

Abstract
Photodynamic therapy (PDT) can destroy local tumors and minimize normal tissue damage, but is ineffective at eliminating metastases. Checkpoint blockade immunotherapy has enjoyed recent success in the clinic, but only elicits limited rates of systemic antitumor response for most cancers due to insufficient activation of the host immune system. Here we describe a treatment strategy that combines PDT by a new chlorin-based nanoscale metal–organic framework (nMOF), TBC-Hf, and a small-molecule immunotherapy agent that inhibits indoleamine 2,3-dioxygenase (IDO), encapsulated in the nMOF channels to induce systemic antitumor immunity. The synergistic combination therapy achieved effective local and distant tumor rejection in colorectal cancer models. We detected increased T cell infiltration in the tumor microenvironment after activation of the immune system with the combination of IDO inhibition by the small-molecule immunotherapy agent and immunogenic cell death induced by PDT. We also elucidated the underlying immunological mechanisms and revealed compensatory roles of neutrophils and B cells in presenting tumor-associated antigens to T cells in this combination therapy. We believe that nMOF-enabled PDT has the potential to significantly enhance checkpoint blockade cancer immunotherapy, affording clinical benefits for the treatment of many difficult-to-treat cancers.
Graphical Abstract
INTRODUCTION
Photodynamic therapy (PDT) combines nontoxic photosensitizers (PSs), light, and tissue oxygen to generate reactive oxygen species (ROS) such as 1O2 to achieve local tumor control/rejection.1 PDT is highly effective for the treatment of esophageal cancer and skin lesions but cannot completely eradicate deep-seated tumors due to shallow penetration of light through tissues.2 The localized light irradiation also renders PDT ineffective for treating systemically disseminated disease. Therefore, more efficient PSs and new treatment strategies are needed to enhance the efficacy of PDT in both eliminating local tumors and controlling distant metastases.
Significant efforts have been devoted to developing nano-particle-based PSs to enhance PDT in the past decade.3 We first reported the use of porphyrin- and chlorin-based nanoscale metal–organic frameworks (nMOFs) as efficient PSs for PDT of cancer. The stable framework and crystalline structure of nMOFs allow for high PS loadings, whereas the heavy metal cluster secondary building units (SBUs) facilitate intersystem crossing to enhance 1O2 generation.4 We further demonstrated in a recent study that both apoptosis and immunogenic cell death (ICD) contribute to the anticancer efficacy of nMOF-enabled PDT.4b The acute inflammatory response of PDT can alter the immunosuppressive microenvironment of the local tumor to prime the host immune system.5 We hypothesized that the porous nMOF structure could be leveraged to accentuate immune response by delivering small-molecule immunostimulatory agents in the open channels, combining PDT and immunotherapy to both eradicate local, treated tumors and reject distant, untreated tumors.
Immunotherapy has recently emerged as a highly effective cancer treatment strategy, with high overall response rates in multiple cancer types and durable tumor control enjoyed by a small subset of patients.6 Of particular interest is checkpoint blockade immunotherapy which uses small molecules or antibodies to stimulate the immunosuppressive microenvironment of tumors by modulating protein expressions and/or functions at dysregulated immune checkpoints.6b,7 Indoleamine 2,3-dioxygenase (IDO) is one such checkpoint, an immunoregulatory enzyme highly expressed in tumors that catalyzes the oxidative catabolism of tryptophan (Trp) to kynurenine (Kyn).8 This catalytic process leads to depletion of Trp and production of Kyn, thus preventing the clonal expansion of T cells and promoting T cell anergy and apoptosis.9 Small-molecule IDO inhibitors such as 1-methyl-tryptophan,9b,10 INCB24360,11 and NLG91912 can effectively block Trp catabolism but show modest effects as monotherapies, due in part to ineffective antigen presentation and insufficient antitumor immunity.
We herein report the design of a new nMOF, TBC-Hf, constructed from a chlorin derivative, 5,10,15,20-tetra(p-benzoato)chlorin (H4TBC), and Hf6(μ3-O)4(μ3-OH)4 SBUs. We took advantage of the highly porous structure of the nMOF to load an IDO inhibitor (IDOi) into TBC-Hf to afford IDOi@ TBC-Hf. The IDOi@TBC-Hf system enables a novel cancer treatment strategy by combining nMOF-enabled PDT and IDOi-based immunotherapy (Figure 1). We demonstrated the eradication of the primary, treated tumors and the rejection of the distant, untreated tumors upon IDOi@TBC-Hf administration and light irradiation using two syngeneic colorectal cancer models, CT26 and MC38, and investigated the underlying immunological mechanisms.
Figure 1.
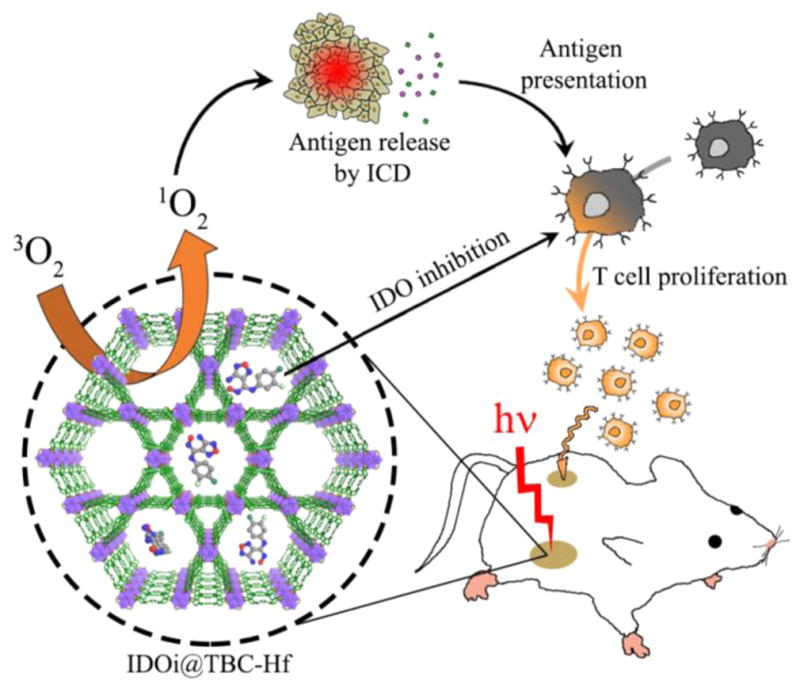
Schematic presentation of combined PDT and immunotherapy by IDOi@TBC-Hf. Local injection of IDOi@TBC-Hf and light irradiation generate reactive oxygen species, causing immunogenic cell death (ICD) and releasing tumor-associated antigens which are presented to T cells. Meanwhile, the IDO inhibitor released from IDOi@TBC-Hf modulates tryptophan/kynurenine catabolism to activate the immunosuppressive tumor microenvironment. The combination of antigen presentation from PDT and checkpoint blockade by IDO inhibition causes T cell proliferation and infiltration, leading to not only eradication of local, treated tumors but also rejection of distant, untreated tumors.
