

Abstract
Fatty acid desaturase (FADS) genes encode rate-limiting enzymes for the biosynthesis of omega-6 and omega-3 long chain polyunsaturated fatty acids (LCPUFAs). This biosynthesis is essential for individuals subsisting on LCPUFAs-poor diets (e.g. plant-based). Positive selection on FADS genes has been reported in multiple populations, but its presence and pattern in Europeans remain elusive. Here, using ancient and modern DNA, we demonstrate that positive selection acted on the same FADS variants both before and after the advent of farming in Europe, but on opposite (i.e. alternative) alleles. Selection in recent farmers also varied geographically, with the strongest signal in Southern Europe. These varying selection patterns concur with anthropological evidence of varying diets, and with the association of farming-adaptive alleles with higher FADS1 expression and thus enhanced LCPUFAs biosynthesis. Genome-wide association studies reveal that farming-adaptive alleles not only increase LCPUFAs, but also affect other lipid levels and protect against several inflammatory diseases.
Identifying genetic adaptations to local environment, including historical diets, and elucidating their implication in human health and disease are of central interest in human evolutionary genomics1. The fatty acid desaturase (FADS) gene family consists of FADS1, FADS2 and FADS3, which evolved by gene duplication2. FADS1 and FADS2 encode rate-limiting enzymes for the biosynthesis of omega-3 and omega-6 long-chain polyunsaturated fatty acids (LCPUFAs) from plant-sourced shorter-chain precursors (Supplementary Fig. 1). LCPUFAs are indispensable for proper human brain development, cognitive function and immune response3,4. While omega-3 and omega-6 LCPUFAs can be obtained from animal-based diets, their biosynthesis is essential to compensate for their absence from plant-based diets. Positive selection on the FADS locus, a 100 kilobase (kb) region containing all three genes (Supplementary Fig. 2), has been identified in multiple populations5–10. Our recent study showed that a 22 bp insertion-deletion polymorphism (indel, rs66698963) within FADS2, which is associated with FADS1 expression11, has been adaptive in Africa, South Asia and parts of East Asia, possibly driven by local historical plant-based diets8. We further supported this hypothesis by association of the adaptive insertion allele with more efficient biosynthesis8. In Greenlandic Inuit, who traditionally subsisted on a LCPUFAs-rich marine diet9, as well as in Native Americans10, adaptation signals were also observed for FADS genes, with adaptive alleles associated with less efficient biosynthesis9.
In Europeans, positive selection on FADS genes was only reported recently in a study based on ancient DNA (aDNA)12. Evidence from modern DNA is still lacking even though most above studies also performed similarly-powered tests in Europeans5–8. Moreover, although there are well-established differences in the Neolithization process and in dietary patterns across Europe13–15, geographical differences of selection within Europe have not been investigated. Furthermore, before the advent of farming, pre-Neolithic hunter-gatherers throughout Europe had been subsisting on animal-based diets with significant aquatic contribution16–18, in contrast to the plant-heavy diets of recent European farmers19–21. We hypothesized that these drastic dietary differences exerted different selection pressure on FADS genes. In this study, we combined analyses on ancient and modern DNA to investigate positive selection on FADS genes in Europe and to examine whether it exhibits geographical and temporal differences. We presented evidence for positive selection on opposite (i.e. alternative) alleles of the same variants before and after the Neolithic revolution, and for varying selection signals between Northern and Southern Europeans in recent history. We interpreted the functional significance of adaptive alleles with expression quantitative trait loci (eQTLs) analysis and genome-wide association studies (GWAS), both pointing to selection for diminishing LCPUFAs biosynthesis in pre-Neolithic hunter-gatherers but for enhancing biosynthesis in recent farmers. Anthropological findings indicate that these selection patterns were likely driven by varying and changing dietary practices.
Results
Evidence of recent positive selection in Europe from both ancient and modern DNA
To systematically evaluate the presence of recent positive selection on FADS genes in Europe, we performed an array of selection tests using both ancient and modern samples. We generated a uniform set of variants across the locus in a variety of aDNA datasets (Supplementary Table 1) via imputation. We first conducted an aDNA-based test, which identifies variants with extreme frequency change between three ancient and four modern samples, suggesting positive selection during recent European history (not more ancient than 8,500 years ago (ya))12. The three ancient samples represent the three major ancestry sources of most present-day Europeans12: Western and Scandinavian hunter-gatherers (WSHG), early European farmers (EF), and Steppe-Ancestry pastoralists (SA). The four modern samples were drawn from the 1000 Genomes Project (1000GP), representing Tuscans (TSI), Iberians (IBS), British (GBR) and Utah residents with Northern and Western European ancestry (CEU). We confirmed significant selection signals on many variants in FADS genes (Fig. 1), including the previously identified peak single nucleotide polymorphism (SNP) rs174546 (p = 1.04e-21)12. The most significant signal is at an imputed SNP, rs174594 (p = 1.29e-24), which was not included in the original study12. SNP rs174570, reported as adaptive in Greenlandic Inuit9, also exhibits a significant signal (p = 7.64e-18) while indel rs66698963 shows no evidence (p = 3.62e-3, likely due to data quality, see Supplementary Notes). Overall, the entire peak of selection signals coincides with a linkage disequilibrium (LD) block (referred to as the FADS1–FADS2 LD block) in Europeans, which extends over a long genomic region of 85 kb, covering the entire FADS1 and most of FADS2 (Supplementary Figs 2–3). The dominant haplotype of this block (haplotype D) has a frequency of 63% in modern Europeans and is composed of alleles under positive selection as revealed by the above test. Of note, some alleles on this haplotype are derived (i.e. the new mutation relative to primates) while others are ancestral (Supplementary Fig. 4). Thus, the large number of variants with significant signals might result from strong selection on one or a few variants, with extensive hitchhiking of nearby neutral variants.
Fig. 1. Ancient DNA-based test for recent positive selection.
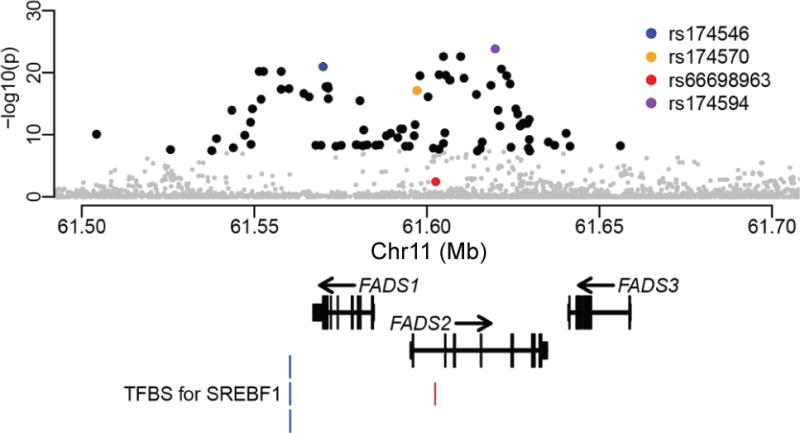
Each point represents a variant with its genomic location on the x-axis and its p value (genomic control corrected and at a negative logarithmic scale) on the y-axis. Four variants are highlighted: the most significant SNP (purple); the top SNP reported by Mathieson et al. based on the same test but without imputation12 (blue); one of the top adaptive SNPs reported in Greenlandic Inuit9 (orange); the adaptive indel reported in multiple populations with historical plant-based diets8 (red). Other variants are in black if exceeding the genome-wide significance level (5e-8), otherwise in gray. The overall pattern is consistent with that previously described12 (Supplementary Fig. 31). At the bottom of the plot are the representative transcript models for the three FADS genes and the four transcription factor binding sites for SREBF1 from ENCODE68 (blue) and another previous study11 (red).
