1006/bbrc.1999.0254. [DOI] [PubMed] [Google Scholar]
- Bisogno T, MacCarrone M, De Petrocellis L, Jarrahian A, Finazzi-Agro A, Hillard C, Di Marzo V. The uptake by cells of 2-arachidonoylglycerol, an endogenous agonist of cannabinoid receptors. Eur J Biochem. 2001;268:1982–1989. doi: 10.1046/j.1432-1327.2001.02072.x. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Martire A, Petrosino S, Popoli P, Di Marzo V. Symptom-related changes of endocannabinoid and palmitoylethanolamide levels in brain areas of R6/2 mice, a transgenic model of Huntington’s disease. Neurochem Int. 2008;52:307–313. doi: 10.1016/j.neuint.2007.06.031. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Melck D, De Petrocellis L, Di Marzo V. Phosphatidic acid as the biosynthetic precursor of the endocannabinoid 2-arachidonoylglycerol in intact mouse neuroblastoma cells stimulated with ionomycin. Journal of Neurochemistry. 1999b;72:2113–2119. doi: 10.1046/j.1471-4159.1999.0722113.x. [DOI] [PubMed] [Google Scholar]
- Blazquez C, Chiarlone A, Sagredo O, et al. Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington’s disease. Brain. 2011;134:119–136. doi: 10.1093/brain/awq278. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Poinot-Chazel C, Marchand J, Canat X, Bourrie B, Rinaldi-Carmona M, Calandra B, Le Fur G, Casellas P. Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur J Biochem. 1996;237:704–711. doi: 10.1111/j.1432-1033.1996.0704p.x. [DOI] [PubMed] [Google Scholar]
- Branchi I, D’Andrea I, Armida M, et al. Striatal 6-OHDA lesion in mice: Investigating early neurochemical changes underlying Parkinson’s disease. Behav Brain Res. 2010;208:137–143. doi: 10.1016/j.bbr.2009.11.020. [DOI] [PubMed] [Google Scholar]
- Branchi I, D’Andrea I, Armida M, Cassano T, Pezzola A, Potenza RL, Morgese MG, Popoli P, Alleva E. Nonmotor symptoms in Parkinson’s disease: investigating early-phase onset of behavioral dysfunction in the 6-hydroxydopamine-lesioned rat model. J Neurosci Res. 2008;86:2050–2061. doi: 10.1002/jnr.21642. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR. Cannabinoid agonist signal transduction in rat brain: comparison of cannabinoid agonists in receptor binding, G-protein activation, and adenylyl cyclase inhibition. J Pharmacol Exp Ther. 2000;295:328–336. [PubMed] [Google Scholar]
- Brenneisen R. Chemistry and analysis of phytocannabinoids and other Cannabis constituents. In: ElSohly MA, editor. In Marijuana and the Cannabinoids. Humana Press; Totowa, NJ: 2007. pp. 17–49. [Google Scholar]
- Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington’s disease. Brain Pathol. 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NE, Hansson S, Harta G, Mezey E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience. 1998;82:1131–1149. doi: 10.1016/s0306-4522(97)00348-5. [DOI] [PubMed] [Google Scholar]
- Calne D, Schulzer M, Mak E, Stoessl AJ. Treatment for the progression of Parkinson’s disease. The Lancet Neurology. 2005;4:206. doi: 10.1016/S1474-4422(05)70026-0. [DOI] [PubMed] [Google Scholar]
- Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65:664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Li Y, Liu H, Bai G, Mayl J, Lin X, Sutherland K, Nabar N, Cai J. The potential therapeutic effects of THC on Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2014;42:973–984. doi: 10.3233/JAD-140093. [DOI] [PubMed] [Google Scholar]
- Cao X, Liang L, Hadcock JR, Iredale PA, Griffith DA, Menniti FS, Factor S, Greenamyre JT, Papa SM. Blockade of cannabinoid type 1 receptors augments the antiparkinsonian action of levodopa without affecting dyskinesias in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated rhesus monkeys. J Pharmacol Exp Ther. 2007;323:318–326. doi: 10.1124/jpet.107.125666. [DOI] [PubMed] [Google Scholar]
- Carnevale D, Perrotta M, Lembo G, Trimarco B. Pathophysiological Links Among Hypertension and Alzheimer’s Disease. High blood pressure & cardiovascular prevention: the official journal of the Italian Society of Hypertension. 2016;23:3–7. doi: 10.1007/s40292-015-0108-1. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Gironella M, Lorente M, Garcia S, Guzman M, Velasco G, Iovanna JL. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006a;66:6748–6755. doi: 10.1158/0008-5472.CAN-06-0169. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Lorente M, Egia A, et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell. 2006b;9:301–312. doi: 10.1016/j.ccr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipatikom K, Pfister SL, Campbell WB, Hillard CJ. Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol Pharmacol. 2004;65:999–1007. doi: 10.1124/mol.65.4.999. [DOI] [PubMed] [Google Scholar]
- Casarejos MJ, Perucho J, Gomez A, et al. Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. Journal of Alzheimer’s disease: JAD. 2013;35:525–539. doi: 10.3233/JAD-130050. [DOI] [PubMed] [Google Scholar]
- Caulfield MP, Brown DA. Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. Br J Pharmacol. 1992;106:231–232. doi: 10.1111/j.1476-5381.1992.tb14321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celorrio M, Fernandez-Suarez D, Rojo-Bustamante E, et al. Fatty acid amide hydrolase inhibition for the symptomatic relief of Parkinson’s disease. Brain Behav Immun. 2016;57:94–105. doi: 10.1016/j.bbi.2016.06.010. [DOI] [PubMed] [Google Scholar]
- Centonze D, Rossi S, Prosperetti C, Tscherter A, Bernardi G, Maccarrone M, Calabresi P. Abnormal sensitivity to cannabinoid receptor stimulation might contribute to altered gamma-aminobutyric acid transmission in the striatum of R6/2 Huntington’s disease mice. Biol Psychiatry. 2005;57:1583–1589. doi: 10.1016/j.biopsych.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Cerri S, Levandis G, Ambrosi G, et al. Neuroprotective potential of adenosine A2A and cannabinoid CB1 receptor antagonists in an animal model of Parkinson disease. J Neuropathol Exp Neurol. 2014;73:414–424. doi: 10.1097/NEN.0000000000000064. [DOI] [PubMed] [Google Scholar]
- Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang YP, Teng Z, Chen C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell reports. 2012;2:1329–1339. doi: 10.1016/j.celrep.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang J, Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons against beta-amyloid insults. Neuroscience. 2011;178:159–168. doi: 10.1016/j.neuroscience.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers SR, Sexton T, Roy MB. Effects of anandamide on cannabinoid receptors in rat brain membranes. Biochem Pharmacol. 1994;47:711–715. doi: 10.1016/0006-2952(94)90134-1. [DOI] [PubMed] [Google Scholar]