RESULTS
Synthesis and Characterization of nMOFs
The new chlorin derivative 5,10,15,20-tetra(p-benzoato)chlorin (H4TBC) was synthesized as depicted in Scheme 1. Briefly, 5,10,15,20-tetra(p-methylbenzoato)chlorin (Me4TBC) was synthesized by a partial reduction of 5,10,15,20-tetra(p-methylbenzoato)porphyrin (Me4TBP) with toluenesulfonhydrazide in 16% yield. Saponification of Me4TBC afforded H4TBC in 78% yield. Me4TBC and H4TBC were characterized by NMR and mass spectrometry (Figures S1–S5, Supporting Information).
Scheme 1.
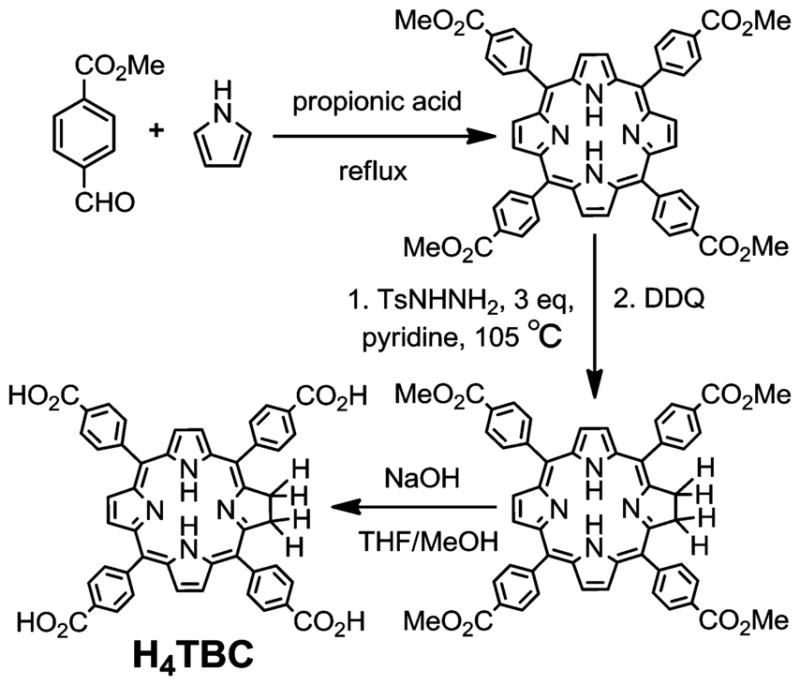
Synthesis of 5,10,15,20-Tetra(p-benzoato)chlorin (H4TBC)
A solvothermal reaction between HfCl4 and H4TBC in N,N-dimethylformamide (DMF) at 80 °C afforded the purple powder of TBC-Hf in 87% yield. TBC-Hf was washed with copious amounts of DMF, 1% triethylamine in ethanol (v/v), and ethanol successively and stored in ethanol as a stock suspension. As a control, TBP-Hf with the corresponding porphyrin ligand 5,10,15,20-tetra(p-benzoato)porphyrin (H4TBP) was synthesized by a solvothermal reaction between HfCl4 and H4TBP in N,N-diethylformamide (DEF) at 120 °C and washed in the same fashion as for TBC-Hf.
TBC-Hf and TBP-Hf adopt the same structure as the previously reported TBP-Zr analogue (MOF-545), based on powder X-ray diffraction (PXRD) (Figure 2a).13 The carboxylate moieties from the TBC ligands bridge 8 out of 12 edges of octahedral Hf6(μ3-O)4(μ3-OH)4 SBUs. The remaining coordination sites on the SBUs are occupied by 4 water and 4 terminal hydroxy groups on the equatorial plane to afford a hexagonal framework of Hf6(μ3-O)4(μ3-OH)4(OH)4-(H2O)4(TBC)2 (Figure S6). The nMOFs therefore feature large one-dimensional channels with diameters of 3.5 nm along the c axis, along with smaller channels with diameters of 1.0 nm along the c axis and 1.1 nm × 0.9 nm windows perpendicular to the c axis. TBC-Hf and TBP-Hf exhibit BET surface areas of 1077 and 1462 m2/g, respectively (Figure S7). During the course of this study, a few reports of TBP-based nMOFs with a cubic structure for PDT have appeared.14 However, we expect that our highly porous TBP-Hf control will outperform the cubic TBP-based nMOFs due to enhanced 1O2 diffusion through the nMOF channels. In TBC-Hf and TBP-Hf, the porphyrin and chlorin rings are highly rigid in the nMOF structures to minimize energy transfer and self-quenching.
Figure 2.

Morphology and structure of TBP-Hf and TBC-Hf. (a) PXRD patterns of TBP-Hf, TBC-Hf, and IDOi@TBC-Hf in comparison to MOF-545. TEM images of TBP-Hf (b,c), TBC-Hf (d,e), and TBC-Hf after incubation in Hank’s balanced salt solution for 24 h (f).
Transmission electron microscopy (TEM) revealed nanorod/nanorice morphologies for TBP-Hf and TBC-Hf (Figure 2b,d). The rods are typically 50–100 nm long and 30–60 nm wide for TBC-Hf, and 50–100 nm long and 20–30 nm wide for TBP-Hf. High-resolution TEM images show the lattice fringes with distances matching the d200 and d001 spacings of the crystal structure (Figures 2c,e, S8, and S9). The average diameters of TBC-Hf and TBP-Hf are 83.2 and 72.7 nm, respectively, as measured by dynamic light scattering (DLS) (Figure S10). The nMOFs are stable in biological environments, as evidenced by the lack of significant changes in PXRD pattern or morphology after incubation in Hank’s balanced salt solution (HBSS) for 24 h (Figure 2a,f).
The Hf content was determined by inductively coupled plasma-mass spectrometry (ICP-MS) to be 30.4% (36.6% calculated). The TBC weight loss of 61.7% (56.8% calculated) determined by thermogravimetric analysis (TGA) further confirms the framework formula (Figure S11a). We encapsulated an INCB24360 analogue,11 4-amino-N-(3-chloro-4-fluorophenyl)-N′-hydroxy-1,2,5-oxadiazole-3-carboximidamide (Figure S11b), into the TBC-Hf channels to afford IDOi@ TBC-Hf for the co-delivery of IDOi with PSs. The IDOi weight percentage after loading was determined to be 4.7% by TGA and 1H NMR (Figures S11a and S12). The release of IDOi from IDOi@TBC-Hf was studied in HBSS (Figure S13). IDOi slowly leached from TBC-Hf, reaching 83.3% release after incubation in HBSS for 24 h.