We next performed several selection tests solely based on extant populations. Considering five European populations from 1000GP, including the four mentioned above and Finns (FIN), a haplotype-based selection test, nSL22, revealed positive selection on many SNPs in the FADS1–FADS2 LD block. Importantly, it unraveled the same adaptive alleles as in the aDNA-based test and the same general trend of stronger signal towards rs174594 (Fig. 2a, Supplementary Fig. 5). For rs174594, nSL values are significant in all five populations and exhibit a gradient of being stronger towards the South (Fig. 2a, Supplementary Fig. 6): TSI (p = 0.00044), IBS (p = 0.0020), CEU (p = 0.0039), GBR (p = 0.0093), and FIN (p = 0.017). Of note, nSL values have been normalized separately in each population to remove demographic effects22. The other three variants of interest exhibit no selection signals, except rs174570 showing borderline significance in the two southernmost populations (TSI: p = 0.022; IBS: p = 0.050, Fig. 2a). Signals were also observed with nSL in two whole-genome sequencing cohorts from the UK10K project (Supplementary Fig. 7). Another test for positive selection in very recent history (during the past ~2,000–3,000 years), Singleton Density Score (SDS)23, applied in the UK10K dataset, also revealed significant signals in the FADS1–FADS2 LD block, with the same adaptive alleles and general trend of localized signals as in the two above tests (Fig. 2b, Supplementary Fig. 8). Significant SDS was observed for rs174594 (p = 0.045) and rs174570 (p = 0.045), but not rs174546. It is the derived allele for rs174594 that was under selection, while it is the ancestral allele for rs174570. Interestingly, selection on the alternative, derived allele of rs174570 has been shown in Greenlandic Inuit9. Additional tests of selection consistently revealed positive selection signals (Supplementary Figs 5, 7–10). Taken together, standard tests on modern DNA support the aDNA-based result of recent positive selection on the D haplotype of the FADS1–FADS2 LD block.
Fig. 2. Tests for recent positive selection based solely on modern DNA.
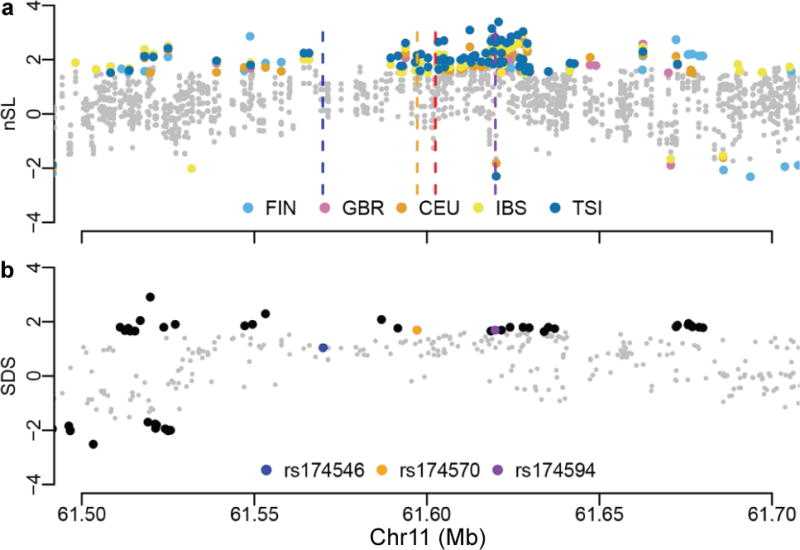
a, Haplotype-based selection test (nSL22) in modern Europeans from 1000GP. The test was performed separately for each of the five European populations. Only variants with significant values are shown with population-specific colors as indicated in the legend. The positions for four variants of interest were indicated with vertical dashed lines, colored as in Fig. 1. For presentation purpose, the sign was set so that being positive indicates that the adaptive allele revealed by nSL is consistent with that revealed by the aDNA-based test in Fig. 1. Original statistics for 1000GP and UK10K are shown in Supplementary Figs 5 and 7. b, Singleton Density Score (SDS23) in modern Europeans from UK10K. Variants under significance level are in gray except for highlighted ones. Three variants of interest were highlighted with colors as indicated in the legend. The indel rs66698963 was not present in the original UK10K dataset. The sign of SDS was set as in nSL. Original statistics are shown in Supplementary Fig. 8.
Geographical differences of recent positive selection signals across Europe
To evaluate geographical differences of recent positive selection on FADS genes across Europe, we revisited the aDNA-based selection test12. We first decomposed the original test for four representative SNPs (Fig. 3a) and then performed the test separately in Northern and Southern Europe for all variants around FADS genes (Fig. 3b). The original test evaluates the frequencies of an allele in both ancient and modern samples under two hypotheses (H0 and H1). Under H1, maximum likelihood estimates (MLEs) of frequencies in all samples are constrained only by observed allele counts and thus equivalent to the observed frequencies (Fig. 3a, blue bars). The observed adaptive allele frequencies for all four SNPs exhibit a South-North gradient in modern samples, with the highest in Tuscans and the lowest in Finns, consistent with the gradient of selection signals observed before. Among ancient samples, the observed allele frequencies, equivalent to the frequencies upon admixture (Fig. 3a, orange bars), are always the lowest and often zero in the WSHG sample.
Fig. 3. Varying selection and frequency patterns between Southern and Northern Europe.
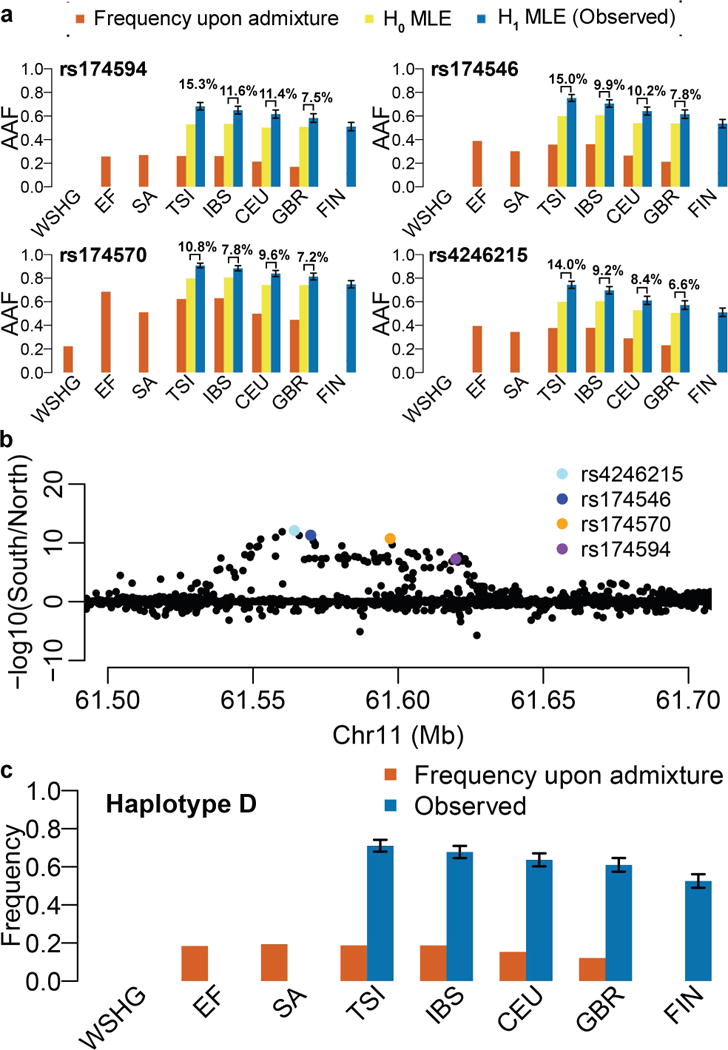
a, South-North frequency gradient for adaptive alleles of four representative SNPs under different scenarios of frequency estimation. Three SNPs (rs174594, rs174546, and rs174570) are top SNPs from this and previous studies9,12, while the fourth (rs4246215) is the one showing the biggest difference in the upcoming South-North comparison analysis. The indel rs66698963 is not highlighted in this and all upcoming analyses because it has no significant selection signals in Europe. AAF refers to adaptive allele frequency. Orange bars represent frequencies upon admixture, which were directly observed in ancient groups and predicted for extant populations based on linear mixture of frequencies in ancient groups. Yellow bars represent frequencies estimated under H0. Estimates for ancient groups were not shown because they are not relevant here. Blue bars represent frequencies estimated under H1, whose only constraint is the observed data and therefore the MLEs are just the observed means. The estimates for ancient groups are the same as their frequencies upon admixture and are omitted on the plot. The absolute difference between H0 and H1 estimates are indicated above the corresponding bars. Please note that the frequencies upon admixture in WSHG are 0 for rs174594, rs174546 and rs4246215 and no bars were plotted. b, Comparison of aDNA-based selection signals between Southern and Northern Europe. aDNA-based selection tests were performed separately for Southern (TSI and IBS) and Northern (CEU and GBR) Europeans. For each variant, the p values from these two tests were compared at a −log10 scale (y-axis). SNPs of interest were colored as indicated. c, South-North frequency gradient for the adaptive haplotype in extant populations. The two frequency types are just as in a. The frequency upon admixture for WSHG is 0. In a and c, FIN has only observed values. If values are not shown or not available, signs of “//” are indicated at corresponding positions. Error bars stand for standard errors.