- Choi IY, Ju C, Anthony Jalin AM, Lee da I, Prather PL, Kim WK. Activation of cannabinoid CB2 receptor-mediated AMPK/CREB pathway reduces cerebral ischemic injury. Am J Pathol. 2013;182:928–939. doi: 10.1016/j.ajpath.2012.11.024. [DOI] [PubMed] [Google Scholar]
- Chung YC, Bok E, Huh SH, et al. Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J Immunol. 2011;187:6508–6517. doi: 10.4049/jimmunol.1102435. [DOI] [PubMed] [Google Scholar]
- Clifford DB. Tetrahydrocannabinol for tremor in multiple sclerosis. Ann Neurol. 1983;13:669–671. doi: 10.1002/ana.410130616. [DOI] [PubMed] [Google Scholar]
- Concannon RM, Okine BN, Finn DP, Dowd E. Upregulation of the cannabinoid CB2 receptor in environmental and viral inflammation-driven rat models of Parkinson’s disease. Exp Neurol. 2016;283:204–212. doi: 10.1016/j.expneurol.2016.06.014. [DOI] [PubMed] [Google Scholar]
- Consroe P, Laguna J, Allender J, Snider S, Stern L, Sandyk R, Kennedy K, Schram K. Controlled clinical trial of cannabidiol in Huntington’s disease. Pharmacol Biochem Behav. 1991;40:701–708. doi: 10.1016/0091-3057(91)90386-g. [DOI] [PubMed] [Google Scholar]
- Consroe P, Musty R, Rein J, Tillery W, Pertwee R. The perceived effects of smoked cannabis on patients with multiple sclerosis. Eur Neurol. 1997;38:44–48. doi: 10.1159/000112901. [DOI] [PubMed] [Google Scholar]
- Consroe P, Sandyk R, Snider SR. Open label evaluation of cannabidiol in dystonic movement disorders. Int J Neurosci. 1986;30:277–282. doi: 10.3109/00207458608985678. [DOI] [PubMed] [Google Scholar]
- Currais A, Quehenberger O, Armando AM, Daugherty D, Maher P, Schubert D. Amyloid proteotoxicity initiates an inflammatory response blocked by cannabinoids. npj Aging and Mechanisms of Disease. 2016;16012:1–8. doi: 10.1038/npjamd.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis A, Mitchell I, Patel S, Ives N, Rickards H. A pilot study using nabilone for symptomatic treatment in Huntington’s disease. Mov Disord. 2009;24:2254–2259. doi: 10.1002/mds.22809. [DOI] [PubMed] [Google Scholar]
- Curtis A, Rickards H. Nabilone could treat chorea and irritability in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2006;18:553–554. doi: 10.1176/jnp.2006.18.4.553. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Faull RL, Glass M. A novel population of progenitor cells expressing cannabinoid receptors in the subependymal layer of the adult normal and Huntington’s disease human brain. J Chem Neuroanat. 2006;31:210–215. doi: 10.1016/j.jchemneu.2006.01.005. [DOI] [PubMed] [Google Scholar]
- D’Addario C, Di Francesco A, Arosio B, et al. Epigenetic regulation of fatty acid amide hydrolase in Alzheimer disease. PloS one. 2012;7:e39186. doi: 10.1371/journal.pone.0039186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TA, Rakhshan F, Deutsch DG, Barker EL. Role of fatty acid amide hydrolase in the transport of the endogenous cannabinoid anandamide. Mol Pharmacol. 2001;59:1369–1375. doi: 10.1124/mol.59.6.1369. [DOI] [PubMed] [Google Scholar]
- de Lago E, de Miguel R, Lastres-Becker I, Ramos JA, Fernandez-Ruiz J. Involvement of vanilloid-like receptors in the effects of anandamide on motor behavior and nigrostriatal dopaminergic activity: in vivo and in vitro evidence. Brain Res. 2004;1007:152–159. doi: 10.1016/j.brainres.2004.02.016. [DOI] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. The Lancet Neurology. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- Denora N, Cassano T, Laquintana V, Lopalco A, Trapani A, Cimmino CS, Laconca L, Giuffrida A, Trapani G. Novel codrugs with GABAergic activity for dopamine delivery in the brain. Int J Pharm. 2012;437:221–231. doi: 10.1016/j.ijpharm.2012.08.023. [DOI] [PubMed] [Google Scholar]
- Denovan-Wright EM, Robertson HA. Cannabinoid receptor messenger RNA levels decrease in a subset of neurons of the lateral striatum, cortex and hippocampus of transgenic Huntington’s disease mice. Neuroscience. 2000;98:705–713. doi: 10.1016/s0306-4522(00)00157-3. [DOI] [PubMed] [Google Scholar]
- Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C, Trzaskos J, Caboche J, Girault JA. Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci. 2003;23:2371–2382. doi: 10.1523/JNEUROSCI.23-06-02371.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch DG, Glaser ST, Howell JM, Kunz JS, Puffenbarger RA, Hillard CJ, Abumrad N. The cellular uptake of anandamide is coupled to its breakdown by fatty-acid amide hydrolase. J Biol Chem. 2001;276:6967–6973. doi: 10.1074/jbc.M003161200. [DOI] [PubMed] [Google Scholar]
- Devane WA, Axelrod J. Enzymatic synthesis of anandamide, an endogenous ligand for the cannabinoid receptor, by brain membranes. Proc Natl Acad Sci USA. 1994;91:6698–6701. doi: 10.1073/pnas.91.14.6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey WL. Cannabinoid pharmacology. Pharmacol Rev. 1986;38:151–178. [PubMed] [Google Scholar]
- Di Gioia S, Trapani A, Mandracchia D, et al. Intranasal delivery of dopamine to the striatum using glycol chitosan/sulfobutylether-beta-cyclodextrin based nanoparticles. Eur J Pharm Biopharm. 2015;94:180–193. doi: 10.1016/j.ejpb.2015.05.019. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. ‘Endocannabinoids’ and other fatty acid derivatives with cannabimimetic properties: biochemistry and possible. Biochim Biophys Acta. 1998;1392:153–175. doi: 10.1016/s0005-2760(98)00042-3. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Hill MP, Bisogno T, Crossman AR, Brotchie JM. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. FASEB J. 2000;14:1432–1438. doi: 10.1096/fj.14.10.1432. [DOI] [PubMed] [Google Scholar]