UV–visible absorption spectroscopy indicates that TBC-Hf more effectively absorbs red light compared to TBP-Hf (Figure 3a). H4TBC has a major Soret peak at λmax = 420 nm and four Q-bands at 518, 546, 600, and 652 nm. The Soret absorption of TBC-Hf almost overlaps with that of the free ligand (λmax = 421 nm), while the Q-bands slightly red-shift to 520, 548, 600, and 653 nm. In comparison, the Soret peaks of H4TBP and TBP-Hf are at λmax = 420 and 418 nm, respectively, and the Q-bands are at 516, 549, 592, and 646 nm for H4TBP and 517, 550, 593, and 647 nm for TBP-Hf. The lowest energy Q-band of the chlorin therefore red-shifts by 6 nm after reduction from the porphyrin counterpart (652 nm compared to 646 nm). The extinction coefficient of H4TBC at the lowest energy absorption (ε = 44 700 M−1·cm−1) is ~9 fold greater than that of H4TBP (ε = 4800 M−1·cm−1), while the lowest energy absorption of TBC-Hf (ε = 38 500 M−1·cm−1) is 6-fold greater than that of TBP-Hf (ε = 6400 M−1·cm−1). Notably, the lowest energy absorption of TBC-Hf (653 nm) is at a wavelength of 7 nm longer than that of our previously reported DBC-UiO, and the ε of TBC-Hf is ~1.6 fold higher than that of DBC-UiO.
Figure 3.
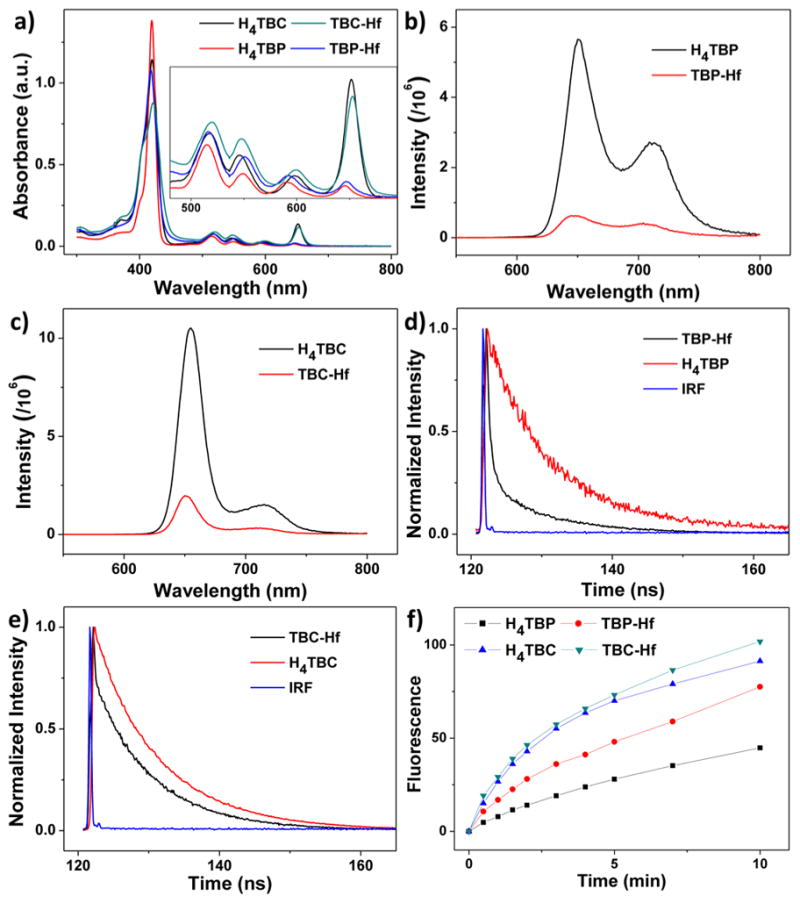
Photophysics and photochemistry of the ligands and nMOFs. (a) UV–visible absorption spectra of H4TBP, H4TBC, TBP-Hf, and TBC-Hf. Inset shows expanded views of the Q-band regions. Fluorescence spectra of H4TBP and TBP-Hf (b), and H4TBC and TBC-Hf (c), with excitation at 420 nm. Time-resolved fluorescence decay traces of H4TBP and TBP-Hf (d), and H4TBC and TBC-Hf (e), along with instrument response function (IRF). (f) Singlet oxygen generation by H4TBP, H4TBC, TBP-Hf, and TBC-Hf with a 650 nm LED irradiation at 20 mW/cm2, detected by Singlet Oxygen Sensor Green assay.
H4TBC exhibits two fluorescence emissions at 655 and 715 nm with an intensity ratio of ~6.8 when excited at 420 nm (Figure 3c). H4TBP fluorescence emissions are close to those of H4TBC, though the 651 nm/713 nm peak intensity ratio is ~2 (Figure 3b). After forming nMOFs, the fluorescence intensities drop significantly, consistent with other nMOF systems.4 For TBC-Hf, the fluorescence peaks slightly blue-shift to 650 and 712 nm and the intensities decrease by 5.3-fold and 4.7-fold, respectively; for TBP-Hf, the fluorescence peaks blue-shift to 648 and 704 nm, and the fluorescence intensities drop by 9.0-fold and 6.5-fold, respectively. The decrease in fluorescence intensity likely results from the enhanced intersystem crossing after coordination of the chlorin/porphyrin ligands to Hf on the SBUs. Time-correlated single photon counting measurement results also supported this interpretation: the fluorescence lifetime at 650 nm dropped from 9.56 ns for H4TBC to 8.25 ns for TBC-Hf and from 9.25 ns for H4TBP to 8.38 ns for TBP-Hf (Figure 3d,e and Table S1).
Singlet Oxygen Generation
The 1O2 generation efficiencies of H4TBC, TBC-Hf, H4TBP, and TBP-Hf were determined by Singlet Oxygen Sensor Green (SOSG) assay. The nMOF samples and ligand controls were irradiated with a LED (λmax = 650 nm, 20 mW/cm2), and the fluorescence enhancement upon reaction of SOSG with 1O2 was measured by a fluorimeter (excitation/emission at 504/525 nm). The fluorescence intensity plotted against irradiation time fit exponential decay equations, as we had previously observed in other nMOF systems,4 indicating a pseudo-first-order 1O2 generation process (Figure 3f). TBC-Hf outperforms both TBP-Hf and H4TBC in terms of 1O2 generation efficiency.