Under H0, the MLEs of frequencies are constrained by observed allele counts and an additional assumption that an allele’s frequencies in the four modern samples are each a linear combination of its frequencies in the three ancient samples. Considering the later assumption alone, we can predict the frequencies of adaptive alleles right after admixture for each modern population. The admixture contribution of WSHG, as estimated genome-wide, is higher towards the North12. Thus, the predicted adaptive allele frequencies upon admixture for modern populations are usually lower in the North (Fig. 3a, orange bars), suggesting higher starting frequencies in the South at the onset of selection. Further considering observed allele counts, we obtained the MLEs of frequencies under H0 (Fig. 3a, yellow bars). As expected, they are higher in the South. But more importantly, the differences between H0 and H1 estimates in modern populations (Fig. 3a, indicated differences between yellow and blue bars) are higher in the South, suggesting that in addition to population-specific admixture proportions and different starting frequencies, more recent factors, such as stronger selection pressure, earlier onset of selection, or unmodeled recent demographic history, might contribute to the observed stronger selection signals in the South.
To examine potential confounding effects of varying demographic history that is not captured by the model, we evaluated all variants in a 3 Mb region surrounding FADS genes. We applied the aDNA-based test separately for Southern and Northern populations. All variants that were significant in the combined analyses (Fig. 1) were also significant in each of the two separate analyses, but many exhibited much stronger signals in Southern populations (Fig. 3b; Supplementary Fig. 11). The maximum difference was found for rs4246215, whose p value in Southern populations is 12 orders of magnitude stronger than that in Northern populations. SNPs rs174594, rs174546 and rs174570 also have signals that are several orders of magnitude stronger in the South. A further decomposition of the test and comparing maximum likelihoods under H0 and H1 between South and North revealed that a stronger deviation under H0 in the South is driving the signal (Supplementary Fig. 12). The pattern of stronger signal in the South is observed only for some but not all SNPs, excluding the possibility of systemic bias and pointing at variant-specific properties, likely for variants that were under selection and the nearby variants in LD. Indeed, the candidate adaptive haplotype D also exhibits frequency patterns that are consistent with adaptive alleles of the four representative SNPs (Fig. 3c). Hence, these results suggest that there might be stronger selection pressure or earlier onset of positive selection on the FADS1–FADS2 LD block in Southern Europeans.
Opposite selection signals in pre-Neolithic European hunter-gatherers
Motivated by the very different diet of pre-Neolithic hunter-gatherers, we set to investigate natural selection on FADS genes before the Neolithic revolution. We examined the frequency trajectory of haplotype D, the candidate adaptive haplotype in recent European history. In stark contrast to its drastic frequency increase after the Neolithic revolution (Fig. 3c), its frequency decreased over time among pre-Neolithic hunter-gatherers24 (Fig. 4a): starting from 32% in the ~30,000-year-old (yo) “Věstonice cluster”, through 21% in the ~15,000 yo “El Mirón cluster”, to 13% in the ~10,000 yo “Villabruna cluster”, and to being absent in the ~7,500 yo WSHG. We hypothesized that there was positive selection on alleles alternative to recently adaptive ones on haplotype D.
Fig. 4. Temporal frequency pattern and selection signals in pre-Neolithic European hunter-gatherers.
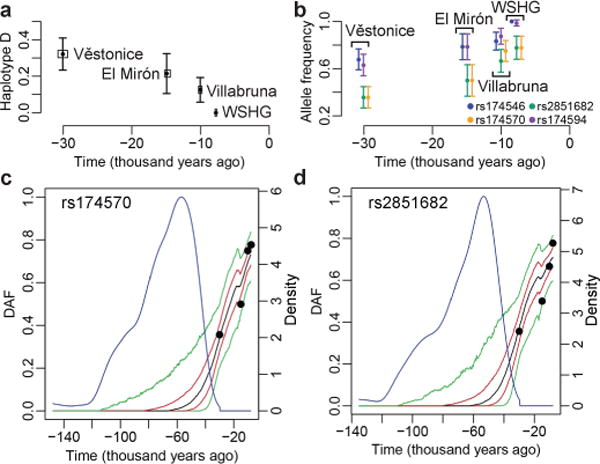
a, The observed frequency of haplotype D over time in four groups of hunter-gatherers. Frequency for each group is plotted as a black point at the median age of samples. The horizontal box surrounding the point represents the medians of lower- and upper-bound estimates of sample ages. Error bars are standard errors. Group names are indicated next to their frequencies. The frequency for WSHG is 0. b, Observed allele frequencies for four SNPs. It has similar format as in a except that small arbitrary values were added on their x coordinates in order to visualize all SNPs, which were colored as indicated in the legend. The alleles chosen are the ones increasing frequency over time. They are derived alleles for rs174570 and rs2851682, and ancestral alleles for rs174546 and rs174594. c and d, Inferences of positive selection based on observed allele frequency time series with the Schraiber et al. method25, for rs174570 and rs2851682, respectively. The observed frequencies are indicated with black points, which are the same point estimates as in b. The posterior distribution of the derived allele frequency (DAF) change over time was estimated with 1,000 Markov chain Monte Carlo samples. The median, 25% and 75% quantiles, and 5% and 95% quantiles of the distribution are indicated respectively with black, red and green lines. The posterior distribution on the age of derived allele is shown with a blue line, with values on the right y-axis. Selection coefficients simultaneously estimated in the analysis were significant for both SNPs (Supplementary Figs 16–17).
To search for variants under positive selection during the pre-Neolithic period, we considered the allele frequency time series for all variants around FADS genes. We applied two rigorous, recently-published Bayesian methods25,26 to infer selection coefficients. Under a simple demographic model of constant population size, both methods highlighted two SNPs (rs174570 and rs2851682) within the FADS1–FADS2 LD block to be under positive selection during the period tested, approximately 30,000-7,500 ya (Supplementary Figs 13–14). The Schraiber et al. method is capable of processing more complicated demographic models25. With this method and considering a more realistic demographic model, the same two SNPs were highlighted (Supplementary Fig. 15). The derived alleles of these two SNPs both increased from 36% to 78% (Fig. 4b). Estimated selection coefficients for homozygotes of adaptive allele (s) for these two SNPs are similar across methods and demographic models. With the Schraiber et al. method and the realistic demographic model, the marginal maximum a posteriori estimate of s for rs174570 is 0.38% (95% credible interval (CI): 0.038% – 0.92%) while the estimated derived allele age is 57,380 years (95% CI: 157,690 – 41,930 years) (Fig. 4c, Supplementary Fig. 16). For rs2851682, the estimated s is 0.40% (95% CI: 0.028% – 1.12%) while its derived allele age is 53,440 years (95% CI: 139,620 – 39,320 years) (Fig. 4d, Supplementary Fig. 17). In addition to these two SNPs, ApproxWF26 revealed significant signals for 44 SNPs in the FADS1–FADS2 LD block (Supplementary Fig. 14), including rs174546 and rs174594, whose ancestral allele frequencies increased from about 65% to almost fixation (Fig. 4b). Importantly, these SNPs have similar estimated s (0.28% - 0.62%) and their adaptive alleles are alternative to the ones under selection in recent history.
Considering haplotype structure of the FADS1–FADS2 LD block (Fig. 5a), we identified a haplotype (referred to as M2), which is comprised of alleles that are mostly alternative to those on haplotype D (Supplementary Fig. 4). M2 appears in modern Europeans at a frequency of 10% but is much more common in Eskimos from Eastern Siberia, presumably for the same reason that the derived allele of rs174570 is prevalent in Greenlandic Inuit. M2 exhibits increasing frequency in pre-Neolithic hunter-gatherers (Supplementary Table 2), suggesting that allele(s) targeted by selection during that period are likely on M2.
Fig. 5. Haplotype network and geographical frequency distribution.
