

Supplemental Digital Content is available in the text.
Keywords: cholesterol, hydroxymethylglutaryl CoA reductases, mammalian haploid genetics, oxysterols, sterols
Abstract
Objective—
The cellular demand for cholesterol requires control of its biosynthesis by the mevalonate pathway. Regulation of HMGCR (3-hydroxy-3-methylglutaryl coenzyme A reductase), a rate-limiting enzyme in this pathway and the target of statins, is a key control point herein. Accordingly, HMGCR is subject to negative and positive regulation. In particular, the ability of oxysterols and intermediates of the mevalonate pathway to stimulate its proteasomal degradation is an exquisite example of metabolically controlled feedback regulation. To define the genetic determinants that govern this process, we conducted an unbiased haploid mammalian genetic screen.
Approach and Results—
We generated human haploid cells with mNeon fused to endogenous HMGCR using CRISPR/Cas9 and used these cells to interrogate regulation of HMGCR abundance in live cells. This resulted in identification of known and new regulators of HMGCR, and among the latter, UBXD8 (ubiquitin regulatory X domain-containing protein 8), a gene that has not been previously implicated in this process. We demonstrate that UBXD8 is an essential determinant of metabolically stimulated degradation of HMGCR and of cholesterol biosynthesis in multiple cell types. Accordingly, UBXD8 ablation leads to aberrant cholesterol synthesis due to loss of feedback control. Mechanistically, we show that UBXD8 is necessary for sterol-stimulated dislocation of ubiquitylated HMGCR from the endoplasmic reticulum membrane en route to proteasomal degradation, a function dependent on its UBX domain.
Conclusions—
We establish UBXD8 as a previously unrecognized determinant that couples flux across the mevalonate pathway to control of cholesterol synthesis and demonstrate the feasibility of applying mammalian haploid genetics to study metabolic traits.
Cholesterol is an essential component of biological membranes and serves as a precursor for a multitude of bioactive molecules. The biosynthesis of cholesterol by the mevalonate pathway, a process requiring the coordinated activity of >20 enzymes, is transcriptionally regulated by the SREBPs (sterol-regulatory element–binding proteins).1,2 Retained in the endoplasmic reticulum (ER) in their membrane-bound precursor form, SREBPs form a tripartite complex with SCAP and INSIGs.3 Decreased content of cholesterol in the ER membrane induces a conformational change in SCAP that releases it from INSIGs and allows the SCAP–SREBP complex to travel to the Golgi, where SREBPs are sequentially cleaved by site-1 and -2 proteases (MBTPS1 and -2, respectively). The released, transcriptionally active N-terminal domain of SREBP translocates to the nucleus and induces expression of sterol-responsive genes, including, among others, that of the low-density lipoprotein receptor, and of HMGCR (3-hydroxy-3-methylglutaryl-coenzyme A reductase).3,4
The mevalonate pathway is subject to multiple feedback mechanisms that ensure that production of cholesterol meets, but does not exceed, cellular needs.5 Among these, the acute, sterol-stimulated degradation of the rate-limiting enzyme HMGCR has received much attention.6,7 HMGCR is a polytopic ER membrane protein, and its degradation in response to oxysterols or intermediates of the cholesterol biosynthetic pathway exemplifies metabolically regulated ER-associated protein degradation (ERAD), where the enzyme is ubiquitylated on conserved lysine residues, extracted from the ER membrane in a VCP/p97-dependent manner, and ultimately degraded by the proteasome, thus effectively shutting down metabolic flux through the pathway.6,8,9 In addition to controlling SREBP-dependent transcription of HMGCR, INSIG proteins play a key role in controlling the metabolically regulated degradation of the enzyme. It has been proposed that in response to metabolic cues, INSIGs bridge HMGCR with the ER-resident E3 ligases gp78 and TRC8 to promote ubiquitylation of the enzyme.10–14 Yet many of the steps in this complex process are not fully elucidated. In fact, a recent study contends the involvement of both gp78 and TRC8 in HMGCR ubiquitylation,15 and we have recently demonstrated that another ER-resident E3 ligase, MARCH6, may also control the abundance of HMGCR.16,17
Because the ensemble of factors that play a role in the metabolically regulated ERAD of HMGCR is far from being complete, we decided to use a haploid genetic screen in human cells in an attempt to identify genes that are required for sterol-stimulated degradation of HMGCR. This recently developed methodology is based on the use of a unique, human haploid cell line (Hap1) that can be readily mutagenized using a viral-based gene-trap cassette.18 Since only a single copy of each gene is present in Hap1 cells, trapping of a genetic locus by integration of this cassette typically results in inactivation of the encoded protein, thus providing a powerful means to establish genotype–phenotype association. This methodology has not been applied yet to studies of cholesterol homeostasis. However, because the cholesterol synthesis pathway is highly conserved, Hap1 cells provide an eminently suitable system for investigating fundamental questions in cholesterol metabolism. With this approach, we identify the UBXD8 (ubiquitin regulatory X domain–containing protein 8; also known as FAF2), as a key determinant of sterol-stimulated degradation of HMGCR and of flux across the mevalonate pathway in mammalian cells.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Cell Culture
Hap1, HepG2, IHH, Hepa1-6, and primary rat hepatocytes were obtained and cultured as previously reported.17,19 Where indicated, cells were sterol depleted by culture in sterol-depletion medium (DMEM or IMDM [Iscove’s Modified Dulbecco’s Medium] with 10% lipoprotein-deficient serum, 2.5 μg/mL simvastatin, and 100 μmol/L mevalonate to support isoprenoid synthesis) or in β-methyl-cyclodextrin–containing medium (DMEM with 3 mmol/L β-methyl-cyclodextrin and 10% lipoprotein-deficient serum).
CRISPR/Cas9-Mediated Genome Editing
Knockout cells were generated by CRISPR/Cas9-mediated genome editing as described.20 To insert the mNeon-2A-PURO reporter cassette into the endogenous HMGCR locus, we used microhomology-based CRISPR/Cas9-CRIS-PITCh, as reported by Nakade et al.21 The donor fragment and sgRNA guides are shown in Tables I and II in the online-only Data Supplement. Independent clones were obtained, and genome editing confirmed by sequencing, immunoblotting, and immunofluorescence.
Plasmids and Expression Constructs
px330 (#42230) and pENTR/pTER+ (430–1; #17453) were from Addgene. Lentiviral constructs encoding N-terminally FLAG-tagged UBXD8 (wild type [WT], ΔUBA, and ΔUBX) were used to generate lentiviral particles and to obtain Zeocin-resistant target clones.
Antibodies and Immunoblot Analysis
Total cell lysates were prepared in RIPA buffer supplemented with protease inhibitors (Roche). Lysates were cleared by centrifugation and samples separated on NuPAGE Novex 4% to 12% Bis-Tris gels (Invitrogen) and then transferred to nitrocellulose membranes. The primary antibodies used are listed in the Methods section in the online-only Data Supplement. Secondary horseradish peroxidase-conjugated antibodies (Invitrogen) were used and visualized with chemiluminescence. All immunoblots shown are representative of at least 3 independent experiments with similar results.