In Vitro PDT Efficacy and Mechanistic Study
We first confirmed efficient cellular uptake of TBC-Hf and TBP-Hf by cancer cells. TBC-Hf and TBP-Hf were incubated with colorectal cancer CT26 cells at a ligand concentration of 25 μM for 24 h. The Hf contents inside cells were determined to be 2.07 ± 0.26 and 1.79 ± 0.17 nmol/104 cells, respectively, by ICP-MS analysis (Figure S14). The in vitro PDT efficacy of TBP-Hf, TBC-Hf, and both ligands against CT26 cells was next investigated. TBC-Hf outperformed TBP-Hf under 90 J/cm2 light irradiation. Both nMOFs and ligands outperformed protoporphyrin IX (PpIX), a commonly used PS, upon irradiation, while no cytotoxicity was observed in dark control groups (Figure S15). The IC50 values of TBC-Hf, TBP-Hf, H4TBC, H4TBP, and PpIX for CT26 cells were 4.82 ± 0.50, 7.09 ± 0.39, 10.04 ± 1.22, 12.83 ± 1.59, and 23.72 ± 0.60 μM, respectively, at an irradiation dose of 90 J/cm2 (Figure 4a). These results were confirmed by a second colorectal cancer MC38 cell with IC50 values of 2.57 ± 0.85 and 17.9 ± 5.0 μM for TBC-Hf and H4TBC, respectively. The IC50 values exceeded 50 μM for TBP-Hf, H4TBP, and PpIX for MC38 cells (Figure 4b). To demonstrate the versatility of our nMOF system, unrelated B16F10 melanoma cells were employed to confirm the PDT efficacy, yielding IC50 values of 5.48 ± 0.70, 9.72 ± 0.78, 11.03 ± 1.57, 20.10 ± 4.28, and 16.48 ± 0.77 μM for TBC-Hf, TBP-Hf, H4TBC, H4TBP, and PpIX, respectively (Figure S16).
Figure 4.
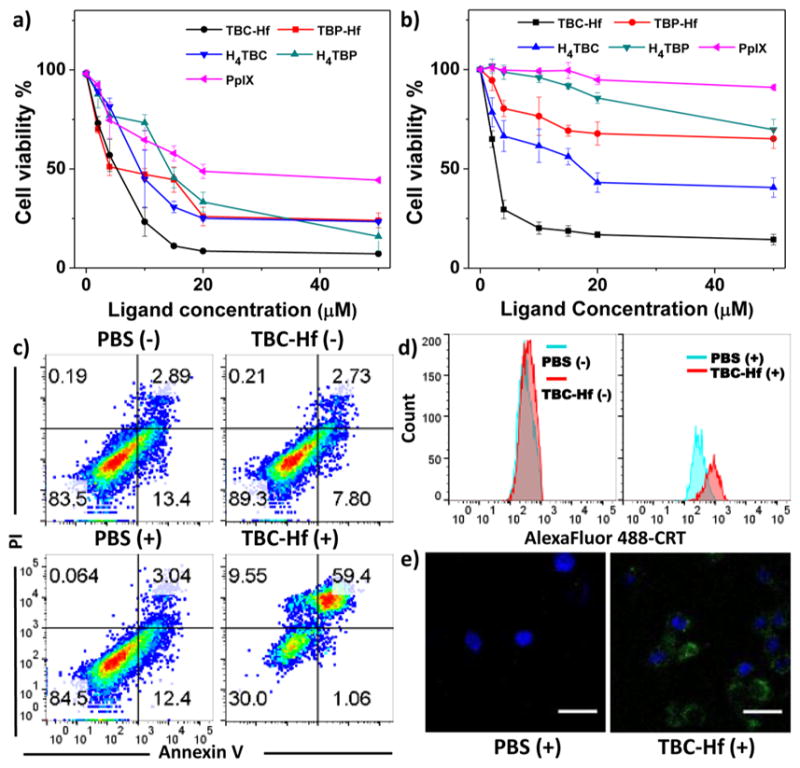
In vitro PDT efficacy and mechanistic study. PDT cytotoxicity of H4TBP, TBP-Hf, H4TBC, TBC-Hf, and PpIX at different PS concentrations in CT26 (a) and MC38 cells (b). (c) Annexin V/PI analysis of MC38 cells incubated with TBC-Hf or PBS, with or without irradiation (90 J/cm2). The quadrants from lower left to upper left (counter clockwise) represent healthy, early apoptotic, late apoptotic, and necrotic cells, respectively. The percentage of cells in each quadrant is shown on each graph. (+) and (−) refer to with and without irradiation, respectively. (d) CRT exposure on the cell surface of MC38 was assessed after incubation with PBS and TBC-Hf, with or without light irradiation (90 J/cm2), by flow cytometry analysis. The fluorescence intensity was gated on PI-negative cells. (e) Immunofluorescence microscopy of CRT expression on the cell surface of MC38 treated with PBS and TBC-Hf upon irradiation (90 J/cm2).
Unlike most conventional cytotoxic agents, PDT is known to induce ICD via apoptosis and necrosis, thereby mediating antitumor immune response.5 We confirmed that both apoptosis and necrosis occur upon treatment with PDT by flow cytometry of Alexa Fluor 488 Annexin V-labeled dead cells. MC38 cells were incubated with TBC-Hf, TBP-Hf, H4TBC, and H4TBP at 1 μM (ligand concentration of 2 μM), followed by light irradiation at 90 J/cm2 (650 nm, 100 mW/cm2). Significant amounts of cells underwent apoptosis/necrosis when treated with PDT of TBC-Hf, TBP-Hf, H4TBC, and H4TBP, with only 30.0%, 55.5%, 32.7%, and 57.6% healthy cells, respectively, compared to 80–90% healthy cells in dark control or PBS-treated cells with irradiation (Figures 4c and S17a). These results were further confirmed in CT26 cells under the same conditions (Figure S17b). Taken together, the chlorin-based TBC-Hf is a more efficient PS than the porphyrin-based TBP-Hf at equivalent nMOF and light doses.
We also investigated the ability of PDT treatment to induce ICD by determining cell-surface expression of calreticulin (CRT). MC38 cells were incubated with TBC-Hf, TBP-Hf, H4TBC, and H4TBP at 1 μM (ligand concentration of 2 μM) followed by light irradiation at 90 J/cm2. For flow cytometry analysis, cells were collected and stained with Alexa Fluor 488-CRT antibody and propidium iodide (PI, Figures 4d and S18). The fluorescence intensity of stained cells was gated on PI-negative cells. For immunostaining analysis, the cells were stained with AlexaFluor 488-CRT and DAPI, and observed under confocal laser scanning microscopy (CLSM, Figures 4e and S19). In both instances, cells treated with nMOF or ligand showed significant CRT expression on the cell-surface upon irradiation but none with PBS control or without irradiation. Due to the higher 1O2 generation efficiency and in vitro PDT efficacy of TBC-Hf, we focus our subsequent in vivo studies on TBC-Hf and use TBP-Hf as a control wherever appropriate.