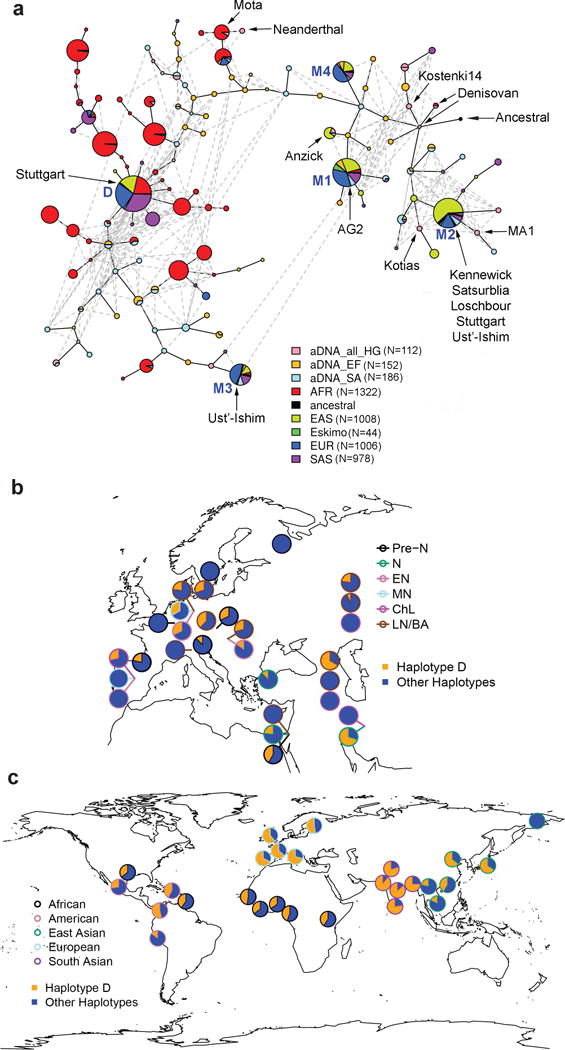
a, Haplotype network for 1000GP samples (2,157 individuals, excluding admixed American samples), 22 modern Eskimos and 225 aDNAs. Each pie chart represents one haplotype and its size is proportional to log2(# of haplotype) plus a minimum size to visualize rare haplotypes. Sections in the pie provide the breakdown by groups. Detailed haplotype frequencies are in Supplementary Table 2. The edges connecting haplotypes are of arbitrary length. Haplotypes for some well-known ancient samples are labelled. The top five haplotypes in modern Europeans, referred to as D, M1, M2, M3, and M4 from the most to least frequent common (63%, 15%, 10%, 5%, 4%, respectively), are indicated with their names in blue. M1, M2 and M4 are closer to the consensus ancestral haplotype observed in primates while D and M3 are more distant. b, Frequency of haplotype D in Eurasian ancient DNAs. Each pie represents one sampled group and is placed at the sampling location or nearby with a line pointing at the sampling location. The color of the pie chart border indicates the archaeological period. If multiple samples of different periods were collected at the same geographical location, these samples are ordered vertically with the older samples at the bottom. Hunter-gatherer groups are indicated with black arrows and pastoralist groups with gray arrows, while others are farmers. Geographical locations for some hunter-gatherer groups (e.g. the Věstonice, El Mirón and Villabruna clusters) are only from representative samples. Detailed frequencies are in Supplementary Table 3. Pre-N: Pre-Neolithic; N: Neolithic; EN: Early Neolithic; MN: Mid-Neolithic; ChL: Chalcolithic; LN/BA: Late Neolithic/Bronze Age. c, Frequency of haplotype D in present-day global populations. All 26 populations from 1000GP and one Eskimo group are included. The color of the pie chart border represents the genetic ancestry. It is noteworthy that there are two samples in America that are actually of African ancestry. Detailed frequencies are in Supplementary Table 4.
The temporal and global evolutionary trajectory of FADS haplotypes
To study different haplotypes in the FADS1–FADS2 LD block, their frequency changes over time and their current global distributions, we performed haplotype network and frequency analysis on 450 and 5,052 haplotypes from ancient and modern DNA, respectively (Fig. 5, Supplementary Fig. 18, Supplementary Tables 2–4). The top five haplotypes in modern Europeans, designated as D, M1, M2, M3 and M4 from the most to the least common, were all present in aDNA and modern Africans. Among the Out-of-Africa ancestors, the frequencies of D and M2 were probably around 35% and 27%, respectively, because these were observed in both the oldest European hunter-gatherer group, the ~30,000 yo “Věstonice cluster”, and the ~14,500 yo Natufian hunter-gatherers in the Levant (Fig. 5b, Supplementary Table 3). Among pre-Neolithic European hunter-gatherers, positive selection on M2 increased its frequency from 29% to 56% from approximately 30,000 to 7,500 ya, while the D haplotype practically disappeared by the advent of farming (Figs 4a, 5b). With the arrival of farmers and Steppe-Ancestry pastoralists, D was re-introduced into Europe. Since the Neolithic revolution, positive selection on D increased its frequency dramatically to 63% while the M2 frequency decreased to 10% among present-day Europeans. Globally, D is present at high frequency in South Asia (82%) but absent in modern-day Eskimos (Fig. 5c). In contract, M2 has very low frequency in South Asia (3%) but moderate frequency in Eskimos (27%). Detailed description of evolutionary trajectories of FADS haplotypes could be found in Supplementary Notes.
The geographical frequency patterns of representative variants (Fig. 6, Supplementary Figs 19–23) mostly mirror those of key haplotypes, but with discrepancies providing insights into casual variants and allele ages. One major discrepancy was found in Africa. The derived alleles of rs174570 and rs2851682 remain almost absent in Africa, consistent with their allele age estimates of ~55,000 years (Figs 4c, d) and ruling out their involvement in the positive selection on FADS genes in Africa5,6,8. Considering the much weaker LD structure of the FADS locus in Africa (Supplementary Fig. 24), it is possible that selection in Africa may be on haplotypes and variants that are different from those in Europe.
Fig. 6. Geographical frequency distribution for SNPs rs174594 and rs174570 in present-day global populations.
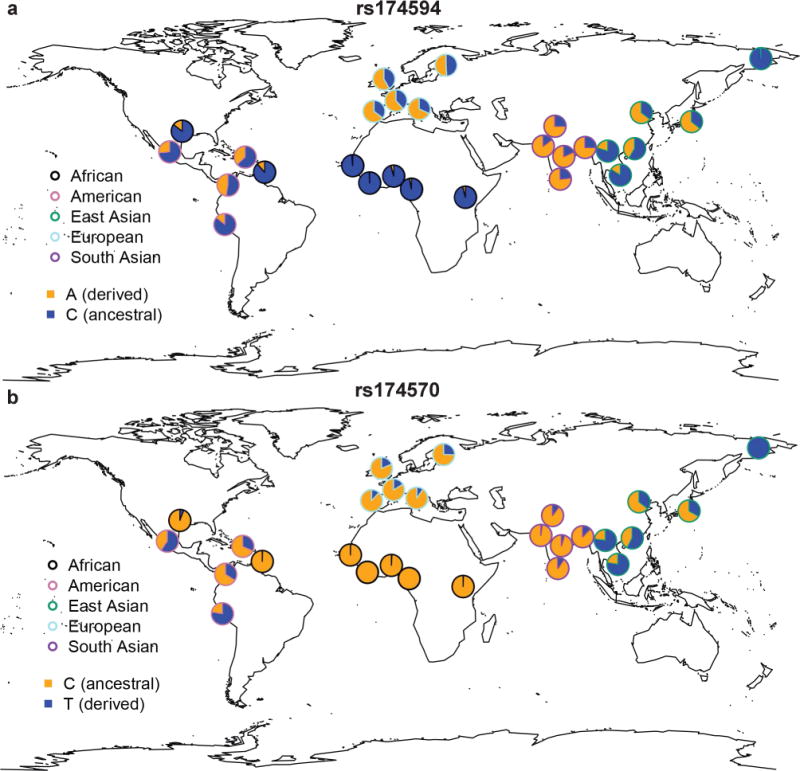
Adaptive alleles in recent European history are colored in orange. All 26 populations from 1000GP and one Eskimo group are included. The color of the pie chart border represents the genetic ancestry. It is noteworthy that there are two samples in America that are actually of African ancestry. Similar global patterns were observed with HGDP samples (Supplementary Figs 19 and 21).
Functional and medical implications of adaptive variants
With data from the Genotype-Tissue Expression (GTEx) project27, we identified many SNPs on the FADS1–FADS2 LD block being eQTLs of FADS genes. Out of a total of 44 tissues, these eQTLs at genome-wide significance level are associated with the expression of FADS1, FADS2, and FADS3 in 12, 23, and 4 tissues, respectively, for a total of 27 tissues (Supplementary Figs 25–27). Considering rs174594, nominally significant associations with these three genes were found in 29, 28 and 4 tissues, respectively. Importantly, out of these tissues, the recently adaptive allele is associated with higher FADS1, lower FADS2 and higher FADS3 expression in 28, 27 and 4 tissues, respectively. This general trend was observed for other recently adaptive alleles on haplotype D.