Generation and Amplification of Adenoviral Particles
Oligonucleotides targeting 3 different regions of Ubxd8 were cloned into pTER+/pENTR (Table II in the online-only Data Supplement), which had been modified by addition of a CMV-GFP cassette. The resulting pTER+/pENTR-GFP-mUbxd8shRNA or pTER+/pENTR-GFP-ScrambledshRNA were recombined into pAD-BLOCK-iT (Invitrogen), and adenoviral particles were generated and amplified as reported.22
RNA Isolation and Quantitative Polymerase Chain Reaction
Isolation of total RNA and subsequent real-time quantitative polymerase chain reaction was done as previously reported.17 Sequences of quantitative polymerase chain reaction primers are available on request.
Fluorescence-Activated Cell Sorter Analysis and Imaging of HMGCR-mNeon
Cells were treated as indicated and analyzed on a CytoFLEX Flow cytometer (Beckman Coulter). Viable cells were gated and 10 000 events per condition acquired and analyzed using the FlowJo software package. Relative HMGCR-mNeon intensity was calculated from GEOmean values and presented as mean±SD. For confocal imaging, Hap1-HMGCR-mNeon cells were fixed with 4% paraformaldehyde for 10 minutes and viewed with A Leica TCS SP8 confocal microscope equipped with a ×63 objective. For live cell imaging, we used a Leica IR-BE inverted wide field microscope equipped with a ×63 objective and a custom-built incubator.
Determination of Sterol Synthesis Rate
Cells were sterol depleted by culturing in β-methyl-cyclodextrin–containing medium for 16 hours. Subsequently, cells were pulse labeled for 45 minutes with 0.5 μCi [14C]-acetate or for 2 hours with [14C]-pyruvate in the presence of 10 μmol/L 25-hydroxycholesterol (25-HC) and 2.5 μg/mL simvastatin, as indicated. The nonsaponifiable lipid fraction was subjected to scintillation counting to determine radioactive content, as previously described.16
Haploid Genetic Screen for Regulators of HMGCR Levels
To identify regulators of HMGCR expression and degradation, we prepared a library of mutagenized Hap1-HMGCR-mNeon-2A-Puro cells using a gene-trap retrovirus expressing blue fluorescent protein, as described previously.23 Briefly, 5×108 Hap1-HMGCR-mNeon cells were seeded and transduced with virus from 2 combined harvests on 3 consecutive days in the presence of 8 μg/mL protamine sulfate (Sigma). Mutagenized cells were expanded to 30 T175 flasks at a confluence of ≈80%. Subsequently, cells were cultured in sterol-depletion medium for 24 hours and with 10 μmol/L 25-HC and 5 mmol/L mevalonate during the last 2 hours to stimulate degradation of HMGCR-mNeon. At the end of this treatment, the cells were washed twice with PBS, dissociated with trypsin, pelleted, and fixed with BD Fix Buffer I (BD Biosciences) for 10 minutes at 37°C. After washing twice with PBS containing 10% FCS, the cells were filtered through a 40-μm strainer (BD Falcon) before sorting 2 populations of cells (ie, HMGCR-mNeonLOW and HMGCR-mNeonHIGH) that constitute ≈5% of the lowest and highest HMGCR-mNeon–expressing cells from the total cell population, respectively (as reported in Brockman et al24). In addition, to reduce potential confounding effects of diploid cells, which are heterozygous for alleles carrying gene-trap integrations, the cells were sorted in parallel for DNA content (1n) by staining with 3 μmol/L 4′,6-diamidino-2-phenylindole. Cell sorting was performed on a Biorad S3 Cell sorter until ≈10 million cells of each population were collected. Sorted cells were pelleted, and genomic DNA was isolated using a DNA mini kit (Qiagen). To assist decrosslinking of genomic DNA, the cell pellets were resuspended in PBS supplemented with proteinase K (Qiagen) followed by overnight incubation at 56°C with lysis buffer AL (Qiagen) with agitation. Viral insertion sites of each sorted cell population were amplified and mapped as described previously using a Linear AMplification polymerase chain reaction on the total yield of isolated genomic DNA.23 Samples were subsequently submitted for deep sequencing, and gene-trap insertion sites were mapped and analyzed as previously described.24 Briefly, insertion sites were retrieved from trimmed reads (50b) that aligned unambiguously to HG19 using Bowtie allowing for 1 mismatch.25 Using intersectBed, aligned reads were mapped to nonoverlapping Refseq gene coordinates. Intragenic gene-trap insertions in sense orientation with its gene were considered disruptive and kept for further analysis. For each gene, the number of unique disruptive insertions was compared between the mNeonlow and mNeonhigh population. Genes that were significantly enriched for insertions in either of the 2 populations (2-sided Fisher exact test with Benjamini–Hochberg multiple testing correction, P<0.05) were considered as regulators of HMGCR-mNeon abundance. To reflect the directionality of the effect on HMGCR-mNeon abundance, a mutational index score was calculated by counting the number of unique inserts as follows:
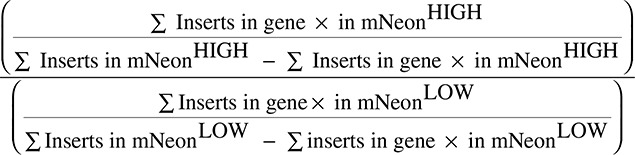 |
For those genes where in only 1 of the 2 populations disruptive insertions were identified, 1 insertion was assigned to the other population to prevent these genes to be omitted from the plots.
Cell Fractionation
Cell fractionation to obtain cytosolic and membrane fractions was done as previously reported.26 The protein content of the different fractions was determined with the BCA (bicinchoninic acid) assay and the indicated amounts analyzed by immunoblotting.
Statistics
Statistical analyses were performed with the Prism software package. Results were evaluated by Student t test when comparing 2 groups or by 1-way ANOVA for grouped analysis. SD is indicated by error bars, and P values are indicated by asterisks: *P<0.05, **P<0.01, and ***P<0.001.
Results
To identify novel genes involved in HMGCR regulation, we developed a Hap1 reporter cell line in which CRISPR/Cas9-dependent microhomology-mediated end-joining was used to integrate a mNeon-2A-PURO cassette (Table I in the online-only Data Supplement) into the last exon of HMGCR (exon 18; Figure 1A).21 Correct integration of this cassette resulted in expression from the endogenous genomic locus of an in-frame fusion protein between HMGCR and a C-terminally appended mNeon fluorescent protein (HMGCR-mNeon). We identified independent clones in which the endogenous HMGCR locus was correctly targeted, as confirmed by sequencing of both the 5′ and 3′ genomic DNA region bordering the integration site (Figure IA in the online-only Data Supplement). Fluorescence-activated cell sorter analysis of these clones demonstrated that HMGCR-mNeon levels were low in cells maintained in lipoprotein-containing medium but that fluorescence intensity increases ≈30-fold after 24 hours incubation in sterol-depletion medium (Figure IB in the online-only Data Supplement). Under these conditions, the chimeric HMGCR-mNeon protein, which colocalizes with the ER-resident protein calnexin, was readily visible by fluorescent microscopy (Figure 1B; Figure IC in the online-only Data Supplement). Consistent with having a single targeted HMGCR allele, the engineered Hap1 cells expressed only a single protein species corresponding in size to HMGCR-mNeon (Figure 1C). Moreover, similar to HMGCR in HepG2 cells (Figure ID in the online-only Data Supplement), the levels of HMGCR-mNeon protein in Hap1 cells were exquisitely sensitive to negative regulation by oxysterols; short-term treatment with 10 μmol/L of the oxysterol 25-HC resulted in its near complete disappearance, as assessed by both immunoblotting (Figure 1C) and by fluorescence-activated cell sorter analysis (Figure 1D; Figure IIA in the online-only Data Supplement; Movie I in the online-only Data Supplement). Importantly, in addition to promoting HMGCR degradation, 25-HC prevents de novo HMGCR transcription because it is a potent inhibitor of SREBP processing, also in these cells. Therefore, the reduction in HMGCR in response to acute treatment with 25-HC reflects the proteasomal degradation of existing HMGCR-mNeon because it was blocked when the cells were also treated with the proteasomal inhibitor MG-132 (Figure 1C and 1D; Figure IIA and IIB in the online-only Data Supplement). Collectively, these results establish the Hap1-HMGCR-mNeon cells as a bona fide reporter system to study the physiological regulation of HMGCR.