Abscopal Effect of IDOi@TBC-Hf
The immunoregulatory enzyme IDO is often overexpressed in the tumor micro-environment, where it facilitates the survival and growth of tumor cells by suppressing antitumor immune responses.15 The small-molecule IDO inhibitor INCB24360, developed by Incyte, can reverse immunosuppression and inhibit tumor growth upon oral administration.16 We propose that, by encapsulation of IDOi into the nMOF channels, IDOi@TBC-Hf can both release IDOi into the local tumor environment and enter blood circulation for systemic IDO inhibition. The effects of IDOi will alter the suppressive tumor microenvironment of both the treated and untreated tumors. We hypothesize that synergizing local PDT of IDOi@TBC-Hf with checkpoint blockade therapy can promote an efficient abscopal effect, regression of an untreated tumor at a distant site following the local treatment of another tumor in the same organism.
We evaluated the abscopal effect of IDOi@TBC-Hf and light irradiation in two immunocompetent mouse models using bilateral tumor models of colorectal cancers CT26 and MC38 in the flank regions of BALB/c mice and C57BL/6 mice, respectively. When the primary tumors reached ~100 mm3, mice received a single injection of H4TBC, H4TBC plus IDOi, TBC-Hf, or IDOi@TBC-Hf into the primary tumors at a ligand dose of 20 μmol/kg or a corresponding IDOi dose of 1.5 mg/kg. Twelve hours after injection, the primary tumors were irradiated at a light dose of 90 J/cm2 (650 nm, 100 mW/cm2). Mice treated with IDOi@TBC-Hf without light irradiation served as dark controls. The primary tumor receiving intratumoral injection with or without irradiation was designated as the “treated tumor”, while the contralateral tumor which received neither direct injection nor irradiation was designated as the “untreated tumor”. As depicted in Figures 5 and S20–S22, local nMOF injection with light irradiation led to near elimination of the treated primary tumors. At the end point, CT26 tumor-bearing mice treated with IDOi@TBC-Hf or TBC-Hf and PDT therapy had tumors only 1.1 ± 0.2% and 0.7 ± 0.4% the size of PBS-treated tumors, respectively. Similarly, MC38 tumor-bearing mice had tumors only 0.8 ± 0.3% and 0.9 ± 0.4% of the size of PBS-treated tumors, respectively. H4TBC with light irradiation and IDOi@TBC-Hf dark group failed to inhibit the tumor growth, while H4TBC plus IDOi with light irradiation slightly inhibits the tumor growth (Table S2). Moreover, only PDT treatment of IDOi@ TBC-Hf successfully reduced the sizes of the untreated distant tumors. Tumors began shrinking on Day 6 and 5 after treatment in the CT26 and MC38 models, respectively, suggesting the treatment evoked systemic antitumor immunity in mice (Table S2–S3). TBC-Hf with light irradiation and IDOi@TBC-Hf dark control slightly inhibited the distant tumor growth, showing ineffectiveness of monotherapies.
Figure 5.
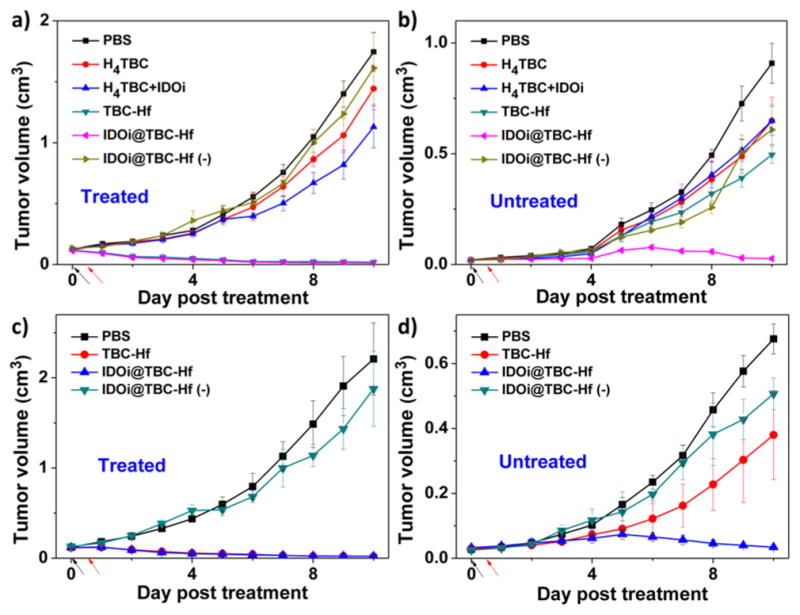
In vivo anticancer efficacy showing abscopal effect. Growth curves for treated (a,c) and untreated (b,d) tumors of CT26 (a,b) or MC38 (c,d) tumor-bearing mice after PDT treatment. Black and red arrows refer to the time of injection and irradiation, respectively.
Antitumor Immunity
We have shown that PDT of IDOi@TBC-Hf caused effective tumor regression of both primary, treated tumors and distant, untreated tumors in two syngeneic mouse models of colorectal cancer. We further investigated the underlying immunological mechanisms in the MC38 model by Enzyme-Linked ImmunoSpot (ELISPOT) assay and flow cytometry. We first performed an ELISPOT assay to detect the presence of tumor antigen-specific T cells 14 days after treatment. As shown in Figure 6a, the number of antigen-specific IFN-γ producing T cells increased to 799 per million splenocytes in mice treated with IDOi@TBC-Hf (vs 128 per million splenocytes in PBS group, P = 0.0879), indicating that PDT of IDOi@TBC-Hf induced in situ tumor vaccination to effectively generate tumor-specific T cell response.
Figure 6.

Antitumor immunity of PDT by IDOi@TBC-Hf. (a) Antigen-specific IFN-γ spot forming cells (SFC) 14 days after IDOi@ TBC-Hf PDT treatment. The percentage of tumor-infiltrating neutrophils (b) and B cells (c) with respect to the total number of cells in the tumor compared to PBS, 12h after IDOi@TBC-Hf PDT treatment. The percentage of tumor-infiltrating CD8+ T cells (d), CD4+ T cells (e), and CD45+ cells (f) with respect to the total number of cells in the tumors 12 days after IDOi@TBC-Hf PDT treatment. Growth curves for primary, treated (g) and distant, untreated (h) tumors after PDT treatment (+) with IDOi@TBC-Hf and injections of Mouse IgG, anti-CD4, or anti-CD8 antibody, compared to PBS control without PDT treatment (−). Black, red, and blue arrows refer to the time of IDOi@TBC-Hf injection, light irradiation, and antibody injection, respectively.
Further analysis of the immune response was carried out by investigating the tumor-infiltrating leukocyte profiles in each tumor by flow cytometry. Previous research has shown that PDT can evoke acute inflammation,5 so we first evaluated the population of leukocytes responsible for innate immune response (neutrophils, B cells, dendritic cells, and macrophages) in both primary and distant tumors 12 h after PDT treatment. As shown in Figures 6b,c and S23, IDOi@TBC-Hf administration and light irradiation led to significant increases in the percentage of tumor-infiltrating neutrophils and B cells with respect to the total number of cells in the tumor compared to PBS (neutrophils: P = 0.0369 vs PBS; B cells: P = 0.0215 vs PBS). To our surprise, the percentage of infiltrating dendritic cells appeared to decrease compared to PBS control in the primary tumor (Figure S23), although the difference is not of statistical significance.