- Diaz-Alonso J, Paraiso-Luna J, Navarrete C, et al. VCE-003.2, a novel cannabigerol derivative, enhances neuronal progenitor cell survival and alleviates symptomatology in murine models of Huntington’s disease. Scientific reports. 2016;6:29789. doi: 10.1038/srep29789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowie MJ, Bradshaw HB, Howard ML, Nicholson LF, Faull RL, Hannan AJ, Glass M. Altered CB1 receptor and endocannabinoid levels precede motor symptom onset in a transgenic mouse model of Huntington’s disease. Neuroscience. 2009;163:456–465. doi: 10.1016/j.neuroscience.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, et al. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. Journal of neuroinflammation. 2005;2:29. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Banoua F, Caraballo I, Flores JA, Galan-Rodriguez B, Fernandez-Espejo E. Effects on turning of microinjections into basal ganglia of D(1) and D(2) dopamine receptors agonists and the cannabinoid CB(1) antagonist SR141716A in a rat Parkinson’s model. Neurobiol Dis. 2004;16:377–385. doi: 10.1016/j.nbd.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Esposito G, De Filippis D, Steardo L, Scuderi C, Savani C, Cuomo V, Iuvone T. CB1 receptor selective activation inhibits beta-amyloid-induced iNOS protein expression in C6 cells and subsequently blunts tau protein hyperphosphorylation in co-cultured neurons. Neurosci Lett. 2006;404:342–346. doi: 10.1016/j.neulet.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Esposito G, Iuvone T, Savani C, Scuderi C, De Filippis D, Papa M, Di Marzo V, Steardo L. Opposing control of cannabinoid receptor stimulation on amyloid-beta-induced reactive gliosis: in vitro and in vivo evidence. J Pharmacol Exp Ther. 2007;322:1144–1152. doi: 10.1124/jpet.107.121566. [DOI] [PubMed] [Google Scholar]
- Eubanks LM, Rogers CJ, Beuscher AE, IV, Koob GF, Olson AJ, Dickerson TJ, Janda KD. A Molecular link between the active component of Marijuana and Alzheimer’s Disease pathology. Molecula Pharmaceutics. 2006;3:773–777. doi: 10.1021/mp060066m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhfouri G, Ahmadiani A, Rahimian R, Grolla AA, Moradi F, Haeri A. WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-gamma pathway. Neuropharmacology. 2012;63:653–666. doi: 10.1016/j.neuropharm.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Liss L, Horrocks LA. Neurochemical aspects of Alzheimer’s disease: involvement of membrane phospholipids. Metab Brain Dis. 1988;3:19–35. doi: 10.1007/BF01001351. [DOI] [PubMed] [Google Scholar]
- Fernandez-Espejo E, Caraballo I, de Fonseca FR, El Banoua F, Ferrer B, Flores JA, Galan-Rodriguez B. Cannabinoid CB1 antagonists possess antiparkinsonian efficacy only in rats with very severe nigral lesion in experimental parkinsonism. Neurobiol Dis. 2005;18:591–601. doi: 10.1016/j.nbd.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ruiz J, Pazos MR, Garcia-Arencibia M, Sagredo O, Ramos JA. Role of CB2 receptors in neuroprotective effects of cannabinoids. Mol Cell Endocrinol. 2008;286:S91–96. doi: 10.1016/j.mce.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ruiz J, Romero J, Velasco G, Tolon RM, Ramos JA, Guzman M. Cannabinoid CB2 receptor: a new target for controlling neural cell survival? Trends Pharmacol Sci. 2007;28:39–45. doi: 10.1016/j.tips.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Ferrer B, Asbrock N, Kathuria S, Piomelli D, Giuffrida A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: implications for the treatment of levodopa-induced dyskinesias. Eur J Neurosci. 2003;18:1607–1614. doi: 10.1046/j.1460-9568.2003.02896.x. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Gajardo-Gomez R, Labra VC, Maturana CJ, Shoji KF, Santibanez CA, Saez JC, Giaume C, Orellana JA. Cannabinoids prevent the amyloid beta-induced activation of astroglial hemichannels: A neuroprotective mechanism. Glia. 2017;65:122–137. doi: 10.1002/glia.23080. [DOI] [PubMed] [Google Scholar]
- Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–7698. doi: 10.1523/JNEUROSCI.0143-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gapen P. THC or analogues may alleviate some tremors. JAMA. 1982;248:2215. [PubMed] [Google Scholar]
- Garcia-Arencibia M, Garcia C, Kurz A, Rodriguez-Navarro JA, Gispert-Sachez S, Mena MA, Auburger G, de Yebenes JG, Fernandez-Ruiz J. Cannabinoid CB1 receptors are early downregulated followed by a further upregulation in the basal ganglia of mice with deletion of specific park genes. J Neural Transm Suppl. 2009:269–275. doi: 10.1007/978-3-211-92660-4_22. [DOI] [PubMed] [Google Scholar]
- Garcia-Arencibia M, Gonzalez S, de Lago E, Ramos JA, Mechoulam R, Fernandez-Ruiz J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007;1134:162–170. doi: 10.1016/j.brainres.2006.11.063. [DOI] [PubMed] [Google Scholar]
- Garcia-Gutierrez MS, Ortega-Alvaro A, Busquets-Garcia A, Perez-Ortiz JM, Caltana L, Ricatti MJ, Brusco A, Maldonado R, Manzanares J. Synaptic plasticity alterations associated with memory impairment induced by deletion of CB2 cannabinoid receptors. Neuropharmacology. 2013;73:388–396. doi: 10.1016/j.neuropharm.2013.05.034. [DOI] [PubMed] [Google Scholar]
- Garcia C, Palomo-Garo C, Garcia-Arencibia M, Ramos J, Pertwee R, Fernandez-Ruiz J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Delta(9)-THCV in animal models of Parkinson’s disease. Br J Pharmacol. 2011;163:1495–1506. doi: 10.1111/j.1476-5381.2011.01278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Gertsch J, Schoop R, Kuenzle U, Suter A. Echinacea alkylamides modulate TNF-alpha gene expression via cannabinoid receptor CB2 and multiple signal transduction pathways. FEBS Lett. 2004;577:563–569. doi: 10.1016/j.febslet.2004.10.064. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Beltramo M, Piomelli D. Mechanisms of endocannabinoid inactivation: biochemistry and pharmacology. J Pharmacol Exp Ther. 2001;298:7–14. [PubMed] [Google Scholar]
- Glaser ST, Abumrad NA, Fatade F, Kaczocha M, Studholme KM, Deutsch DG. Evidence against the presence of an anandamide transporter. Proc Natl Acad Sci U S A. 2003;100:4269–4274. doi: 10.1073/pnas.0730816100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RL. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77:299–318. doi: 10.1016/s0306-4522(96)00428-9. [DOI] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RL. The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience. 2000;97:505–519. doi: 10.1016/s0306-4522(00)00008-7. [DOI] [PubMed] [Google Scholar]
- Glass M, Faull RL, Dragunow M. Loss of cannabinoid receptors in the substantia nigra in Huntington’s disease. Neuroscience. 1993;56:523–527. doi: 10.1016/0306-4522(93)90352-g. [DOI] [PubMed] [Google Scholar]