GWAS have revealed 178 associations with 44 traits in the FADS1–FADS2 LD block, as recorded in the GWAS Catalog (Supplementary Tables 5–9, Supplementary Notes and references therein)28. We report here the direction of associations for recently adaptive alleles, while the direction is opposite for adaptive alleles in pre-Neolithic hunter-gatherers. (1) The most prominent group of associated traits are polyunsaturated fatty acids (PUFAs, Supplementary Fig. 1), including LCPUFAs and their precursors. Recently adaptive alleles are associated with higher levels of arachidonic acid (AA), adrenic acid (AdrA), eicosapentaenoic acid (EPA) and docosapentaenoic acid (DPA), but with lower levels of dihomo-gamma-linolenic acid (DGLA), all of which suggest increased activity of delta-5 desaturase encoded by FADS1. This is consistent with the association of recently adaptive alleles with higher FADS1 expression. Surprisingly, these alleles are associated with higher levels of gamma-linolenic acid (GLA) and stearidonic acid (SDA), but with lower levels of linoleic acid (LA) and alpha- linolenic acid (ALA), suggesting increased activity of delta-6 desaturase encoded by FADS2. However, the above eQTL analysis suggested that recently adaptive alleles are associated with lower FADS2 expression. Some of these associations have been replicated across Europeans, Africans, East Asians, and Hispanic/Latino. (2) Besides PUFAs, recently adaptive alleles are associated with decreased cis/trans-18:2 fatty acids, which in turn is associated with lower risk of systemic inflammation and cardiac death29. Consistently, these alleles are also associated with decreased resting heart rate, which reduces risk of cardiovascular diseases30. (3) Regarding other lipids, recently adaptive alleles are associated with higher levels of high-density lipoprotein cholesterol (HDL), low-density lipoprotein cholesterol (LDL) and total cholesterol (TC), but lower levels of triglycerides. (4) In terms of disease risk, these alleles are associated with lower risk of inflammatory bowel diseases, both Crohn’s disease and ulcerative colitis, and of bipolar disorder.
Going beyond known associations, we analyzed the UK10K datasets with a focus on rs174594. We confirmed the association of the recently adaptive allele with higher levels of TC, LDL, and HDL. We further revealed its association with higher levels of Apo A1 and Apo B (Supplementary Fig. 28). Taken together, recently adaptive alleles, beyond their associations with fatty acid levels, are associated with factors protective against inflammatory and cardiovascular diseases, and indeed show direct associations with decreased risk of inflammatory bowel diseases.
Discussion
Positive selection on FADS genes after the Neolithic revolution in Europe has been previously reported12. A study conducted in parallel to ours tried to identify targets of recent positive selection in Europe by comparing allele frequency changes between present-day and Bronze Age (5,000 – 3,000 ya) Europeans and concluded that they might be different in Europe from those in South Asia and Greenland31. In this study, we provided a detailed view of the recent selection in Europe and revealed that it varied geographically, between the North and the South (Figs 1–3). We further discovered a unique phenomenon that before the Neolithic revolution, the same variants were also subject to positive selection, but with the alternative alleles being selected (Fig. 4). We showed that alleles diminishing LCPUFAs biosynthesis were adaptive before the Neolithic revolution, while alleles enhancing biosynthesis were adaptive after the Neolithic revolution. In Supplementary Notes, we provided detailed discussions, including 1) interpreting results from different selection tests, especially considering the complications of selection on alternative alleles in two historic periods and selection on standing variations in recent history; 2) interpreting results concerning South-North differences, with consideration of potential geographical differences in demographic history; 3) interpreting eQTLs and GWAS results; and 4) examining the role of SNP rs174557, which has been functionally highlighted in another parallel study32. Here, we focus on interpreting the selection patterns in light of anthropological findings.
The dispersal of the Neolithic package into Europe about 8,500 ya caused a sharp dietary shift from an animal-based diet with significant aquatic contribution to a terrestrial plant-heavy diet including dairy products16–21. For pre-Neolithic European hunter-gatherers, the significant role of aquatic food, either marine or freshwater, has been established in sites along the Atlantic coast18,33–35, around the Baltic sea18, and along the Danube river36. The content of LCPUFAs is usually the highest in aquatic foods, lower in animal meat and milk, and almost negligible in most plants37. Consistent with the dietary pattern, positive selection in pre-Neolithic hunter-gatherers was on alleles associated with less efficient LCPUFAs biosynthesis, possibly compensating for the high dietary input. In addition to obtaining sufficient amounts of LCPUFAs, maintaining a balanced ratio of omega-6 to omega-3 is critical for human health38. Hence, it is also plausible that positive selection in hunter-gatherers was in response to an unbalanced omega-6 to omega-3 ratio (i.e. too much omega-3 LCPUFAs). Positive selection on FADS genes was also observed in modern Greenlandic Inuit, who subsist on a seafood diet9. It is noteworthy that aquatic food was less prevalent among pre-Neolithic hunter-gatherers around the Mediterranean basin, possibly due to the low productivity of the Mediterranean Sea39–41. It would be interesting to examine the geographical differences of selection in pre-Neolithic Europe. However, pre-Neolithic aDNA is still scarce, prohibiting such an analysis at present.
The Neolithization of Europe13,42,43 started around 8,500 ya when farming and herding spread into the Aegean and the Balkans. Despite a few temporary stops, it continued spreading into Central and Northern Europe following the Danube River and its tributaries, and along the Mediterranean coast. It arrived at the Italian Peninsula about 8,000 ya and reached Iberia by 7,500 ya. While farming rapidly spread across the loess plains of Central Europe and reached the Paris Basin by 7,000 ya, it took another 1,000 or more years before it spread into Britain and Northern Europe around 6,000 ya. From then on, European farmers relied heavily on domesticated animals and plants. Compared to pre-Neolithic hunter-gatherers, farmers consumed much more plants and less aquatic foods19–21,44. Consistent with the lack of LCPUFAs in plant-based diets, positive selection on FADS genes during recent European history was on alleles associated with enhanced LCPUFAs biosynthesis from plant-derived precursors (LA and ALA). Positive selection for enhanced LCPUFAs synthesis has also been observed in Africans, South Asians and some East Asians, possibly driven by their traditional plant-based diets5,6,8.
Despite the overall trend of relying heavily on domesticated plants, there are geographical differences of dietary patterns among European farmers. In addition to the 2,000-year-late arrival of farming at Northern Europe, animal husbandry and the consumption of animal milk became gradually more prevalent as Neolithic farmers spread to the Northwest19,43,45–47. Moreover, similar to their pre-Neolithic predecessors, Northwestern European farmers close to the Atlantic Ocean or the Baltic Sea still consumed more marine food than their Southern counterparts in the Mediterranean basin48,49. It is noteworthy that historic dairying practice in Northwestern Europe has driven the adaptive evolution of lactase persistence in Europe to reach the highest prevalence in this region46. In this study, we observed that recent selection signals for alleles enhancing LCPUFAs biosynthesis are stronger in Southern than in Northern Europeans, even after considering the later arrival of farming and the lower starting allele frequencies in the North. The higher aquatic contribution and stronger reliance on animal meat and milk might be responsible for a weaker selection pressure in the North. However, since GWAS have unraveled many traits and diseases associated with FADS genes, it is possible that other environmental factors were involved.
Conclusions
We presented several lines of evidence for positive selection on FADS genes in Europe and for its geographically and temporally varying patterns. These patterns concur with mounting anthropological evidence of geographical variability and historical change in diet. Specifically, in pre-Neolithic hunter-gatherers subsisting on animal-based diets with significant aquatic contribution, LCPUFAs-synthesis-diminishing alleles were adaptive. In recent European farmers subsisting on plant-heavy diets, LCPUFAs-synthesis-enhancing alleles were adaptive. Importantly, these are not simply any alleles with opposite functional consequence, but are alternative alleles of the same variants such that when one is under selection and increases in frequency, the other will decrease in frequency. To the best of our knowledge, this is the first example of its kind in humans. Moreover, we reported geographically varying patterns of recent selection that are in line with a stronger dietary reliance on plants in Southern European farmers. These unique, varying patterns of positive selection in different dietary environments, together with the large number of traits and diseases associated with the adaptive region, highlight the importance and potential of matching diet to genome in the future nutritional practice.