To better understand the roles these types of cells played in the antigen presentation after PDT, we further detected the major histocompatibility complex class II (MHC-II) expression on these cells 12 h after PDT treatment. MHC-II mediates establishment of specific immunity by presenting tumor antigens to T cell receptors on the surface of CD4+ T helper cells.17 We found a significant decrease in MHC-II expression levels on dendritic cells, macrophages, and neutrophils in the primary treated tumors (DC: P < 0.0001 vs PBS; macrophages: P = 0.0004 vs PBS; neutrophils: P = 0.006 vs PBS, Figure S24). These results imply that TBC-Hf-mediated PDT may have an impact on the method of antigen presentation, which is typically the role of dendritic cells.
We profiled the tumor-infiltrating leukocytes on 7 days post PDT treatment. The mice treated with IDOi@TBC-Hf plus PDT did not show significant difference in all cell types except B cells from the control mice (Figure S25). The percentage of B cells significantly increased with respect to the total number of cells in the primary tumors of IDOi@TBC-Hf- and PDT-treated mice (P = 0.0018, Figure S25c), while the percentage of dendritic cells significantly decreased in the distant tumors of treated mice (P = 0.0083, Figure S25f). We also found that the percentage of NK cells was extremely low on day 7 post PDT treatment both in IDOi@TBC-Hf-treated mice and PBS-treated mice (both <0.05%, Figure S25e).
We further determined the tumor-infiltrating leukocyte profiles 12 days post PDT treatment. As shown in Figure 6d–f, IDOi@TBC-Hf significantly increased the proportion of infiltrating CD8+ T cells relative to the total number of cells in the distant tumor (P = 0.0012 vs PBS), an essential step to mounting an antitumor immune response to induce the abscopal effect. In both primary and distant tumors, the percentages of infiltrating CD45+ leukocytes and CD4+ T cells with respect to the total number of cells in the tumors were significantly increased in mice treated with PDT of IDOi@ TBC-Hf (Treated tumor, CD45+, P = 0.0061 vs PBS; CD4+ T cells, P = 0.0206 vs PBS; Untreated tumor, CD45+, P = 0.0001 vs PBS; CD4+ T cells, P = 0.0388 vs PBS). Furthermore, the percentage of tumor-infiltrating NK cells significantly increased in the distant tumors of mice treated with IDOi@TBP-Hf and PDT (P = 0.0034 vs PBS, Figure S26). Interestingly, IDOi@ TBP-Hf administration and PDT also led to a significant increase in the percentages of tumor-infiltrating B cells and a decrease in the percentages of tumor-infiltrating dendritic cells with respect to the total number of cells in the primary tumors 12 days post the treatment (B cells, P = 0.0017 vs PBS; DCs, P = 0.0041 vs PBS, Figure S27).
We also performed T cell blocking experiments to confirm the involvement of T cells in the efficient abscopal response on a bilateral subcutaneous MC38 mouse model. MC38 tumor-bearing mice were treated with IDOi@TBC-Hf and light irradiation as described before and received i.p. injections of Mouse IgG, anti-CD4, or anti-CD8 antibody at a dose of 200 μg/mouse/injection on Day 0 and Day 5 post the PDT treatment. While mice treated with Mouse IgG showed regression of both primary and distant tumors, no abscopal effect was observed on mice with blocking of CD4+ or CD8+ T cells (Figure 6g,h). Furthermore, blocking of CD4+ and CD8+ T cells also diminished the effect of IDOi@TBC-Hf in regressing the primary tumors. These results indicated that CD4+ and CD8+ T cells were essential not only to the abscopal effect but also to primary tumor rejection.
DISCUSSION
Approximately 140 000 patients are diagnosed with colorectal cancer in the U.S. annually, with one-third dying from metastasis.18 Stimulation of the host immune system has been shown to generate an antitumor immunity capable of controlling metastatic tumor growth, and thus represents a promising treatment strategy for metastatic colon cancer.8 Clinical results suggest that immunotherapies have the potential to achieve systemic and adaptable cancer control. However, combination with other treatment modalities is necessary to maximize the benefits of immunotherapy and increase the tumor-specific immune response rate.6b Here we report a new treatment strategy that combines local PDT of a new nMOF, TBC-Hf, in combination with IDO inhibition with nMOF-delivered small molecules to achieve effective and consistent abscopal responses in mouse models of colorectal cancers. We have shown that IDOi released from locally injected IDOi@ TBC-Hf reversed the suppressive tumor microenvironment in both treated and untreated tumors, which further synergized with TBC-Hf-mediated PDT to stimulate the immune system for activating both acute innate and prolonged adaptive immune response to achieve efficient local tumor regression and a consistent abscopal response. Our treatment method maximizes the benefits of local treatment with systemic immune response for the rejection of both primary and distant tumors while minimizing side effects, potentially affording an effective systemic therapy for metastatic colorectal cancers.
Owing to the outstanding photophysical properties of the chlorin-based TBC ligand, TBC-Hf shows potent PDT efficacy and outperforms its porphyrin counterpart TBP-Hf. The tetracarboxylate ligand constructs a robust framework with very large channels for small-molecular inhibitor loading and ROS diffusion, and the coordination to Hf6 SBUs enhances intersystem crossing of TBC to enhance 1O2 generation. As we19 and others20 have demonstrated the synthetic tunability and potential biomedical applications of nMOFs in the past decade, we believe that the nMOF compositions and structures can be further optimized in order to enhance light absorption and 1O2 generation/diffusion. We thus believe that nMOF-based synergistic PDT and immunotherapy have the potential for future clinical translation for the treatment of metastatic cancers.
We propose that the combination of IDOi@TBC-Hf administration and light irradiation causes highly efficient tumor regression of both primary treated tumors and distant untreated tumors owing to two factors. First, TBC-Hf based PDT causes ICD of cancer cells in the primary tumors, which activates innate immune system and promotes antigen presentation (Figure 1). The massive stressed and dying necrotic tumor cells in the PDT-treated primary tumor sites are engulfed by the innate immune effector cells followed by presenting tumor-derived antigenic peptides to T cells, thus stimulating a tumor-specific T cell response. Second, the IDOi is released from intratumorally injected IDOi@TBC-Hf to systemically inhibit IDO activity to reverse the immunosuppressive tumor environments. Alternatively, the recruited immune cells likely undergo IDO inhibition in the treated tumors and migrate to the distant tumors to cause an abscopal effect. The two treatment modalities, PDT and IDOi checkpoint blockade therapy, synergize with each other to kill cancer cells locally and create an immunogenic tumor microenvironment systemically, leading to strong and consistent abscopal effects.