Figure 1.
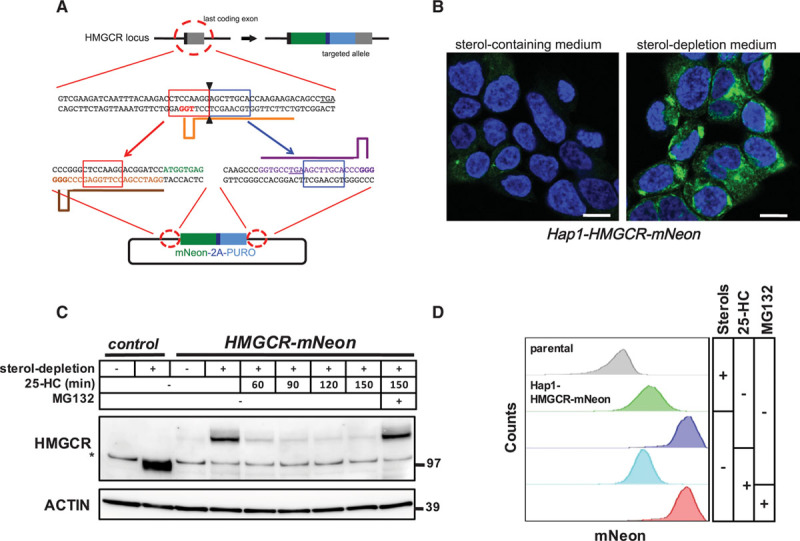
CRISPR/Cas9-mediated targeting of the endogenous HMGCR (3-hydroxy-3-methylglutaryl coenzyme A reductase) locus. A, Schematic illustration of CRISPR/Cas9-mediated targeting of the endogenous HMGCR locus for in-frame integration of mNeon-2A-PURO. Orange, brown, and purple bars correspond to the gRNA target sites. Red- and blue-boxed sequences indicate the microhomology sequences. The PAM sites are highlighted in bold, and the stop codons are underlined. B, Hap1-HMGCR-mNeon cells were grown on coverslips and cultured in sterol-containing or sterol-depletion medium for 24 h. Subsequently, cells were fixed, counterstained with 4′,6-diamidino-2-phenylindole, and imaged by confocal fluorescence microscopy. Representative images are shown (scale bar, 7.5 μm). C, Control Hap1 cells and Hap1-HMGCR-mNeon cells were grown in sterol-depletion or sterol-containing medium for 24 h, before treatment with 10 μmol/L 25-hydroxycholesterol (25-HC) and 25 μmol/L MG132 for the indicated time. Total cell lysates were immunoblotted as indicated. Immunoblot is representative of 3 independent experiments. The asterisk (*) indicates a nonspecific band. D, Hap1-HMGCR-mNeon cells were cultured as in (C) before addition of 10 μmol/L 25-HC and 25 μmol/L MG132 for 1 h after which the intensity of mNeon fluorescence was quantified by fluorescence-activated cell sorter analysis.
To identify genes that are essential for sterol-stimulated degradation of HMGCR, we used a haploid genetic screening approach (Figure 2A). A total of 3×109 mutagenized Hap1-HMGCR-mNeon cells were subjected to sterol depletion, followed by a 2-hour treatment with 25-HC and mevalonate (10 μmol/L and 5 mmol/L, respectively) to promote HMGCR-mNeon degradation. Subsequently, cell populations were sorted based on mNeon intensity, isolating cells with low and high levels of HMGCR-mNeon to enrich for positive and negative regulators of HMGCR abundance, respectively. Gene-trap insertion sites were mapped in both populations individually, and the mutation index (ie, ratio of disruptive mutations in the mNeonHIGH versus mNeonLOW population) was plotted (Figure 2B; Table III in the online-only Data Supplement). As expected, HMGCR itself was identified as the strongest positive regulator. Likewise, SREBP2, SCAP, MBTPS1, and MBTPS2 showed increased mutation load in the mNeonLOW population, indicating that also regulators of HMGCR transcription were identified (Figure III in the online-only Data Supplement).3 Reciprocally, INSIG1, which has been implicated in HMGCR degradation,10 was one of the strongest identified genes in the mNeonHIGH cell population, as was the ERAD-associated ubiquitin-conjugating E2 UBE2G2. Overall, our haploid screen revealed both positive and negative regulators of HMGCR levels.
Figure 2.
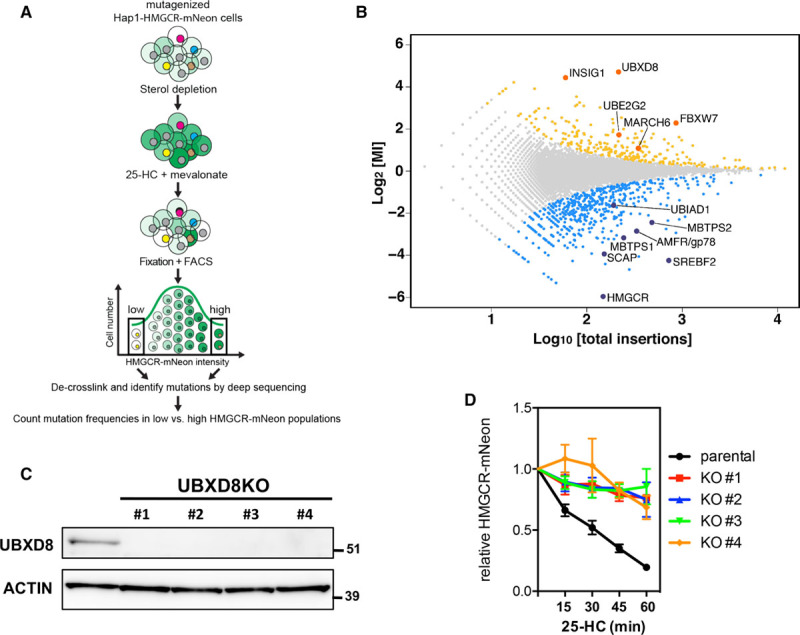
A mammalian haploid genetic screen identifies regulators of HMGCR (3-hydroxy-3-methylglutaryl coenzyme A reductase). A, Schematic illustration of the haploid genetic screen. A library of mutant Hap1-HMGCR-mNeon cells was generated by random, virus-mediated integration of a gene-trap cassette. To induce expression of HMGCR-mNeon, cells were cultured in sterol-depletion medium for 24 h. During the last 2 h, cells were treated with 10 μmol/L 25-hydroxycholesterol (25-HC) and 5 mmol/L mevalonate to initiate sterol-stimulated degradation of HMGCR. Genes that regulate HMGCR-mNeon abundance can be identified after cell sorting and recovery of the trapped loci. B, The log mutational index (MI) scores (see Materials and Methods in the online-only Data Supplement) were plotted against the number of trapped alleles per gene. Statistically significant hits (P<0.05) in the mNeonHIGH and mNeonLOW populations are indicated in yellow and blue, respectively (C). Total cell lysates of control and 4 independent UBXD8KO Hap1-HMGCR-mNeon cells were immunoblotted as indicated. Immunoblot is representative of 3 independent experiments. D, Control and UBXD8KO Hap1-HMGCR-mNeon cells were cultured in sterol-depletion medium for 24 h and subsequently treated with 10 μmol/L 25-HC for the indicated time. The intensity of HMGCR-mNeon was determined by fluorescence-activated cell sorter analysis, and the relative HMGCR-mNeon intensity is presented as mean±SD (n=3). KO indicates knockout; and UBXD8, ubiquitin regulatory X domain–containing protein 8.