Methods
Ancient DNA
The ancient DNA (aDNA) dataset was compiled from two previous studies24,50, which in turn were assembled from many studies, in addition to new sequenced samples. These two datasets were merged by removing overlapping samples. In total, there are 325 ancient samples included in this study. Information about these samples and their original references could be found in Supplementary Table 1. For the aDNA-based test for recent selection, a subset of 178 ancient samples were used and clustered into three groups as in the original study12, representing the three major ancestral sources for most present-day European populations. These three groups are: West and Scandinavian hunter-gatherers (WSHG, N=9), early European farmers (EF, N=76), and individuals of Steppe-pastoralist Ancestry (SA, N=93). Three samples in the EF group in the original study were excluded from our analysis because they are genetic outliers based on additional analysis50. For aDNA-based tests for ancient selection in pre-Neolithic European hunter-gatherers, a subset of 42 ancient samples were used and four groups were defined. In addition to the WSHG (N=9), the other three groups were as originally defined in a previous study24: the “Věstonice cluster”, composed of 14 pre-Last Glacial Maximum individuals from 34,000-26,000 ya; the “El Mirón cluster”, composed of 7 post-Last Glacial Maximum individuals from 19,000-14,000 ya; the “Villabruna cluster”, composed of 12 post-Last Glacial Maximum individuals from 14,000-7,000 ya. There were three Western hunter-gatherers that were originally included in the “Villabruna cluster”24, but we included them in WSHG in the current study because of their similar ages in addition to genetic affinity12. In haplotype network analysis, all aDNAs included in the two aDNA-based selection tests were also included. In addition, we included some well-known ancient samples, such as the Neanderthal, Denisovan, and Ust’-Ishim. In total, there were 225 ancient samples (450 haplotypes). For geographical frequency distribution analysis, a total of 300 ancient samples were used and classified into 29 previously defined groups12,24,50 based on their genetic affinity, sampling locations and estimated ages.
Modern DNA
The 1000 Genomes Project (1000GP, phase 3)7 has sequencing-based genome-wide SNPs for 2,504 individuals from 5 continental regions and 26 global populations. Detailed description of these populations and their sample sizes are in Supplementary Methods. The Human Genome Diversity Project (HGDP)51 has genotyping-based genome-wide SNPs for 939 unrelated individuals from 51 populations. The data from the Population Reference Sample (POPRES)52 were retrieved from dbGaP with permission. Only 3,192 Europeans were included in our analysis. The 22 Eskimo samples were extracted from the Human Origins dataset53.
The two sequencing cohorts of UK10K were obtained from European Genome-phenome Archive with permission54. These two cohorts, called ALSPAC and TwinsUK, included low-depth whole-genome sequencing data and a range of quantitative traits for 3,781 British individuals of European ancestry (N=1,927 and 1,854 for ALSPAC and TwinsUK, respectively)54.
Imputation for ancient and modern DNA
Genotype imputation was performed using Beagle 4.155 separately for datasets of aDNA, HGDP and POPRES. The 1000GP phase 3 data were used as the reference panel7. Imputation was performed for a 5 Mb region surrounding the FADS locus (hg19:chr11: 59,100,000–64,100,000), although most of our analysis was restricted to a 200 kb region (hg19:chr11:61,500,000–61,700,000). For most of our analysis (e.g. estimated allele count or frequency for each group), genotype probabilities were taken into account without setting a specific cutoff. For haplotype-based analysis (e.g. estimated haplotype frequency for each group), a cutoff of 0.8 was enforced and haplotypes were defined with missing data and following the phasing information from imputation.
Genotype imputation for aDNA has been shown to be desirable and reliable56. We also evaluated the imputation quality for aDNA by comparing with the two modern datasets (Supplementary Fig. 29). Overall, the imputation accuracy for ungenotyped SNPs, measured with allelic R2 and dosage R2, is comparable between aDNA and HGDP, but is higher in aDNA when compared with POPRES. Note that sample sizes are much larger for HGDP (N=939) and POPRES (N=3,192), compared to aDNA (N=325). The comparable or even higher imputation quality in aDNA was achieved because of the higher density of genotyped SNPs in the region.
Linkage disequilibrium and haplotype network analysis
Linkage disequilibrium (LD) analysis was performed with the Haploview software (version 4.2)57. Analysis was performed on a 200 kb region (chr11:61,500,000–61,700,000), covering all three FADS genes. Variants were included in the analysis if they fulfilled the following criteria: 1) biallelic; 2) minor allele frequency (MAF) in the sample not less than 5%; 3) with rsID; 4) p value for Hardy-Weinberg equilibrium test larger than 0.001. Analysis was performed separately for the combined UK10K cohort and each of the five European populations in 1000GP.
Haplotype network analysis was performed with an R software package, pegas58. To reduce the number of SNPs and thus the number of haplotypes included in the analysis, we restricted this analysis to part of the 85 kb FADS1–FADS2 LD block, starting 5 kb downstream of FDAS1 to the end of the LD block (a 60-kb region). To further reduce the number of SNPs, in the analysis with all 1000GP European samples, we applied an iterative algorithm59 to merge haplotypes that have no more than three nucleotide differences by removing the differing SNPs. The algorithm stops when all remaining haplotypes are more than 3 nucleotides away. With this procedure, we were able to reduce the number of total haplotypes from 81 to 12, with the number of SNPs decreased from 88 to 34 (Supplementary Fig. 30). This set of 34 representative SNPs was used in all haplotype-based analysis in aDNA, 1000GP, HGDP and POPRES. Missing data (e.g. from a low imputation genotype probability) were included in the haplotype network analysis.
Of note, for the 12 haplotypes identified in 1000GP European samples, only five of them have frequency higher than 1% (Supplementary Table 2). These five haplotypes were designated as D, M1, M2, M3 and M4, from the most common to the least.
Ancient DNA-based test for recent selection in Europe
The test was performed as described before12. Briefly, most European populations could be modelled as a mixture of three ancient source populations at fixed proportions12,60. The three ancient source populations are West or Scandinavian hunter-gatherers (WSHG), early European farmers (EF), and Steppe-Ancestry pastoralists (SA) (Supplementary Table 1). For modern European populations in 1000GP, the proportions of these three ancestral sources estimated at genome-wide level are (0.196, 0.257, 0.547) for CEU, (0.362, 0.229, 0.409) for GBR, (0, 0.686, 0.314) for IBS, and (0, 0.645, 0.355) for TSI. FIN was not used because it does not fit this three-population model12. Under neutrality, the frequencies of a SNP (e.g. reference allele) in present-day European populations are expected to be the linear combination of its frequencies in the three ancient source populations. This serves as the null hypothesis: pmod = Cpanc, where pmod is the frequencies in A modern populations, panc is the frequencies in B ancient source populations while C is an AxB matrix with each row representing the estimated ancestral proportions for one modern population. The alternative hypothesis is that pmod is unconstrained by panc. The frequency in each population is modelled with binomial distribution: L(p; D) = B(X, 2N, p), where X is the number of designated allele observed while N is the sample size. In ancient populations, X is the expected number of designated allele observed, taking into account uncertainty in imputation. We write ℓ(p; D) for the log-likelihood. The log-likelihood for SNP frequencies in all three ancient populations and four modern populations are: . Under the null hypothesis, there are B parameters in the model, corresponding to the frequencies in B ancient populations. Under the alternative hypothesis, there are A+B parameters, corresponding to the frequencies in A modern populations and B ancestral populations. We numerically maximized the likelihood separately under each hypothesis and evaluate the statistic (twice the difference in log-likelihood) with the null distribution. Inflation was observed with this statistic in a previous genome-wide analysis and a λ = 1.38 was used for correction12. Following this, we applied the same factor in correcting the p values in our analysis. For genotyped SNPs previously tested, similar scales of statistical significance were observed as in the previous study (Supplementary Fig. 31). We note that for the purpose of refining the selection signal with imputed variants, only relative significance levels across variants are informative.
In addition to combining signals from four present-day European populations, we further performed tests separately in the two South European populations (IBS and TSI) and in the two North European populations (CEU and GBR). In these two cases, A = 2 and the null distribution is . For comparison between the North and the South, we used three statistics: the final p value, the maximum likelihood under the null hypothesis, and the maximum likelihood under the alternative hypothesis.
Ancient DNA-based test for ancient selection in pre-Neolithic European hunter-gatherers
Two Bayesian methods, the Schraiber et al. method25 and the ApproxWF26, were applied to infer natural selection from allele frequency time series data. The two software were downloaded from https://github.com/Schraiber/selection and https://bitbucket.org/phaentu/approxwf/downloads/, respectively. The Schraiber et al. method models the evolutionary trajectory of an allele under a specified demographic history and estimates selection coefficients (s1 and s2) for heterozygotes and homozygotes of the allele under study. This method has two modes, with or without the simultaneous estimation of allele age. Without the estimation of allele age, this method models the frequency trajectory only between the first and last time points provided and its estimates of selection coefficients describe the selection force during this period only. With the simultaneous estimation of allele age, this method models the frequency trajectory starting from the first appearance of the allele to the last time point provided. In this case, the selection coefficients describe the selection force starting from the mutation of the allele, which therefore should be the derived allele. For demographic history, we used two models: a constant population size model with Ne=10,000 and a more realistic model with two historic epochs of bottleneck and recent exponential growth61. However, the recent epoch of exponential growth does not have an impact on our analysis because for our analysis the most recent sample, WSHG, has an age estimate of ~7500 years, predating the onset of exponential growth (3520 ya, assuming 25 years per generation). ApproxWF can simultaneously estimate selection coefficient and demographic history (only for constant population size model). For our purpose, we set the demographic history as Ne=10,000. It estimates selection coefficient for homozygotes, s, and dominance coefficient, h. The selection coefficient estimated is for the time points specified by the input data.