- Gomez-Galvez Y, Palomo-Garo C, Fernandez-Ruiz J, Garcia C. Potential of the cannabinoid CB(2) receptor as a pharmacological target against inflammation in Parkinson’s disease. Prog Neuropsychopharmacol Bol Psychiatry. 2016;64:200–208. doi: 10.1016/j.pnpbp.2015.03.017. [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Scorticati C, Garcia-Arencibia M, de Miguel R, Ramos JA, Fernandez-Ruiz J. Effects of rimonabant, a selective cannabinoid CB1 receptor antagonist, in a rat model of Parkinson’s disease. Brain Res. 2006:1073–1074. doi: 10.1016/j.brainres.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Graham ES, Ball N, Scotter EL, Narayan P, Dragunow M, Glass M. Induction of Krox-24 by endogenous cannabinoid type 1 receptors in Neuro2A cells is mediated by the MEK-ERK MAPK pathway and is suppressed by the phosphatidylinositol 3-kinase pathway. J Biol Chem. 2006;281:29085–29095. doi: 10.1074/jbc.M602516200. [DOI] [PubMed] [Google Scholar]
- Grammas P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. Journal of neuroinflammation. 2011;8:26. doi: 10.1186/1742-2094-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, Bernardi G, Finazzi-Agro A, Maccarrone M. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci. 2002;22:6900–6907. doi: 10.1523/JNEUROSCI.22-16-06900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E, Royea J, Ongali B, Tong XK. Neurovascular and Cognitive failure in Alzheimer’s Disease: Benefits of Cardiovascular Therapy. Cell Mol Neurobiol. 2016;36:219–232. doi: 10.1007/s10571-015-0285-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen HH, Hansen SH, Schousboe A, Hansen HS. Determination of the phospholipid precursor of anandamide and other N-acylethanolamine phospholipids before and after sodium azide-induced toxicity in cultured neocortical neurons. J Neurochem. 2000;75:861–871. doi: 10.1046/j.1471-4159.2000.0750861.x. [DOI] [PubMed] [Google Scholar]
- Hardy J. Molecular genetics of Alzheimer’s disease. Acta Neurol Scand Suppl. 1996a;165:13–17. doi: 10.1111/j.1600-0404.1996.tb05867.x. [DOI] [PubMed] [Google Scholar]
- Hardy J. New insights into the genetics of Alzheimer’s disease. Ann Med. 1996b;28:255–258. doi: 10.3109/07853899609033127. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harkany T, Mackie K, Doherty P. Wiring and firing neuronal networks: endocannabinoids take center stage. Curr Opin Neurobiol. 2008;18:338–345. doi: 10.1016/j.conb.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267–274. doi: 10.1016/0006-8993(91)90970-7. [DOI] [PubMed] [Google Scholar]
- Herrera B, Carracedo A, Diez-Zaera M, Guzman M, Velasco G. p38 MAPK is involved in CB2 receptor-induced apoptosis of human leukaemia cells. FEBS Lett. 2005;579:5084–5088. doi: 10.1016/j.febslet.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Chesselet MF. Apoptosis in Huntington’s disease. Prog Neuropsychopharmacol Bol Psychiatry. 2003;27:255–265. doi: 10.1016/S0278-5846(03)00021-6. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem. 1997;69:631–638. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A. The movement of N-arachidonoylethanolamine (anandamide) across cellular membranes. Chem Phys Lipids. 2000;108:123–134. doi: 10.1016/s0009-3084(00)00191-2. [DOI] [PubMed] [Google Scholar]
- Hollister LE. Health aspects of cannabis. Pharmacol Rev. 1986;38:1–20. [PubMed] [Google Scholar]
- Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- Howlett AC. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002:68–69. doi: 10.1016/s0090-6980(02)00060-6. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Bidaut-Russell M, Devane WA, Melvin LS, Johnson MR, Herkenham M. The cannabinoid receptor: biochemical, anatomical and behavioral characterization. Trends Neurosci. 1990;13:420–423. doi: 10.1016/0166-2236(90)90124-s. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Mukhopadhyay S. Cellular signal transduction by anandamide and 2-arachidonoylglycerol. Chem Phys Lipids. 2000;108:53–70. doi: 10.1016/s0009-3084(00)00187-0. [DOI] [PubMed] [Google Scholar]
- Hurley MJ, Mash DC, Jenner P. Expression of cannabinoid CB1 receptor mRNA in basal ganglia of normal and parkinsonian human brain. J Neural Transm. 2003;110:1279–1288. doi: 10.1007/s00702-003-0033-7. [DOI] [PubMed] [Google Scholar]
- Iuvone T, Esposito G, Esposito R, Santamaria R, Di Rosa M, Izzo AA. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J Neurochem. 2004;89:134–141. doi: 10.1111/j.1471-4159.2003.02327.x. [DOI] [PubMed] [Google Scholar]
- Janefjord E, Maag JL, Harvey BS, Smid SD. Cannabinoid effects on beta amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell Mol Neurobiol. 2014;34:31–42. doi: 10.1007/s10571-013-9984-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javed H, Azimullah S, Haque ME, Ojha SK. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Frontiers in neuroscience. 2016;10:321. doi: 10.3389/fnins.2016.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayant S, Sharma BM, Bansal R, Sharma B. Pharmacological benefits of selective modulation of cannabinoid receptor type 2 (CB2) in experimental Alzheimer’s disease. Pharmacol Biochem Behav. 2016;140:39–50. doi: 10.1016/j.pbb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Urban GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsey JE, Harris O, Cassin J. The CB(1) antagonist rimonabant is adjunctively therapeutic as well as monotherapeutic in an animal model of Parkinson’s disease. Behav Brain Res. 2009;203:304–307. doi: 10.1016/j.bbr.2009.04.035. [DOI] [PubMed] [Google Scholar]
- Klegeris A, Bissonnette CJ, McGeer PL. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br J Pharmacol. 2003;139:775–786. doi: 10.1038/sj.bjp.0705304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klegeris A, Pelech S, Giasson BI, Maguire J, Zhang H, McGeer EG, McGeer PL. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol Aging. 2008;29:739–752. doi: 10.1016/j.neurobiolaging.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Kofalvi A, Lemos C, Martin-Moreno AM, et al. Stimulation of brain glucose uptake by cannabinoid CB2 receptors and its therapeutic potential in Alzheimer’s disease. Neuropharmacology. 2016;110:519–529. doi: 10.1016/j.neuropharm.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Koppel J, Vingtdeux V, Marambaud P, d’Abramo C, Jimenez H, Stauber M, Friedman R, Davies P. CB2 receptor deficiency increases amyloid pathology and alters tau processing in a transgenic mouse model of Alzheimer’s disease. Mol Med. 2014;20:29–36. doi: 10.2119/molmed.2013.00140.revised. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME, Denovan-Wright EM. Biased Type 1 Cannabinoid Receptor Signaling Influences Neuronal Viability in a Cell Culture Model of Huntington Disease. Mol Pharmacol. 2016;89:364–375. doi: 10.1124/mol.115.101980. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Bizat N, Boyer F, Hantraye P, Brouillet E, Fernandez-Ruiz J. Effects of cannabinoids in the rat model of Huntington’s disease generated by an intrastriatal injection of malonate. Neuroreport. 2003;14:813–816. doi: 10.1097/00001756-200305060-00007. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Bizat N, Boyer F, Hantraye P, Fernandez-Ruiz J, Brouillet E. Potential involvement of cannabinoid receptors in 3-nitropropionic acid toxicity in vivo. Neuroreport. 2004;15:2375–2379. doi: 10.1097/00001756-200410250-00015. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Cebeira M, de Ceballos ML, Zeng BY, Jenner P, Ramos JA, Fernandez-Ruiz JJ. Increased cannabinoid CB1 receptor binding and activation of GTP-binding proteins in the basal ganglia of patients with Parkinson’s syndrome and of MPTP-treated marmosets. Eur J Neurosci. 2001a;14:1827–1832. doi: 10.1046/j.0953-816x.2001.01812.x. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Fezza F, Cebeira M, Bisogno T, Ramos JA, Milone A, Fernandez-Ruiz J, Di Marzo V. Changes in endocannabinoid transmission in the basal ganglia in a rat model of Huntington’s disease. Neuroreport. 2001b;12:2125–2129. doi: 10.1097/00001756-200107200-00017. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Hansen HH, Berrendero F, De Miguel R, Perez-Rosado A, Manzanares J, Ramos JA, Fernandez-Ruiz J. Alleviation of motor hyperactivity and neurochemical deficits by endocannabinoid uptake inhibition in a rat model of Huntington’s disease. Synapse. 2002;44:23–35. doi: 10.1002/syn.10054. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Molina-Holgado F, Ramos JA, Mechoulam R, Fernandez-Ruiz J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson’s disease. Neurobiol Dis. 2005;19:96–107. doi: 10.1016/j.nbd.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry (Mosc) 2006;45:4720–4726. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kim J. CB2 Cannabinoid Receptor Knockout in Mice Impairs Contextual Long-Term Memory and Enhances Spatial Working Memory. Neural Plast. 2016a;2016:1–14. doi: 10.1155/2016/9817089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kim J. Deletion of CB2 cannabinoid receptors reduces synaptic transmission and long-term potentiation in the mouse hippocampus. Hippocampus. 2016b;26:275–281. doi: 10.1002/hipo.22558. [DOI] [PubMed] [Google Scholar]
- Libro R, Diomede F, Scionti D, Piattelli A, Grassi G, Pollastro F, Bramanti P, Mazzon E, Trubiani O. Cannabidiol Modulates the Expression of Alzheimer’s Disease-Related Genes in Mesenchymal Stem Cells. International journal of molecular sciences. 2016;18:1–19. doi: 10.3390/ijms18010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology. 2008;54:1–7. doi: 10.1016/j.neuropharm.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, et al. A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sendon Moreno JL, Garcia Caldentey J, Trigo Cubillo P, et al. A double-blind, randomized, cross-over, placebo-controlled, pilot trial with Sativex in Huntington’s disease. J Neurol. 2016;263:1390–1400. doi: 10.1007/s00415-016-8145-9. [DOI] [PubMed] [Google Scholar]
- Lu Y, Anderson HD. Cannabinoid signaling in health and disease. Can J Physiol Pharmacol. 2017;95:311–327. doi: 10.1139/cjpp-2016-0346. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Gubellini P, Bari M, Picconi B, Battista N, Centonze D, Bernardi G, Finazzi-Agro A, Calabresi P. Levodopa treatment reverses endocannabinoid system abnormalities in experimental parkinsonism. J Neurochem. 2003;85:1018–1025. doi: 10.1046/j.1471-4159.2003.01759.x. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, van der Stelt M, Rossi A, Veldink GA, Vliegenthart JF, Agro AF. Anandamide hydrolysis by human cells in culture and brain. J Biol Chem. 1998;273:32332–32339. doi: 10.1074/jbc.273.48.32332. [DOI] [PubMed] [Google Scholar]
- Mackie K. Mechanisms of CB1 receptor signaling: endocannabinoid modulation of synaptic strength. Int J Obes (Lond) 2006;30(Suppl 1):S19–23. doi: 10.1038/sj.ijo.0803273. [DOI] [PubMed] [Google Scholar]
- Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailleux P, Vanderhaeghen JJ. Age-related loss of cannabinoid receptor binding sites and mRNA in the rat striatum. Neurosci Lett. 1992a;147:179–181. doi: 10.1016/0304-3940(92)90589-y. [DOI] [PubMed] [Google Scholar]
- Mailleux P, Vanderhaeghen JJ. Distribution of neuronal cannabinoid receptor in the adult rat brain: a comparative receptor binding radioautography and in situ hybridization histochemistry. Neurosci. 1992b;48:655–668. doi: 10.1016/0306-4522(92)90409-u. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Nash JE, Crossman AR, Brotchie JM. Activation of the cannabinoid receptor by delta 9-tetrahydrocannabinol reduces gamma-aminobutyric acid uptake in the globus pallidus. Eur J Pharmacol. 1996;308:161–164. doi: 10.1016/0014-2999(96)00326-3. [DOI] [PubMed] [Google Scholar]
- Manuel I, Lombardero L, LaFerla FM, Gimenez-Llort L, Rodriguez-Puertas R. Activity of muscarinic, galanin and cannabinoid receptors in the prodromal and advanced stages in the triple transgenic mice model of Alzheimer’s disease. Neuroscience. 2016;329:284–293. doi: 10.1016/j.neuroscience.2016.05.012. [DOI] [PubMed] [Google Scholar]
- Maroof N, Ravipati S, Pardon MC, Barrett DA, Kendall DA. Reductions in endocannabinoid levels and enhanced coupling of cannabinoid receptors in the striatum are accompanied by cognitive impairments in the AbetaPPswe/PS1DeltaE9 mouse model of Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2014;42:227–245. doi: 10.3233/JAD-131961. [DOI] [PubMed] [Google Scholar]
- Martin-Moreno AM, Brera B, Spuch C, et al. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in Tg APP 2576 mice. Journal of neuroinflammation. 2012;9:8. doi: 10.1186/1742-2094-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Moreno AM, Reigada D, Ramirez BG, Mechoulam R, Innamorato N, Cuadrado A, de Ceballos ML. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer’s disease. Mol Pharmacol. 2011;79:964–973. doi: 10.1124/mol.111.071290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Lopez M, Colin-Gonzalez AL, Aguilera G, et al. Neuroprotective effect of WIN55,212–2 against 3-nitropropionic acid-induced toxicity in the rat brain: involvement of CB1 and NMDA receptors. American journal of translational research. 2017;9:261–274. [PMC free article] [PubMed] [Google Scholar]