Our primary goal was to identify genes required for sterol-stimulated degradation of HMGCR, and we therefore focused on genes identified in the mNeonHIGH population. Gene ontology analysis of significant genes (P<0.05) belonging to this group identified several prominent biological processes, including among others, peroxisosomal biogenesis and targeting, ubiquitin-dependent degradation, and cholesterol biosynthesis (Figure IV in the online-only Data Supplement). Next to INSIG1, the highest scoring hit in our screen was UBXD8 (Figure 2B), a member of the ERAD-associated UBX family of p97 adaptors,27 that has not been implicated in either HMGCR degradation or direct control of cholesterol synthesis. To study the role of UBXD8 in HMGCR degradation, we used CRISPR/Cas9-mediated genome editing to generate independent UBXD8-deficient Hap1-HMGCR-mNeon clones and confirmed ablation of UBXD8 by immunoblotting and genomic sequencing (Figure 2C). Functionally, the absence of UBXD8 in each of these clones abolished the sterol-stimulated, time-dependent degradation of HMGCR-mNeon (Figure 2D; Figure V in the online-only Data Supplement), thus confirming the identification of UBXD8 in the mNeonHIGH population in our screen. These results indicate that UBXD8 is a key player in HMGCR physiology.
To generalize the role of UBXD8 beyond haploid Hap1 cells, we next examined whether it played a significant role in HMGCR regulation also in hepatocytes, the major cell type contributing to bulk cholesterol biosynthesis in the body, and the main target cells for the statin class of cholesterol-lowering drugs. CRISPR/Cas9-mediated ablation of UBXD8 in independent HepG2 and IHH clones abolished the rapid degradation of HMGCR after treatment with 25-HC (Figure 3A; Figures VI and VIIA in the online-only Data Supplement). Importantly, mRNA expression of HMGCR itself and of SQLE and low-density lipoprotein receptor, 2 additional established SREBP2-regulated genes, remained unaffected by the loss of UBXD8 (Figure 3E; Figure VIIB in the online-only Data Supplement). Similarly, CRISPR/Cas9-mediated targeting of either exon 1 or exon 2 of murine Ubxd8 in murine hepatocyte-like Hepa1-6 cells also attenuated sterol-stimulated degradation of HMGCR (Figure VIIC in the online-only Data Supplement). To rule out compensatory responses because of UBXD8 ablation and potential confounding effects in our experiments, we also tested the effect of acute silencing of Ubxd8 by adenoviral-mediated delivery of Ubxd8-shRNA. As was the case with CRISPR/Cas9-mediated genetic ablation, effective silencing of Ubxd8 in either mouse Hepa1-6 cells or in primary rat hepatocytes largely prevented sterol-stimulated degradation of HMGCR (Figure 3B and 3C). As such, our results demonstrate that UBXD8 is a key determinant of sterol-stimulated degradation of HMGCR in human and rodent hepatocytes. In these experiments, we used 25-HC to stimulate reductase degradation. However, metabolic intermediates of the mevalonate pathway are also known to promote degradation of HMGCR and as such act in a negative feedback loop to control flux through this pathway. Accordingly, supplementing sterol-depleted cells with high concentrations of mevalonate—the product of HMGCR activity—potently promoted HMGCR degradation in HepG2 cells in a UBXD8-dependent manner (Figure 3D). In aggregate, our results establish UBXD8 as a regulator of HMGCR degradation in hepatocytes in response to metabolic cues.
Figure 3.
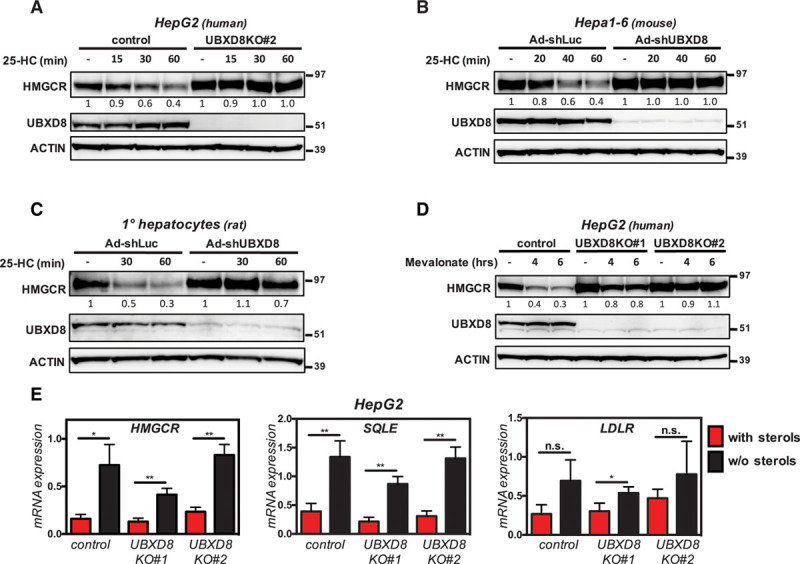
UBXD8 (ubiquitin regulatory X domain–containing protein 8) is essential for oxysterol- and mevalonate-stimulated degradation of HMGCR (3-hydroxy-3-methylglutaryl coenzyme A reductase) in hepatocytes. A, Control and UBXD8KO HepG2 cells, (B) Hepa1-6 cells, and (C) primary rat hepatocytes were cultured in sterol-depletion medium for 24 h and subsequently treated with 10 μmol/L 25-hydroxycholesterol (25-HC) for the indicated time. In B and C, cells were infected with pAD-GFP-shUbxd8 or pAD-GFP-shLuc adenoviruses at a multiplicity of infection of 100 for 72 h before the indicated treatment. D, Control HepG2 cells and 2 independent UBXD8KO clones were sterol depleted by culture in β-methyl-cyclodextrin (β-MCD)–containing medium for 16 h, after which 10 mmol/L mevalonate was added for the indicated time. A–D, Total cell lysates were immunoblotted as indicated. Immunoblots are representative of 3 independent experiments with the mean HMGCR-mNeon intensity shown. E, HepG2 control and UBXD8KO clones were cultured in sterol-containing- (red bars) or sterol-depletion medium (black bars) for 24 h. Total RNA was isolated, and expression of the indicated genes was determined by quantitative polymerase chain reaction. Each bar and error represent the mean±SD (n=3); *P<0.05, **P<0.01; KO indicates knockout; and n.s., not significant.