Dendritic cells are one of the most important antigen-presenting cells and prevalently believed to play a key role in antitumor immune response.21 However, we observed a decrease of dendritic cell population percentage at different time points along with a significant decrease of MHC-II expression level on dendritic cells after PDT treatment with IDOi@TBC-Hf, indicating an altered role for dendritic cells after PDT treatment. Meanwhile, the increase of both neutrophils and B cell population percentages implies a compensating effect of other antigen presenting cells to present tumor-associated antigens and initiate the antitumor immune response. Previous studies suggested that neutrophils, rather than dendritic cells, directly affect T cell proliferation upon PDT treatment.22 We also observed significant increase in the percentage of tumor infiltrating NK cells from 0.02% on day 7 to 3.83% on day 12 post PDT treatment in distant tumors, suggesting the important role of tumor-infiltrating NK cells in tumor rejection in addition to those played by CD8+ T cells, CD4+ T cells, and B cells after IDOi@TBC-Hf and PDT treatment. Our future efforts will be directed toward elucidating the roles of neutrophils and B cells in initiating antitumor immune response after the PDT of IDOi@TBC-Hf.
CONCLUSION
In this work, we have rationally designed a chlorin-based nMOF with large channels for highly efficient PDT, while simultaneously loading an IDO inhibitor into its channels to achieve a combination of PDT and checkpoint blockade immunotherapy. We consistently observed an abscopal effect in mice receiving treatment with PDT of IDOi@TBC-Hf. The in situ vaccination induced by PDT treatment and IDOi immunotherapy synergize with each other and effectively generate systemic antitumor immunity. We believe the present strategy has the potential to significantly increase the systemic tumor-specific immune response rates of checkpoint blockade cancer immunotherapy and lead to clinical benefits for the treatment of metastatic colorectal cancers and other difficult-to-treat cancers.
MATERIALS AND METHODS
Materials, Cell Lines, and Animals
All of the starting materials were purchased from Sigma-Aldrich and Fisher (USA), unless otherwise noted, and used without further purification. INCB24360 analogue, 4-amino-N-(3-chloro-4-fluorophenyl)-N′-hydroxy-1,2,5-oxadiazole-3-carboximidamide was purchased from Medkoo Biosciences, USA.
Murine colon adenocarcinoma cells CT26 and MC38 and murine melanoma cells B16F10 were purchased from the American Type Culture Collection (Rockville, MD, USA) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% FBS.
C57BL/6 female mice (6 weeks, 20–22 g) and BALB/c female mice (6 weeks, 20–22 g) were provided by Harlan Laboratories, Inc. (USA). The study protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Chicago.
Synthesis of TBC-Hf
To a 2-dram glass vial was added 1 mL of HfCl4 solution (2 mg/mL in DMF, 6.2 μmol), 1 mL of the H4TBC solution (1.9 mg/mL in DMF, 2.4 μmol), and 60 μL of 88% formic acid (1.4 mmol). The reaction mixture was kept in an 80 °C oven for 2 days. The purple powder was collected by centrifugation and washed with DMF, 1% triethylamine in ethanol (v/v), and ethanol.
Singlet Oxygen Generation
A light-emitting diode (LED) array with peak emission at 650 nm was used as the light source of singlet oxygen generation test. The irradiance of LED is 20 mW/cm2. SOSG reagent (Life Technologies) was employed for the detection of singlet oxygen. H4TBP, H4TBC, TBP-Hf, and TBC-Hf samples were prepared in 1 μM solutions/suspensions in water (for the nMOFs, the concentration was calculated as ligand equivalents). To 2 mL each of these solutions/suspensions, SOSG stock solution (5 μL at 5 mM) was added (final concentration =12.5 μM) before fluorescence measurement. For a typical measurement, fluorescence intensity was acquired on a spectrofluorophotometer (RF-5301PC, Shimadzu, Japan) with excitation at 504 nm and emission at 525 nm (slit width 1.5 nm/3 nm for ex/em). Fluorescence was measured after irradiation by LED for 0 (as background), 0.5, 1, 1.5, 2, 3, 4, 5, 7, and 10 min.
In Vitro PDT Efficacy
The cytotoxicity of TBC-Hf and TBC was evaluated in murine colorectal cancer cell CT26 and murine melanoma cell B16F10, respectively. CT26 cells or B16F10 cells were seeded on 96-well plates at 1000 cells/well. The cells were treated with TBC-Hf and H4TBC at various ligand concentrations (2, 4, 10, 15, 20, and 50 μM based on ligand concentrations). A further incubation of 4 h was allowed followed by replacing the culture medium with 100 μL of fresh medium. The cells were irradiated with LED light (650 nm) at 100 mW/cm2 for 15 min (total light dose 90 J/cm2) or kept in dark, respectively. The cells were further incubated to achieve a total incubation time of 72 h. The cell viability was detected by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Promega, USA).
Abscopal Effect
The PDT efficacy of IDOi@TBC-Hf was investigated using a bilateral CT26 mouse colorectal cancer model and a bilateral MC38 mouse colorectal cancer model. Tumor-bearing mice were established by subcutaneous inoculation of CT26 or MC38 cell suspension (1 × 106 cells per mouse on the right flank and 2 × 105 cells per mouse on the left flank) into 6-week female BALB/c or 6-week female C57BL/6 mice, respectively. When the right tumors reached 100 mm3, the CT26 tumor-bearing mice received intratumoral injection only into the right tumors of PBS, H4TBC, H4TBC+IDOi, TBC-Hf, or IDOi@TBC-Hf at a ligand dose of 20 μmol/kg and IDOi dose of 1.5 mg/kg. Twelve hours post injection, CT26 tumor-bearing mice were anesthetized with 2% (v/v) isoflurane, and right flank tumors were irradiated with a 650 nm LED at 0.1 W/cm2 for 15 min (90 J/cm2). IDOi@TBC-Hf without light irradiation served as a dark control. The nMOF injection and light irradiations were performed every 3 days for a total two injections and irradiations. For MC38 tumor model, mice received intratumoral injection only into the right tumors of PBS, TBC-Hf, or IDOi@TBC-Hf at a ligand dose of 20 μmol/kg and IDOi dose of 1.5 mg/kg. Twelve hours post injection, MC38 tumor-bearing mice were anesthetized with 2% (v/v) isoflurane, and right flank tumors were irradiated with a 650 nm LED at 0.1 W/cm2 for 15 min (90 J/cm2). IDOi@TBC-Hf without light irradiation served as a dark control. The nMOF injection and light irradiations were performed only once on MC38 tumor-bearing mice. For both CT26 and MC38 models, no nMOF injection or light irradiation was performed on the left tumors.
To evaluate the therapeutic efficacy, the tumor size was measured with a digital caliper every day. Tumor volumes were calculated as follows: (width2 × length)/2. Finally, all mice were sacrificed when the tumor size of control group exceeded 2 cm3, and the excised tumors were photographed and weighed.