Four groups of pre-Neolithic European hunter-gatherers were included in our test: the Věstonice cluster (median sample age: 30,076 yo), the El Mirón cluster (14,959 yo), the Villabruna cluster (10,059 yo) and WSHG (7,769 yo). To identify SNPs with evidence of positive selection during the historic period from Věstonice to WSHG, we applied both methods on most SNPs in the FADS locus. The Schraiber et al. method was run twice with two demographic models while ApproxWF was run once with the constant size model. For the two candidate SNPs (rs174570 and rs2851682), we further ran the Schraiber et al. method with the more realistic demographic model to simultaneously estimate their selection coefficients and allele ages. Statistical significance was considered if the 95% CI of selection coefficient does not overlap with 0. Details about running the two software were in Supplementary Methods.
Modern DNA-based selection tests
We performed two types of selection tests for modern DNA: site frequency spectrum (SFS)-based and haplotype-based tests. These tests were performed separately in each of the five European populations from 1000GP and each of the two cohorts from UK10K. For SFS-based tests, we calculated genetic diversity (π), Tajima’s D62, and Fay and Wu’s H63, using in-house Perl scripts. We calculated these three statistics with a sliding-window approach (window size = 5 kb and moving step = 1 kb). Statistical significance for these statistics were assessed using the genome-wide empirical distribution. Haplotype-based tests, including iHS64 and nSL22, were calculated using software selscan (version 1.1.0a)65. Only common biallelic variants (MAF > 5%) were included in the analysis. Genetic variants without ancestral information were excluded. These two statistics were normalized in frequency bins (1% interval) and the statistical significance of the normalized iHS and nSL were evaluated with the empirical genome-wide distribution. The haplotype bifurcation diagrams and EHH decay plots were drawn using an R package, rehh66. Singleton Density Score (SDS) based on UK10K was directly retrieved from a previous study23.
Geographical frequency distribution analysis
For plots of geographical frequency distribution, the geographical map was plotted with an R software package, maps (https://CRAN.R-project.org/package=maps) while the pie charts were added with the mapplots package (https://cran.r-project.org/web/packages/mapplots/index.html). Haplotype frequencies were calculated based on haplotype network analysis with pegas58, which groups haplotypes while taking into account missing data. SNP frequencies were either the observed frequency, if the SNP was genotyped, or the expected frequency based on genotype probability, if the SNP was imputed.
Targeted association analysis for SNP rs174594 in UK10K
We performed association analysis for rs174594 in two UK10K datasets – ALSPAC and TwinsUK54. For both datasets, we analyzed height, weight, BMI and lipid-related traits including total cholesterol, low density lipoprotein, very low density lipoprotein, high density lipoprotein, Apolipoprotein A-I (APOA1), Apolipoprotein B (APOB) and triglyceride. We performed principal components analysis using smartpca from EIGENSTRAT software67 with genome-wide autosomal SNPs and we added top 4 principal components as covariates for all association analysis. We also used age as a covariate for all association analysis. Sex was added as a covariate only for ALSPAC dataset since all individuals in TwinsUK dataset are female. For all lipid-related traits, we also added BMI as a covariate.
Data availability
All datasets used in this study are publicly available or available from dbGaP with application. Links or Study Accession are as follows.
Ancient DNA: https://reich.hms.harvard.edu/datasets
1000 Genomes Project: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/
Human Genome Diversity Project (HGDP): http://www.hagsc.org/hgdp/files.html
Population Reference Sample (POPRES): dbGaP Study Accession: phs000145.v4.p2 UK10K: https://www.uk10k.org/data_access.html
Singleton Density Score (SDS): https://github.com/yairf/SDS
Code availability
Most analyses were conducted with available software and packages as described in the respective subsections of Methods. Customized Perl and R scripts were used in performing aDNA-based test for recent positive selection, site frequency spectrum-based selection tests, and for general plotting purposes. These customized scripts were provided in Supplementary Data 1.
Supplementary Material
Acknowledgments
We thank Montgomery Slatkin and Joshua Schraiber for their help in running their software, David Reich and Iain Mathieson for making their data publicly available, Leonardo Arbiza, Charles Liang, Daniel (Alex) Marburgh, Edward Li, Kumar Kothapalli, Tom Brenna, and all members of the Keinan lab for helpful discussion and comments on the manuscript. This work was supported by the National Institutes of Health (Grants R01HG006849 and R01GM108805 to AK) and the Edward Mallinckrodt, Jr. Foundation (AK).
Footnotes
Author contributions
A.K. and K.Y. conceived and designed the project. K.Y. performed data collection and analysis, with contributions from D.W. and F.G.. K.Y. and A.K. interpreted the results, with contribution from O.B. on the anthropological perspective. K.Y. and A.K. wrote the manuscript. All authors read, edited and approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
References
- 1.Fan S, Hansen MEB, Lo Y, Tishkoff SA. Going global by adapting local: A review of recent human adaptation. Science. 2016;354:54–59. doi: 10.1126/science.aaf5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakamura MT, Nara TY. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu Rev Nutr. 2004;24:345–376. doi: 10.1146/annurev.nutr.24.121803.063211. [DOI] [PubMed] [Google Scholar]
- 3.Raphael W, Sordillo LM. Dietary polyunsaturated fatty acids and inflammation: the role of phospholipid biosynthesis. Int J Mol Sci. 2013;14:21167–21188. doi: 10.3390/ijms141021167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bazinet RP, Laye S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat Rev Neurosci. 2014;15:771–785. doi: 10.1038/nrn3820. [DOI] [PubMed] [Google Scholar]
- 5.Mathias RA, et al. Adaptive evolution of the FADS gene cluster within Africa. PLoS One. 2012;7:e44926. doi: 10.1371/journal.pone.0044926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ameur A, et al. Genetic adaptation of fatty-acid metabolism: a human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am J Hum Genet. 2012;90:809–820. doi: 10.1016/j.ajhg.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kothapalli KS, et al. Positive Selection on a Regulatory Insertion-Deletion Polymorphism in FADS2 Influences Apparent Endogenous Synthesis of Arachidonic Acid. Mol Biol Evol. 2016;33:1726–1739. doi: 10.1093/molbev/msw049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fumagalli M, et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science. 2015;349:1343–1347. doi: 10.1126/science.aab2319. [DOI] [PubMed] [Google Scholar]
- 10.Amorim CEG, et al. Genetic signature of natural selection in first Americans. Proc Natl Acad Sci U S A. 2017;114:2195–2199. doi: 10.1073/pnas.1620541114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reardon HT, et al. Insertion-deletions in a FADS2 intron 1 conserved regulatory locus control expression of fatty acid desaturases 1 and 2 and modulate response to simvastatin. Prostaglandins Leukot Essent Fatty Acids. 2012;87:25–33. doi: 10.1016/j.plefa.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathieson I, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bar-Yosef O. In: On Human Nature: Biology, Psychology, Ethics, Politics, and Religion. Tibayrenc M, Ayala FJ, editors. Academic Press; 2017. pp. 297–331. Ch. 19. [Google Scholar]
- 14.Coward F, Shennan S, Colledge S, Conolly J, Collard M. The spread of Neolithic plant economies from the Near East to northwest Europe: a phylogenetic analysis. Journal of Archaeological Science. 2008;35:42–56. [Google Scholar]
- 15.Bogaard A, et al. Crop manuring and intensive land management by Europe’s first farmers. Proc Natl Acad Sci U S A. 2013;110:12589–12594. doi: 10.1073/pnas.1305918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards MP. In: The Evolution of Hominin Diets: Integrating Approaches to the Study of Palaeolithic Subsistence. Hublin JJ, Richards MP, editors. Springer Science; Business Media; 2009. pp. 251–257. [Google Scholar]
- 17.Richards MP, Schulting RJ, Hedges RE. Archaeology: sharp shift in diet at onset of Neolithic. Nature. 2003;425:366. doi: 10.1038/425366a. [DOI] [PubMed] [Google Scholar]
- 18.Richards MP, Price TD, Koch E. Mesolithic and Neolithic Subsistence in Denmark:New Stable Isotope Data. Current Anthropology. 2003;44:288–295. [Google Scholar]