Because cholesterol synthesis and HMGCR expression are concomitantly increased under sterol-depleting conditions, we next examined the possibility that UBXD8 might be reciprocally regulated. As expected, sterol depletion of Hap1 cells and human (HepG2, IHH) and murine (Hepa1-6) hepatocyte-like cells resulted in a marked increase in the level of HMGCR mRNA and protein (Figure 4A and 4B). Yet under this condition, no changes were observed in UBXD8 mRNA or protein levels, indicating that sterol-dependent changes in UBXD8 expression do not account for its role in HMGCR degradation. UBXD8 has been reported to contribute to ERAD of proteins destined for proteasomal degradation, and given our findings, one could anticipate HMGCR and UBXD8 to interact. However, numerous attempts by different experimental approaches to demonstrate such an interaction were unsuccessful, suggesting that the HMGCR-UBXD8 interaction is either too weak, temporally regulated, or indirect. UBXD8 is a membrane-associated protein with a hairpin topology that partitions and traffics between the ER and lipid droplets.28,29 It harbors a membrane-associating domain, an N-terminal UBA domain, and a C-terminal UBX domain, with the latter 2 facing the cytosol (Figure 4C). To determine which of these protein domains is critical for sterol-stimulated degradation of HMGCR, we reconstituted UBXD8WT, UBXD8ΔUBA, or UBXD8ΔUBX in UBXD8-ablated Hap1 cells (Figure 4D; Figure VIII in the online-only Data Supplement). Reintroducing WT or UBXD8ΔUBA effectively restored sterol-stimulated degradation of HMGCR, yet UBXD8ΔUBX did not, despite being abundantly expressed. Because the UBX domain recruits VCP/p97 to the ER to facilitate extraction of ubiquitylated ERAD substrates from the ER membrane,27 this offers a plausible mechanistic explanation for the inability of UBXD8ΔUBX to support HMGCR degradation since HMGCR also requires VCP/p97 for its extraction.26,30 We, therefore, examined the effect of UBXD8 ablation on HMGCR extraction from the ER membrane in response to 25-HC (Figure 4E and 4F). In control HepG2 cells, 25-HC induced rapid degradation of HMGCR that was severely attenuated by proteasomal inhibition or when UBXD8 was absent (Figure 4E). When membrane and cytosolic fractions were analyzed, a significant decrease in HMGCR was detected in the membrane fraction of control HepG2 cells in response to 25-HC (Figure 4F). This was mirrored by a reciprocal appearance of HMGCR in the cytosolic fraction of control cells when the proteasome was inhibited (Figure 4F). In contrast, UBXD8KO cells were refractory to 25-HC and did not support HMGCR extraction because the reductase remained in the membrane fraction and was not detected in the cytosol even when the proteasome was inhibited. Importantly, this was not because of abrogated ubiquitylation as UBXD8KO cells maintained sterol-stimulated ubiquitylation of HMGCR (Figure IX in the online-only Data Supplement).
Figure 4.
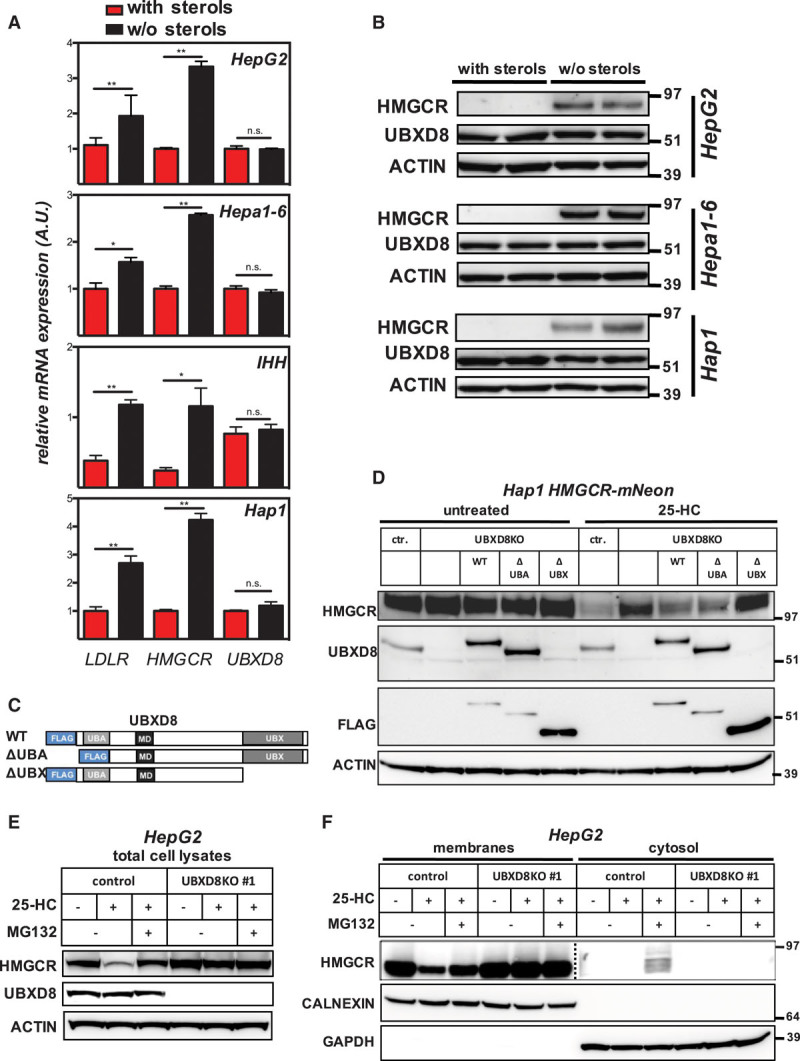
UBXD8 (ubiquitin regulatory X domain–containing protein 8) promotes degradation of HMGCR (3-hydroxy-3-methylglutaryl coenzyme A reductase) in a UBX domain–dependent manner. A, Hap1, and the hepatocyte lines HepG2, Hepa1-6, and IHH were cultured for 24 h in sterol-containing (red bars) or sterol-depletion medium (black bars). Subsequently, (A) total RNA was isolated, and expression of the indicated genes determined by quantitative polymerase chain reaction (n=3), *P<0.05, **P<0.01, or (B) total cell lysates were immunoblotted as indicated. Immunoblots are representative of 3 independent experiments. C, Schematic representation of FLAG-tagged full length wild type (WT) UBXD8 and its variants lacking the UBA or UBX domain; membrane-associating domain (MD). D, Control Hap1 or Hap1-UBXD8KO cells in which the FLAG-tagged full length or indicated variant were stably reintroduced were cultured in sterol-depletion medium for 24 h and then treated with 10 μmol/L 25-hydroxycholesterol (25-HC) for 1 h as shown. Total cell lysates were immunoblotted as indicated and are representative of 3 independent experiments. E, HepG2 control and UBXD8KO cells were cultured in sterol-containing or sterol-depletion medium for 24 h and subsequently treated with 10 μmol/L 25-HC or 25 μmol/L MG132 for 2 h as shown. Total cell lysates were immunoblotted as indicated and are representative of 3 independent experiments. F, HepG2 control and UBXD8KO cells were treated as described in (E), and membrane and cytosolic fractions were prepared, as described in Materials and Methods in the online-only Data Supplement. Subsequently, 20 or 60 μg protein per lane of the membrane and cytosolic fractions were analyzed by immunoblotting as indicated. Immunoblot is representative of 3 independent experiments. Note that the membrane and cytosolic HMGCR immunoblot are from different exposures.