- Mazzola C, Micale V, Drago F. Amnesia induced by beta-amyloid fragments is counteracted by cannabinoid CB1 receptor blockade. Eur J Pharmacol. 2003;477:219–225. doi: 10.1016/j.ejphar.2003.08.026. [DOI] [PubMed] [Google Scholar]
- McCaw EA, Hu H, Gomez GT, Hebb AL, Kelly ME, Denovan-Wright EM. Structure, expression and regulation of the cannabinoid receptor gene (CB1) in Huntington’s disease transgenic mice. Eur J Biochem. 2004;271:4909–4920. doi: 10.1111/j.1432-1033.2004.04460.x. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Mecha M, Feliu A, Carrillo-Salinas FJ, Rueda-Zubiaurre A, Ortega-Gutierrez S, de Sola RG, Guaza C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav Immun. 2015;49:233–245. doi: 10.1016/j.bbi.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Psychol. 2013;64:21–47. doi: 10.1146/annurev-psych-113011-143739. [DOI] [PubMed] [Google Scholar]
- Meschler JP, Howlett AC, Madras BK. Cannabinoid receptor agonist and antagonist effects on motor function in normal and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP)-treated non-human primates. Psychopharmacology (Berl) 2001;156:79–85. doi: 10.1007/s002130100728. [DOI] [PubMed] [Google Scholar]
- Micale V, Cristino L, Tamburella A, Petrosino S, Leggio GM, Di Marzo V, Drago F. Enhanced cognitive performance of dopamine D3 receptor “knock-out” mice in the step-through passive-avoidance test: assessing the role of the endocannabinoid/endovanilloid systems. Pharmacol Res. 2010;61:531–536. doi: 10.1016/j.phrs.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Mievis S, Blum D, Ledent C. Worsening of Huntington disease phenotype in CB1 receptor knockout mice. Neurobiol Dis. 2011;42:524–529. doi: 10.1016/j.nbd.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Milton NG. Anandamide and noladin ether prevent neurotoxicity of the human amyloid-beta peptide. Neurosci Lett. 2002;332:127–130. doi: 10.1016/s0304-3940(02)00936-9. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado E, Vela JM, Arevalo-Martin A, Almazan G, Molina-Holgado F, Borrell J, Guaza C. Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci. 2002a;22:9742–9753. doi: 10.1523/JNEUROSCI.22-22-09742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Holgado F, Molina-Holgado E, Guaza C, Rothwell NJ. Role of CB1 and CB2 receptors in the inhibitory effects of cannabinoids on lipopolysaccharide-induced nitric oxide release in astrocyte cultures. J Neurosci Res. 2002b;67:829–836. doi: 10.1002/jnr.10165. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Pinteaux E, Moore JD, Molina-Holgado E, Guaza C, Gibson RM, Rothwell NJ. Endogenous interleukin-1 receptor antagonist mediates anti-inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J Neurosci. 2003;23:6470–6474. doi: 10.1523/JNEUROSCI.23-16-06470.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E, Chiarlone A, Medrano M, et al. Neuropsychopharmacology. 2017. Singular Location and Signaling Profile of Adenosine A2A-Cannabinoid CB1 Receptor Heteromers in the Dorsal Striatum; pp. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounsey RB, Mustafa S, Robinson L, Ross RA, Riedel G, Pertwee RG, Teismann P. Increasing levels of the endocannabinoid 2-AG is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Exp Neurol. 2015;273:36–44. doi: 10.1016/j.expneurol.2015.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Zhuang SY, Kirby MT, Hampson RE, Deadwyler SA. Cannabinoid receptors differentially modulate potassium A and D currents in hippocampal neurons in culture. J Pharmacol Exp Ther. 1999;291:893–902. [PubMed] [Google Scholar]
- Mulder J, Zilberter M, Pasquare SJ, et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain. 2011;134:1041–1060. doi: 10.1093/brain/awr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Vahl K, Schneider U, Emrich H. Nabilone increases choreatic movements in Huntington’s disease. Mov Disord. 1999;14:1038–1040. doi: 10.1002/1531-8257(199911)14:6<1038::aid-mds1024>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Muller-Vahl KR, Kolbe H, Schneider U, Emrich HM. Cannabis in movement disorders. Forsch Komplementarmed. 1999a;6(Suppl 3):23–27. doi: 10.1159/000057153. [DOI] [PubMed] [Google Scholar]
- Muller-Vahl KR, Schneider U, Kolbe H, Emrich HM. Treatment of Tourette’s syndrome with delta-9-tetrahydrocannabinol. A J Psychiatry. 1999b;156:495. [PubMed] [Google Scholar]
- Nakane S, Oka S, Arai S, Waku K, Ishima Y, Tokumura A, Sugiura T. 2-Arachidonoyl-sn-glycero-3-phosphate, an arachidonic acid-containing lysophosphatidic acid: occurrence and rapid enzymatic conversion to 2-arachidonoyl-sn-glycerol, a cannabinoid receptor ligand, in rat brain. Arch Biochem Biophys. 2002;402:51–58. doi: 10.1016/S0003-9861(02)00038-3. [DOI] [PubMed] [Google Scholar]
- Natarajan V, Reddy PV, Schmid PC, Schmid HH. On the biosynthesis and metabolism of N-acylethanolamine phospholipids in infarcted dog heart. Biochim Biophys Acta. 1981;664:445–448. doi: 10.1016/0005-2760(81)90067-9. [DOI] [PubMed] [Google Scholar]
- Nogueron MI, Porgilsson B, Schneider WE, Stucky CL, Hillard CJ. Cannabinoid receptor agonists inhibit depolarization-induced calcium influx in cerebellar granule neurons. J Neurochem. 2001;79:371–381. doi: 10.1046/j.1471-4159.2001.00567.x. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminal. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Sawada S, Kano M. Heterosynaptic expression of depolarization-induced suppression of inhibition (DSI) in rat hippocampal cultures. Neurosci Res. 2000;36:67–71. doi: 10.1016/s0168-0102(99)00110-8. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Sawada S, Yamamoto C. Properties of depolarization-induced suppression of inhibitory transmission in cultured rat hippocampal neurons. Pflugers Arch. 1998;435:273–279. doi: 10.1007/s004240050512. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci. 2002;22:3864–3872. doi: 10.1523/JNEUROSCI.22-10-03864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onaivi ES, Ishiguro H, Gong JP, et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci. 2006;1074:514–536. doi: 10.1196/annals.1369.052. [DOI] [PubMed] [Google Scholar]
- Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173:649–665. doi: 10.1111/bph.13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- Ozaita A, Puighermanal E, Maldonado R. Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J Neurochem. 2007;102:1105–1114. doi: 10.1111/j.1471-4159.2007.04642.x. [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Aguado T, Egia A, Mechoulam R, Guzman M, Galve-Roperh I. Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J. 2006;20:2405–2407. doi: 10.1096/fj.06-6164fje. [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Aguado T, Pazos MR, et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain. 2009;132:3152–3164. doi: 10.1093/brain/awp239. [DOI] [PubMed] [Google Scholar]
- Palomo-Garo C, Gomez-Galvez Y, Garcia C, Fernandez-Ruiz J. Targeting the cannabinoid CB2 receptor to attenuate the progression of motor deficits in LRRK2-transgenic mice. Pharmacol Res. 2016;110:181–192. doi: 10.1016/j.phrs.2016.04.004. [DOI] [PubMed] [Google Scholar]
- Pan X, Ikeda SR, Lewis DL. Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol Pharmacol. 1996;49:707–714. [PubMed] [Google Scholar]
- Pascual AC, Martin-Moreno AM, Giusto NM, de Ceballos ML, Pasquare SJ. Normal aging in rats and pathological aging in human Alzheimer’s disease decrease FAAH activity: modulation by cannabinoid agonists. Exp Gerontol. 2014;60:92–99. doi: 10.1016/j.exger.2014.10.011. [DOI] [PubMed] [Google Scholar]
- Passmore MJ. The cannabinoid receptor agonist nabilone for the treatment of dementia-related agitation. Int J Geriatr Psychiatry. 2008;23:116–117. doi: 10.1002/gps.1828. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- Pinto JC, Potie F, Rice KC, Boring D, Johnson MR, Evans DM, Wilken GH, Cantrell CH, Howlett AC. Cannabinoid receptor binding and agonist activity of amides and esters of arachidonic acid. Mol Pharmacol. 1994;46:516–522. [PubMed] [Google Scholar]
- Pintor A, Tebano MT, Martire A, et al. The cannabinoid receptor agonist WIN 55,212-2 attenuates the effects induced by quinolinic acid in the rat striatum. Neuropharmacology. 2006;51:1004–1012. doi: 10.1016/j.neuropharm.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Pisani A, Fezza F, Galati S, et al. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson’s disease patients. Ann Neurol. 2005;57:777–779. doi: 10.1002/ana.20462. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Prescott SM, Majerus PW. Characterization of 1,2-diacylglycerol hydrolysis in human platelets. Demonstration of an arachidonoyl-monoacylglycerol intermediate. J Biol Chem. 1983;258:764–769. [PubMed] [Google Scholar]
- Price DA, Martinez AA, Seillier A, et al. WIN55,212–2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur J Neurosci. 2009;29:2177–2186. doi: 10.1111/j.1460-9568.2009.06764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Johnson GV. Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res Bull. 2009;80:242–247. doi: 10.1016/j.brainresbull.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25:1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Glanzer JG, Kadiu I, et al. Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J Neurochem. 2008a;104:1504–1525. doi: 10.1111/j.1471-4159.2007.05087.x. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Kadiu I, Garg SK, Glanzer JG, Nordgren T, Ciborowski P, Banerjee R, Gendelman HE. Nitrated alpha-synuclein and microglial neuroregulatory activities. Journal of neuroimmune pharmacology: the official journal of the Society on NeuroImmune Pharmacology. 2008b;3:59–74. doi: 10.1007/s11481-008-9100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Herkenham M. Selective vulnerability in Huntington’s disease: preferential loss of cannabinoid receptors in lateral globus pallidus. Ann Neurol. 1994;36:577–584. doi: 10.1002/ana.410360406. [DOI] [PubMed] [Google Scholar]
- Rikani AA, Choudhry Z, Choudhry AM, Rizvi N, Ikram H, Mobassarah NJ, Tulli S. The mechanism of degeneration of striatal neuronal subtypes in Huntington disease. Annals of neurosciences. 2014;21:112–114. doi: 10.5214/ans.0972.7531.210308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero J, Berrendero F, Garcia-Gil L, de la Cruz P, Ramos JA, Fernandez-Ruiz JJ. Loss of cannabinoid receptor binding and messenger RNA levels and cannabinoid agonist-stimulated [35S]guanylyl-5′O-(thio)-triphosphate binding in the basal ganglia of aged rats. Neuroscience. 1998;84:1075–1083. doi: 10.1016/s0306-4522(97)00552-6. [DOI] [PubMed] [Google Scholar]
- Sagredo O, Gonzalez S, Aroyo I, et al. Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: relevance for Huntington’s disease. Glia. 2009;57:1154–1167. doi: 10.1002/glia.20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagredo O, Ramos JA, Decio A, Mechoulam R, Fernandez-Ruiz J. Cannabidiol reduced the striatal atrophy caused 3-nitropropionic acid in vivo by mechanisms independent of the activation of cannabinoid, vanilloid TRPV1 and adenosine A2A receptors. Eur J Neurosci. 2007;26:843–851. doi: 10.1111/j.1460-9568.2007.05717.x. [DOI] [PubMed] [Google Scholar]
- Samson MT, Small-Howard A, Shimoda LM, Koblan-Huberson M, Stokes AJ, Turner H. Differential roles of CB1 and CB2 cannabinoid receptors in mast cells. J Immunol. 2003;170:4953–4962. doi: 10.4049/jimmunol.170.10.4953. [DOI] [PubMed] [Google Scholar]
- Sandyk R, Snider SR, Consroe P, Elias SM. Cannabidiol in dystonic movement disorders. Psychiatry Res. 1986;18:291. doi: 10.1016/0165-1781(86)90117-4. [DOI] [PubMed] [Google Scholar]
- Schmid PC, Reddy PV, Natarajan V, Schmid HH. Metabolism of N-acylethanolamine phospholipids by a mammalian phosphodiesterase of the phospholipase D type. J Biol Chem. 1983;258:9302–9306. [PubMed] [Google Scholar]
- Schmole AC, Lundt R, Ternes S, et al. Cannabinoid receptor 2 deficiency results in reduced neuroinflammation in an Alzheimer’s disease mouse model. Neurobiol Aging. 2015;36:710–719. doi: 10.1016/j.neurobiolaging.2014.09.019. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieradzan K, Fox S, Dick J, Brotchie J. The effects of the cannabinoid receptor agonist nabilone on L-dopa induced dyskinesia in patients with idiopathic Parkinson’s disease (PD) Mov Disord. 1998;13:29. [Google Scholar]