- 19.Fraser RA, Bogaard A, Schäfer M, Arbogast R, Heaton THE. Integrating botanical, faunal and human stable carbon and nitrogen isotope values to reconstruct land use and palaeodiet at LBK Vaihingen an der Enz, Baden-Württemberg. World Archaeology. 2013;45:492–517. [Google Scholar]
- 20.Knipper C, et al. What is on the menu in a Celtic town? Iron Age diet reconstructed at Basel-Gasfabrik, Switzerland. Archaeological and Anthropological Sciences. 2016 [Google Scholar]
- 21.López-Costas O, Müldner G, Martínez Cortizas A. Diet and lifestyle in Bronze Age Northwest Spain: the collective burial of Cova do Santo. Journal of Archaeological Science. 2015;55:209–218. [Google Scholar]
- 22.Ferrer-Admetlla A, Liang M, Korneliussen T, Nielsen R. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol Biol Evol. 2014;31:1275–1291. doi: 10.1093/molbev/msu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Field Y, et al. Detection of human adaptation during the past 2000 years. Science. 2016;354:760–764. doi: 10.1126/science.aag0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu Q, et al. The genetic history of Ice Age Europe. Nature. 2016;534:200–205. doi: 10.1038/nature17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schraiber JG, Evans SN, Slatkin M. Bayesian Inference of Natural Selection from Allele Frequency Time Series. Genetics. 2016;203:493–511. doi: 10.1534/genetics.116.187278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrer-Admetlla A, Leuenberger C, Jensen JD, Wegmann D. An Approximate Markov Model for the Wright-Fisher Diffusion and Its Application to Time Series Data. Genetics. 2016;203:831–846. doi: 10.1534/genetics.115.184598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mozaffarian D, et al. Genetic loci associated with circulating phospholipid trans fatty acids: a meta-analysis of genome-wide association studies from the CHARGE Consortium. Am J Clin Nutr. 2015;101:398–406. doi: 10.3945/ajcn.114.094557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.den Hoed M, et al. Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet. 2013;45:621–631. doi: 10.1038/ng.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buckley MT, et al. Selection in Europeans on fatty acid desaturases associated with dietary changes. Mol Biol Evol. 2017 doi: 10.1093/molbev/msx103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan G, et al. PATZ1 down-regulates FADS1 by binding to rs174557 and is opposed by SP1/SREBP1c. Nucleic Acids Res. 2017;45:2408–2422. doi: 10.1093/nar/gkw1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards MP, Hedges REM. Stable Isotope Evidence for Similarities in the Types of Marine Foods Used by Late Mesolithic Humans at Sites Along the Atlantic Coast of Europe. Journal of Archaeological Science. 1999;26:717–722. [Google Scholar]
- 34.Lubell D, Jackes M, Schwarcz H, Knyf M. The Mesolithic-Neolithic Transition in Portugal:Isotopic and Dental Evidence of Diet. Journal of Archaeological Science. 1994;21:201–216. [Google Scholar]
- 35.Richards MP, Mellars PA. Stable isotopes and the seasonality of the Oronsay middens. Antiquity. 1998;72:178–184. [Google Scholar]
- 36.Bonsall C, et al. Mesolithic and Early Neolithic in the Iron Gates: A Palaeodietary Perspective. Journal of European Archaeology. 1997;5:50–92. [Google Scholar]
- 37.Abedi E, Sahari MA. Long-chain polyunsaturated fatty acid sources and evaluation of their nutritional and functional properties. Food Sci Nutr. 2014;2:443–463. doi: 10.1002/fsn3.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simopoulos AP. Evolutionary aspects of diet: the omega-6/omega-3 ratio and the brain. Mol Neurobiol. 2011;44:203–215. doi: 10.1007/s12035-010-8162-0. [DOI] [PubMed] [Google Scholar]
- 39.Mannino MA, Thomas KD, Leng MJ, Di Salvo R, Richards MP. Stuck to the shore? Investigating prehistoric hunter-gatherer subsistence, mobility and territoriality in a Mediterranean coastal landscape through isotope analyses on marine mollusc shell carbonates and human bone collagen. Quaternary International. 2011;244:88–104. [Google Scholar]
- 40.Mannino MA, et al. Origin and diet of the prehistoric hunter-gatherers on the mediterranean island of Favignana (Egadi Islands, Sicily) PLoS One. 2012;7:e49802. doi: 10.1371/journal.pone.0049802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lightfoot E, Boneva B, Miracle PT, Šlaus M, O’Connell TC. Exploring the Mesolithic and Neolithic transition in Croatia through isotopic investigations. Antiquity. 2015;85:73–86. [Google Scholar]
- 42.Bocquet-Appel JP, Naji S, Vander Linden M, Kozlowski J. Understanding the rates of expansion of the farming system in Europe. Journal of Archaeological Science. 2012;39:531–546. [Google Scholar]
- 43.Rowley-Conwy P. Westward Ho! The Spread of Agriculture from Central Europe to the Atlantic. Current Anthropology. 2011;52:S431–S451. [Google Scholar]
- 44.Vigne JD. In: The Neolithic Demographic Transition and its Consequences. Bocquet-Appel J-P, Bar-Yosef O, editors. Springer Science+Business Media B.V.; 2008. pp. 179–205. [Google Scholar]
- 45.Cramp LJ, et al. Immediate replacement of fishing with dairying by the earliest farmers of the Northeast Atlantic archipelagos. Proc Biol Sci. 2014;281:20132372. doi: 10.1098/rspb.2013.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Curry A. Archaeology: The milk revolution. Nature. 2013;500:20–22. doi: 10.1038/500020a. [DOI] [PubMed] [Google Scholar]
- 47.Salque M, et al. Earliest evidence for cheese making in the sixth millennium BC in northern Europe. Nature. 2013;493:522–525. doi: 10.1038/nature11698. [DOI] [PubMed] [Google Scholar]
- 48.Lidén K, Eriksson G, Nordqvist B, Götherström A, Bendixen E. “The wet and the wild followed by the dry and the tame” – or did they occur at the same time? Diet in Mesolithic – Neolithic southern Sweden. Antiquity. 2004;78:23–33. [Google Scholar]
- 49.Rottoli M, Castiglioni E. Prehistory of plant growing and collecting in northern Italy, based on seed remains from the early Neolithic to the Chalcolithic (c. 5600–2100 cal b.c.) Vegetation History and Archaeobotany. 2008;18:91–103. [Google Scholar]
- 50.Lazaridis I, et al. Genomic insights into the origin of farming in the ancient Near East. Nature. 2016;536:419–424. doi: 10.1038/nature19310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li JZ, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 52.Nelson MR, et al. The Population Reference Sample, POPRES: a resource for population, disease, and pharmacological genetics research. Am J Hum Genet. 2008;83:347–358. doi: 10.1016/j.ajhg.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lazaridis I, et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513:409–413. doi: 10.1038/nature13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.The UK10 Consortium et al. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Browning BL, Browning SR. Genotype Imputation with Millions of Reference Samples. Am J Hum Genet. 2016;98:116–126. doi: 10.1016/j.ajhg.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gamba C, et al. Genome flux and stasis in a five millennium transect of European prehistory. Nat Commun. 2014;5:5257. doi: 10.1038/ncomms6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 58.Paradis E. pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics. 2010;26:419–420. doi: 10.1093/bioinformatics/btp696. [DOI] [PubMed] [Google Scholar]
- 59.Dannemann M, Andres AM, Kelso J. Introgression of Neandertal- and Denisovan-like Haplotypes Contributes to Adaptive Variation in Human Toll-like Receptors. Am J Hum Genet. 2016;98:22–33. doi: 10.1016/j.ajhg.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patterson N, et al. Ancient admixture in human history. Genetics. 2012;192:1065–1093. doi: 10.1534/genetics.112.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gazave E, et al. Neutral genomic regions refine models of recent rapid human population growth. Proc Natl Acad Sci U S A. 2014;111:757–762. doi: 10.1073/pnas.1310398110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Szpiech ZA, Hernandez RD. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31:2824–2827. doi: 10.1093/molbev/msu211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gautier M, Vitalis R. rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics. 2012;28:1176–1177. doi: 10.1093/bioinformatics/bts115. [DOI] [PubMed] [Google Scholar]
- 67.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 68.Wang J, et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013;41:D171–176. doi: 10.1093/nar/gks1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets used in this study are publicly available or available from dbGaP with application. Links or Study Accession are as follows.
Ancient DNA: https://reich.hms.harvard.edu/datasets
1000 Genomes Project: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/
Human Genome Diversity Project (HGDP): http://www.hagsc.org/hgdp/files.html
Population Reference Sample (POPRES): dbGaP Study Accession: phs000145.v4.p2 UK10K: https://www.uk10k.org/data_access.html
Singleton Density Score (SDS): https://github.com/yairf/SDS