HMGCR is a rate-limiting step in the mevalonate pathways and controls the rate of cholesterol biosynthesis. Given our results demonstrating that UBXD8 regulates HMGCR abundance, UBXD8 would be predicted to also impinge on cholesterol synthesis. We addressed this assertion in hepatocytes by measuring the incorporation of [14C]-acetate into nonsaponifiable lipids (ie, sterols). The rate of sterol synthesis was comparable in control cells and in 2 independent UBXD8KO clones (Figure 5A). Although short-term treatment with simvastatin, which blocks HMGCR activity, abolished acetate incorporation into sterols in both control and UBXD8KO cells, 25-HC–induced suppression of sterol synthesis was observed only in the control cells; the rate of sterol synthesis, hence HMGCR activity, in the short-term 25-HC–treated UBXD8KO cells remained high and mirrored the effect of UBXD8 ablation on HMGCR protein abundance. Similar results were obtained when we used [14C]-pyruvate as a cellular source for acetyl-CoA generation (Figure X in the online-only Data Supplement). Collectively (Figure 5B), our results demonstrate that UBXD8 is required for extraction of HMGCR from the ER membrane, and that it is a component of a feedback loop that couples lipid-based cues to metabolic flux across the mevalonate pathway.
Figure 5.
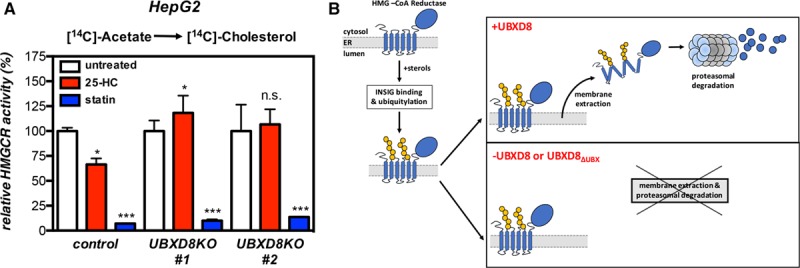
UBXD8 (ubiquitin regulatory X domain–containing protein 8) governs cholesterol synthesis in hepatocytes. A, HepG2 control and 2 independent UBXD8KO clones were sterol-depleted by culture in β-methyl-cyclodextrin (β-MCD)–containing medium for 16 h. Subsequently, cells were labeled for 45 min with [14C]-acetate in the presence of 10 μmol/L 25-HC or 2.5 μg/mL simvastatin, as indicated. The [14C]-labeled nonsaponifiable lipid fraction, extracted from samples containing an equal amount of cell protein, was quantified by scintillation counting. Each bar and error are the mean±SD of percent HMGCR (3-hydroxy-3-methylglutaryl coenzyme A reductase) activity (n=6); *P<0.05, ***P<0.005. B, Schematic representation of the role of UBXD8 in regulating sterol-stimulated degradation of HMGCR. In the absence of UBXD8 or its UBX domain, extraction of HMGCR to the cytosol en route to proteosomal degradation is abolished.
Discussion
In mammalian cells, mevalonate is the committed precursor for a central metabolic pathway in which sterols and a myriad of essential isoprenoids are synthesized.5 Accordingly, the flux of metabolites through the mevalonate pathway is tightly regulated, and the rate limiting and major control point of the pathway is exerted on mevalonate production by the enzyme HMGCR.5 Next to transcriptional and post-transcriptional control of HMGCR, degradation of the HMGCR protein, which is stimulated by mevalonate-derived sterols and nonsterol isoprenoids, constitutes a robust mechanism that couples metabolic flux to the enzyme’s levels and activity.6,7,9 By interrogating this process using an unbiased genetic screen, we identify UBXD8 as a previously unrecognized key determinant of regulated degradation of HMGCR and demonstrate that functional inactivation of UBXD8 deregulates cholesterol synthesis.
Haploid genetic screens have been only recently applied to mammalian cells and provide a powerful means to identify genotype–phenotype associations.18 These screens have been used to investigate, among others, viral entry,31,32 signaling pathways,33 modes of toxin action,18,34 and gene essentiality23 but have not been applied to the study of lipid-related traits. The power of our screen is manifested by the identification of several genes that have been previously implicated in the metabolically regulated degradation of HMGCR. Notably, INSIG1, a protein proposed to couple HMGCR to E3 ligase(s),10,14 was among the strongest hits in the mNeonHIGH population. Similarly, UBIAD1 was found in the mNeonLOW population, consistent with its reported role in limiting ubiquitylation and extraction of HMGCR from the ER membrane.35 Our screen also allowed us to address the identity of the putative E3 ligase(s) involved in stimulated ubiquitylation of HMGCR as long as it was not an essential gene. We reasoned that an E3 that ubiquitylates HMGCR would be present in the mNeonHIGH population. Two candidate E3s in this population were MARCH6 and FBW7, E3s that were previously implicated in regulation of HMGCR and SREBP levels, respectively.16,17,36 However, consistent with these earlier reports, sterol-stimulated degradation of HMGCR remained unaffected in cells lacking these ligases. HRD1 (SYVN1), an E3 ligase implicated in basal degradation of HMGCR, was found in our screen, yet the low number of integrations in this gene (13) may suggest that losing HRD1 impaired cellular fitness. Two additional E3s, AMFR/gp78 and TRC8/RNF139, which have been implicated in sterol-stimulated HMGCR ubiquitylation,11–13 were also detected in our screen. However, TRC8 had no significant mutational index score, indicating that in Hap1 cells this E3 may not contribute to degradation of HMGCR. The second ligase, AMFR/gp78 was found in the mNeonLOW population, which was unexpected because this indicates that ablation of gp78 does not increase but rather decreases abundance of HMGCR in Hap1 cells. This suggests that at least in Hap1 cells, in response to 25-HC, these E3s are dispensable for regulated degradation of HMGCR, in line with a recent report.15 In fact, the reciprocal mutational index of INSIG1 and gp78 is consistent with the notion that gp78 directly promotes ubiquitylation of INSIG1.11,13,37 Accordingly, the level of INSIG1 is elevated in Gp78-ablated primary mouse hepatocytes, in livers of Gp78−/− mice, and after silencing of gp78 in fibroblasts.11,13 In primary hepatocytes, loss of Gp78 elevated the basal level of HMGCR protein and severely attenuated its sterol-stimulated degradation.13 This observation contrasts with our findings in Hap1 cells and with the lack of a role for gp78 in HMGCR degradation recently reported by Tsai et al15 in other cell types. Possibly, flexibility within the ERAD system or an overlapping E3 substrate specificity may limit our ability to conclusively identify a candidate E3 using our genetic approach. Our results, therefore, suggest the possibility that different E3s and yet to be determined cellular factors may be used for this purpose in a cell type and context-dependent manner.