- Silverdale MA, McGuire S, McInnes A, Crossman AR, Brotchie JM. Striatal cannabinoid CB1 receptor mRNA expression is decreased in the reserpine-treated rat model of Parkinson’s disease. Exp Neurol. 2001;169:400–406. doi: 10.1006/exnr.2001.7649. [DOI] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J Biol Chem. 2008;283:9341–9349. doi: 10.1074/jbc.M707807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solas M, Francis PT, Franco R, Ramirez MJ. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol Aging. 2013;34:805–808. doi: 10.1016/j.neurobiolaging.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kishimoto S, Oka S, Gokoh M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res. 2006;45:405–446. doi: 10.1016/j.plipres.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kobayashi Y, Oka S, Waku K. Biosynthesis and degradation of anandamide and 2-arachidonoylglycerol and their possible physiological significance. Prostaglandins Leukot Essent Fatty Acids. 2002;66:173–192. doi: 10.1054/plef.2001.0356. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Sun YX, Tsuboi K, Okamoto Y, Tonai T, Murakami M, Kudo I, Ueda N. Biosynthesis of anandamide and N-palmitoylethanolamine by sequential actions of phospholipase A2 and lysophospholipase D. Biochem J. 2004;380:749–756. doi: 10.1042/BJ20040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadaiesky MT, Dombrowski PA, Da Cunha C, Takahashi RN. Effects of SR141716A on Cognitive and Depression-Related Behavior in an Animal Model of Premotor Parkinson’s Disease. Parkinson’s disease. 2010;2010:238491. doi: 10.4061/2010/238491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–1766. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- Tanveer R, Gowran A, Noonan J, Keating SE, Bowie AG, Campbell VA. The endocannabinoid, anandamide, augments Notch-1 signaling in cultured cortical neurons exposed to amyloid-beta and in the cortex of aged rats. J Biol Chem. 2012;287:34709–34721. doi: 10.1074/jbc.M112.350678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JM, Main BS, Crack PJ. Neuroinflammation and oxidative stress: co-conspirators in the pathology of Parkinson’s disease. Neurochem Int. 2013;62:803–819. doi: 10.1016/j.neuint.2012.12.016. [DOI] [PubMed] [Google Scholar]
- Ternianov A, Perez-Ortiz JM, Solesio ME, Garcia-Gutierrez MS, Ortega-Alvaro A, Navarrete F, Leiva C, Galindo MF, Manzanares J. Overexpression of CB2 cannabinoid receptors results in neuroprotection against behavioral and neurochemical alterations induced by intracaudate administration of 6-hydroxydopamine. Neurobiol Aging. 2012;33:421.e421–421.e416. doi: 10.1016/j.neurobiolaging.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Thomas MP, Chartrand K, Reynolds A, Vitvitsky V, Banerjee R, Gendelman HE. Ion channel blockade attenuates aggregated alpha synuclein induction of microglial reactive oxygen species: relevance for the pathogenesis of Parkinson’s disease. J Neurochem. 2007;100:503–519. doi: 10.1111/j.1471-4159.2006.04315.x. [DOI] [PubMed] [Google Scholar]
- Tolon RM, Nunez E, Pazos MR, Benito C, Castillo AI, Martinez-Orgado JA, Romero J. The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 2009;1283:148–154. doi: 10.1016/j.brainres.2009.05.098. [DOI] [PubMed] [Google Scholar]
- Trapani A, De Giglio E, Cafagna D, Denora N, Agrimi G, Cassano T, Gaetani S, Cuomo V, Trapani G. Characterization and evaluation of chitosan nanoparticles for dopamine brain delivery. Int J Pharm. 2011;419:296–307. doi: 10.1016/j.ijpharm.2011.07.036. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Fox SH, Hill M, Crossman AR, Petrosino S, Di Marzo V, Brotchie JM. A role for endocannabinoids in the generation of parkinsonism and levodopa-induced dyskinesia in MPTP-lesioned non-human primate models of Parkinson’s disease. FASEB J. 2005;19:1140–1142. doi: 10.1096/fj.04-3010fje. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Mazzola C, Esposito G, et al. Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci. 2006;63:1410–1424. doi: 10.1007/s00018-006-6037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel SA, Cravatt BF, Engram AE, Lichtman AH. Fatty acid amide hydrolase (−/−) mice exhibit an increased sensitivity to the disruptive effects of anandamide or oleamide in a working memory water maze task. J Pharmacol Exp Ther. 2006;317:251–257. doi: 10.1124/jpet.105.095059. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Wise LE, Niyuhire F, Cravatt BF, Lichtman AH. Inhibition of fatty-acid amide hydrolase accelerates acquisition and extinction rates in a spatial memory task. Neuropsychopharmacology. 2007;32:1032–1041. doi: 10.1038/sj.npp.1301224. [DOI] [PubMed] [Google Scholar]
- Viveros-Paredes JM, Gonzalez-Castaneda RE, Escalante-Castaneda A, Tejeda-Martinez AR, Castaneda-Achutigui F, Flores-Soto ME. Effect of inhibition of fatty acid amide hydrolase on MPTP-induced dopaminergic neuronal damage. Neurologia. 2017 doi: 10.1016/j.nrl.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Volicer L, Stelly M, Morris J, McLaughlin J, Volicer BJ. Effects of dronabinol on anorexia and disturbed behavior in patients with Alzheimer’s disease. Int J Geriatr Psychiatry. 1997;12:913–919. [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endocannabinoid Signaling in the Brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- Wu J, Bie B, Yang H, Xu JJ, Brown DL, Naguib M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol Aging. 2013;34:791–804. doi: 10.1016/j.neurobiolaging.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Yan W, Yun Y, Ku T, Li G, Sang N. NO2 inhalation promotes Alzheimer’s disease-like progression: cyclooxygenase-2-derived prostaglandin E2 modulation and monoacylglycerol lipase inhibition-targeted medication. Scientific reports. 2016;6:22429. doi: 10.1038/srep22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zeng BY, Dass B, Owen A, Rose S, Cannizzaro C, Tel BC, Jenner P. Chronic L-DOPA treatment increases striatal cannabinoid CB1 receptor mRNA expression in 6-hydroxydopamine-lesioned rats. Neurosci Lett. 1999;276:71–74. doi: 10.1016/s0304-3940(99)00762-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hu M, Teng Z, Tang YP, Chen C. Synaptic and cognitive improvements by inhibition of 2-AG metabolism are through upregulation of microRNA-188-3p in a mouse model of Alzheimer’s disease. J Neurosci. 2014;34:14919–14933. doi: 10.1523/JNEUROSCI.1165-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang T, Pei Z, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]