The highest scoring hit identified in our screen was UBXD8, a membrane-embedded recruitment factor for VCP/p97.9,27 UBXD8 has been reported to participate in ERAD of several substrates, including that of INSIG1 and APOB.38,39 Consistent with this, we find that degradation of HMGCR is dependent on UBXD8’s VCP-interacting UBX domain,27,40 leading us to propose that UBXD8 is required for recruiting VCP to allow the extraction of HMGCR from the ER membrane. Indeed, in the absence of UBXD8, HMGCR accumulates in the membranes and fails to be released to the cytosol (Figure 5B). Recent studies have demonstrated a role for UBXD8 in maintaining fatty acid homeostasis because of its ability to sense the level of unsaturated fatty acids in a UAS domain–dependent manner.29 In response to unsaturated fatty acids, UBXD8 partitions between the ER and lipid droplets, where it governs production of triglycerides and the release of fatty acids from triglycerides, respectively.9,29,41 In line with this notion, mice lacking Ubxd8 have been recently reported to develop hepatosteatosis associated with neutral lipid accumulation.42 The functional link between UBXD8’s role in the metabolically regulated ERAD of HMGCR and cholesterol synthesis and in fatty acid homeostasis is intriguing. Given that sterol-stimulated degradation of HMGCR is associated with translocation of the enzyme to lipid droplet–resembling membranes in the ER,43 it is tempting to speculate that the lipid-dependent association of UBXD8 with these membrane domains functionally couples lipid homeostasis to ERAD of HMGCR. With this in mind, it is intriguing that the most significant biological process that we found to affect degradation of HMGCR is peroxisomal biogenesis and function. A finding that is in line with a recent report demonstrating that the peroxisomal biogenesis genes PEX3 and PEX19, both of which are identified in our screen within the mNeonHIGH population, are required for proper insertion of UBXD8 into the outer leaflet of the ER membrane.44 Thus, the UBXD8-dependent degradation of HMGCR may serve as a suitable model to investigate the functional links between lipid metabolism, peroxisomes, and ERAD.
In conclusion, the current study supports the use of mammalian haploid genetics to interrogate complex metabolic traits, thus enabling the identification of both negative and positive metabolic regulators. We demonstrate that UBXD8 is a key regulator of sterol-stimulated degradation of HMGCR, thereby controlling flux through the mevalonate pathway. This positions UBXD8 as an important regulator of cellular lipid homeostasis because of its ability to concomitantly govern both fatty acid and cholesterol synthesis.
Highlights.
An engineered reporter line allows interrogation of endogenous HMGCR (3-hydroxy-3-methylglutaryl coenzyme A reductase) in live cells.
Metabolically regulated degradation of HMGCR requires UBXD8 (ubiquitin regulatory X domain–containing protein 8).
UBXD8 governs HMGCR activity and cholesterol biosynthesis.
Mammalian haploid genetic screens can be applied to interrogate complex metabolic pathways.
Acknowledgments
We thank Franca Tol, Duco Koenis, members of the Zelcer group, Shoshana Bar-Nun, and Irith Koster for their comments, Albert van Wijk for primary hepatocytes, and Jessica Nelson for graphical editing.
Sources of Funding
J. Roitelman is supported by a grant from the Israel Science Foundation (no. 850/14). A. Loregger is supported by a Dekker grant from the Dutch Heart Foundation (2016T015). N. Zelcer is an Established Investigator of the Dutch Heart Foundation (2013T111) and is supported by an ERC Consolidator grant (617376) from the European Research Council.
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- 25-HC
- 25-hydroxycholesterol
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- HMGCR
- 3-hydroxy-3-methylglutaryl coenzyme A reductase
- SREBP
- sterol-response element–binding protein
- UBXD8
- ubiquitin regulatory X domain–containing protein 8
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.117.310002/-/DC1.
References
- 1.Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, Goldstein JL, Brown MS. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75:187–197. [PubMed] [Google Scholar]
- 2.Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, Wang X. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci USA. 1993;90:11603–11607. doi: 10.1073/pnas.90.24.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 4.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 6.Jo Y, Debose-Boyd RA. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit Rev Biochem Mol Biol. 2010;45:185–198. doi: 10.3109/10409238.2010.485605. doi: 10.3109/10409238.2010.485605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharpe LJ, Cook EC, Zelcer N, Brown AJ. The UPS and downs of cholesterol homeostasis. Trends Biochem Sci. 2014;39:527–535. doi: 10.1016/j.tibs.2014.08.008. doi: 10.1016/j.tibs.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 9.Stevenson J, Huang EY, Olzmann JA. Endoplasmic reticulum-associated degradation and lipid homeostasis. Annu Rev Nutr. 2016;36:511–542. doi: 10.1146/annurev-nutr-071715-051030. doi: 10.1146/annurev-nutr-071715-051030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell. 2003;11:25–33. doi: 10.1016/s1097-2765(02)00822-5. [DOI] [PubMed] [Google Scholar]
- 11.Song BL, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19:829–840. doi: 10.1016/j.molcel.2005.08.009. doi: 10.1016/j.molcel.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Jo Y, Lee PC, Sguigna PV, DeBose-Boyd RA. Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc Natl Acad Sci USA. 2011;108:20503–20508. doi: 10.1073/pnas.1112831108. doi: 10.1073/pnas.1112831108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu TF, Tang JJ, Li PS, Shen Y, Li JG, Miao HH, Li BL, Song BL. Ablation of gp78 in liver improves hyperlipidemia and insulin resistance by inhibiting SREBP to decrease lipid biosynthesis. Cell Metab. 2012;16:213–225. doi: 10.1016/j.cmet.2012.06.014. doi: 10.1016/j.cmet.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 14.Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003;278:52479–52490. doi: 10.1074/jbc.M310053200. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- 15.Tsai YC, Leichner GS, Pearce MM, Wilson GL, Wojcikiewicz RJ, Roitelman J, Weissman AM. Differential regulation of HMG-CoA reductase and Insig-1 by enzymes of the ubiquitin-proteasome system. Mol Biol Cell. 2012;23:4484–4494. doi: 10.1091/mbc.E12-08-0631. doi: 10.1091/mbc.E12-08-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zelcer N, Sharpe LJ, Loregger A, Kristiana I, Cook EC, Phan L, Stevenson J, Brown AJ. The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol Cell Biol. 2014;34:1262–1270. doi: 10.1128/MCB.01140-13. doi: 10.1128/MCB.01140-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loregger A, Cook EC, Nelson JK, Moeton M, Sharpe LJ, Engberg S, Karimova M, Lambert G, Brown AJ, Zelcer N. A MARCH6 and IDOL E3 ubiquitin ligase circuit uncouples cholesterol synthesis from lipoprotein uptake in hepatocytes. Mol Cell Biol. 2015;36:285–294. doi: 10.1128/MCB.00890-15. doi: 10.1128/MCB.00890-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, Brummelkamp TR. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- 19.Seglen PO. Hepatocyte suspensions and cultures as tools in experimental carcinogenesis. J Toxicol Environ Health. 1979;5:551–560. doi: 10.1080/15287397909529766. doi: 10.1080/15287397909529766. [DOI] [PubMed] [Google Scholar]
- 20.Lackner DH, Carré A, Guzzardo PM, Banning C, Mangena R, Henley T, Oberndorfer S, Gapp BV, Nijman SM, Brummelkamp TR, Bürckstümmer T. A generic strategy for CRISPR-Cas9-mediated gene tagging. Nat Commun. 2015;6:10237. doi: 10.1038/ncomms10237. doi: 10.1038/ncomms10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakade S, Tsubota T, Sakane Y, Kume S, Sakamoto N, Obara M, Daimon T, Sezutsu H, Yamamoto T, Sakuma T, Suzuki KT. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat Commun. 2014;5:5560. doi: 10.1038/ncomms6560. doi: 10.1038/ncomms6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blomen VA, Májek P, Jae LT, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–1096. doi: 10.1126/science.aac7557. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- 24.Brockmann M, Blomen VA, Nieuwenhuis J, Stickel E, Raaben M, Bleijerveld OB, Altelaar AFM, Jae LT, Brummelkamp TR. Genetic wiring maps of single-cell protein states reveal an off-switch for GPCR signalling. Nature. 2017;546:307–311. doi: 10.1038/nature22376. doi: 10.1038/nature22376. [DOI] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leichner GS, Avner R, Harats D, Roitelman J. Dislocation of HMG-CoA reductase and Insig-1, two polytopic endoplasmic reticulum proteins, en route to proteasomal degradation. Mol Biol Cell. 2009;20:3330–3341. doi: 10.1091/mbc.E08-09-0953. doi: 10.1091/mbc.E08-09-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kloppsteck P, Ewens CA, Förster A, Zhang X, Freemont PS. Regulation of p97 in the ubiquitin-proteasome system by the UBX protein-family. Biochim Biophys Acta. 2012;1823:125–129. doi: 10.1016/j.bbamcr.2011.09.006. doi: 10.1016/j.bbamcr.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 28.Zehmer JK, Bartz R, Bisel B, Liu P, Seemann J, Anderson RG. Targeting sequences of UBXD8 and AAM-B reveal that the ER has a direct role in the emergence and regression of lipid droplets. J Cell Sci. 2009;122(pt 20):3694–3702. doi: 10.1242/jcs.054700. doi: 10.1242/jcs.054700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JN, Kim H, Yao H, Chen Y, Weng K, Ye J. Identification of Ubxd8 protein as a sensor for unsaturated fatty acids and regulator of triglyceride synthesis. Proc Natl Acad Sci USA. 2010;107:21424–21429. doi: 10.1073/pnas.1011859107. doi: 10.1073/pnas.1011859107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris LL, Hartman IZ, Jun DJ, Seemann J, DeBose-Boyd RA. Sequential actions of the AAA-ATPase valosin-containing protein (VCP)/p97 and the proteasome 19 S regulatory particle in sterol-accelerated, endoplasmic reticulum (ER)-associated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2014;289:19053–19066. doi: 10.1074/jbc.M114.576652. doi: 10.1074/jbc.M114.576652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pillay S, Carette JE. Hunting viral receptors using haploid cells. Annu Rev Virol. 2015;2:219–239. doi: 10.1146/annurev-virology-100114-055119. doi: 10.1146/annurev-virology-100114-055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reiling JH, Olive AJ, Sanyal S, Carette JE, Brummelkamp TR, Ploegh HL, Starnbach MN, Sabatini DM. A CREB3-ARF4 signalling pathway mediates the response to Golgi stress and susceptibility to pathogens. Nat Cell Biol. 2013;15:1473–1485. doi: 10.1038/ncb2865. doi: 10.1038/ncb2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popov LM, Marceau CD, Starkl PM, et al. The adherens junctions control susceptibility to Staphylococcus aureus α-toxin. Proc Natl Acad Sci USA. 2015;112:14337–14342. doi: 10.1073/pnas.1510265112. doi: 10.1073/pnas.1510265112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schumacher MM, Elsabrouty R, Seemann J, Jo Y, DeBose-Boyd RA. The prenyltransferase UBIAD1 is the target of geranylgeraniol in degradation of HMG CoA reductase. Elife. 2015;4:e27336. doi: 10.7554/eLife.05560. doi: 10.7554/eLife.05560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 2005;1:379–391. doi: 10.1016/j.cmet.2005.04.010. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Lee JN, Song B, DeBose-Boyd RA, Ye J. Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J Biol Chem. 2006;281:39308–39315. doi: 10.1074/jbc.M608999200. doi: 10.1074/jbc.M608999200. [DOI] [PubMed] [Google Scholar]
- 38.Lee JN, Zhang X, Feramisco JD, Gong Y, Ye J. Unsaturated fatty acids inhibit proteasomal degradation of Insig-1 at a postubiquitination step. J Biol Chem. 2008;283:33772–33783. doi: 10.1074/jbc.M806108200. doi: 10.1074/jbc.M806108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki M, Otsuka T, Ohsaki Y, Cheng J, Taniguchi T, Hashimoto H, Taniguchi H, Fujimoto T. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol Biol Cell. 2012;23:800–810. doi: 10.1091/mbc.E11-11-0950. doi: 10.1091/mbc.E11-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell. 2008;134:804–816. doi: 10.1016/j.cell.2008.06.048. doi: 10.1016/j.cell.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olzmann JA, Richter CM, Kopito RR. Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc Natl Acad Sci USA. 2013;110:1345–1350. doi: 10.1073/pnas.1213738110. doi: 10.1073/pnas.1213738110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imai N, Suzuki M, Hayashi K, Ishigami M, Hirooka Y, Abe T, Shioi G, Goto H, Fujimoto T. Hepatocyte-specific depletion of UBXD8 induces periportal steatosis in mice fed a high-fat diet. PLoS One. 2015;10:e0127114. doi: 10.1371/journal.pone.0127114. doi: 10.1371/journal.pone.0127114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartman IZ, Liu P, Zehmer JK, Luby-Phelps K, Jo Y, Anderson RG, DeBose-Boyd RA. Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J Biol Chem. 2010;285:19288–19298. doi: 10.1074/jbc.M110.134213. doi: 10.1074/jbc.M110.134213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schrul B, Kopito RR. Peroxin-dependent targeting of a lipid-droplet-destined membrane protein to ER subdomains. Nat Cell Biol. 2016;18:740–751. doi: 10.1038/ncb3373. doi: 10.1038/ncb3373. [DOI] [PMC free article] [PubMed] [Google Scholar]