T Cell Blocking
The abscopal effect of IDOi@TBC-Hf was evaluated on bilateral subcutaneous MC38 model on C57BL/6 mice with CD4+ T cell or CD8+ T cell depletion. When the right tumors reached ~100 mm3, IDOi@TBC-Hf was intratumorally injected to the mice at a dose of 20 μmol/kg. Anti-CD4 (for CD4+ T cell depletion), anti-CD8 (for CD8+ T cell depletion), or mouse IgG (control) were intraperitoneally injected to the mice (200 μg/mouse/injection) on Day 0 and Day 5 post the first treatment. Twelve hours post injection, mice were anesthetized with 2% (v/v) isoflurane, and the right tumors were irradiated with LED light irradiation (100 mW/cm2, 90 J/cm2, 650 nm). Single IDOi@TBC-Hf injections followed by single light irradiations were carried out. To evaluate the therapeutic efficacy, the tumor size was measured with a digital caliper every other day. Tumor volumes were calculated as (width2 × length)/2.
ELISPOT
Tumor-specific immune responses to IFN-γ was measured in vitro by ELISPOT assay (Mouse IFN gamma ELISPOT Ready-SET-Go!; Cat. No. 88-7384-88; eBioscience). A Millipore Multiscreen HTS-IP plate was coated overnight at 4 °C with anti-Mouse IFN-γ capture antibody. Single-cell suspensions of splenocytes were obtained from MC38 tumor-carrying mice and seeded onto the antibody-coated plate at a concentration of 2 × 105 cells/well. Cells were incubated with or without KSPWFTTL (KSP) stimulation (10 μg/mL; purity >95%; PEPTIDE 2.0) for 48 h at 37 °C and then discarded. The plate was then incubated with biotin-conjugated anti-IFN-γ detection antibody at room temperature for 2 h, followed by incubation with Avidin-HRP for 2 h at room temperature. AEC substrate solution (Sigma, Cat. AEC101) was added for cytokine spot detection.
Flow Cytometry
Tumors were harvested, treated with 1 mg/mL collagenase I (Gibco, USA) for 1 h, and ground by the rubber end of a syringe. Cells were filtered through nylon mesh filters and washed with PBS. The single-cell suspension was incubated with anti-CD16/32 (clone 93; eBiosciences) to reduce nonspecific binding to FcRs. Cells were further stained with the following fluorochrome-conjugated antibodies: CD45 (30-F11), CD3e (145-2C11), CD4 (GK1.5), CD8 (53-6.7), Foxp3 (FJK-16s), CD11b (M1/70), Ly6C (HK1.4), Ly6G (RB6-8C5), F4/80 (BM8), B220 (RA3-6B2), NKp46 (29A1.4), and PI staining solution (all from eBioscience). LSR FORTESSA (BD Biosciences) was used for cell acquisition, and data analysis was carried out using FlowJo software (TreeStar, Ashland, OR).
MHC-II Expression
Tumors were harvested 12 h after LED irradiation, treated with 1 mg/mL collagenase I, and ground by the rubber end of a syringe. Cells were filtered through nylon mesh filters and washed with PBS. The single cell suspension was incubated with anti-CD16/32 (clone 93; eBiosciences) for 10 min and then stained with CD45, CD3e, CD11b, Ly6G, B220, F4/80, CD11c, and MHC-II. The expression levels of MHC-II on CD11b+Ly-6G+, CD3e-B220+, CD3e-CD11c+, and CD3e-CD11c-F4/80 populations were determined, respectively.
Supplementary Material
Acknowledgments
We thank Mr. Christopher Poon and Mr. Zekai Lin for experimental help. We acknowledge the National Cancer Institute (U01-CA198989), the University of Chicago Medicine Comprehensive Cancer Center (NIH CCSG: P30 CA014599), the Cancer Research Foundation, and the Ludwig Institute for Metastasis Research for funding support.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06663.
Experimental details and supporting results for the synthesis and characterization of H4TBC, TBC-Hf, and IDOi@TBC-Hf, cellular uptake, in vitro PDT efficacy, abscopal effect, and antitumor immunity, including Figures S1–S27 and Tables S1–S3 (PDF)
References
- 1.(a) Hamblin MR, Hasan T. Photoch Photobio Sci. 2004;3(5):436–450. doi: 10.1039/b311900a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S, Pogue BW, Hasan T. Chem Rev. 2010;110(5):2795–2838. doi: 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dolmans DEJGJ, Fukumura D, Jain RK. Nat Rev Cancer. 2003;3(5):380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]; (d) Ethirajan M, Chen Y, Joshi P, Pandey RK. Chem Soc Rev. 2011;40(1):340–362. doi: 10.1039/b915149b. [DOI] [PubMed] [Google Scholar]
- 2.Stolik S, Delgado J, Perez A, Anasagasti L. J Photochem Photobiol, B. 2000;57(2):90–93. doi: 10.1016/s1011-1344(00)00082-8. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ng KK, Zheng G. Chem Rev. 2015;115(19):11012–11042. doi: 10.1021/acs.chemrev.5b00140. [DOI] [PubMed] [Google Scholar]; (b) Lovell JF, Jin CS, Huynh E, Jin H, Kim C, Rubinstein JL, Chan WC, Cao W, Wang LV, Zheng G. Nat Mater. 2011;10(4):324–332. doi: 10.1038/nmat2986. [DOI] [PubMed] [Google Scholar]; (c) Huynh E, Leung BY, Helfield BL, Shakiba M, Gandier JA, Jin CS, Master ER, Wilson BC, Goertz DE, Zheng G. Nat Nanotechnol. 2015;10(4):325–332. doi: 10.1038/nnano.2015.25. [DOI] [PubMed] [Google Scholar]; (d) Carter KA, Shao S, Hoopes MI, Luo D, Ahsan B, Grigoryants VM, Song W, Huang H, Zhang G, Pandey RK, Geng J, Pfeifer BA, Scholes CP, Ortega J, Karttunen M, Lovell JF. Nat Commun. 2014;5:3546. doi: 10.1038/ncomms4546. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kim J, Piao Y, Hyeon T. Chem Soc Rev. 2009;38(2):372–390. doi: 10.1039/b709883a. [DOI] [PubMed] [Google Scholar]; (f) Wang W, Wang L, Li Z, Xie Z. Chem Commun. 2016;52(31):5402–5405. doi: 10.1039/c6cc01048b. [DOI] [PubMed] [Google Scholar]; (g) Padilla R, Maza WA, Dominijanni AJ, Winkel BS, Morris AJ, Brewer KJ. J Photochem Photobiol, A. 2016;322:67–75. [Google Scholar]; (h) Han K, Wang SB, Lei Q, Zhu JY, Zhang XZ. ACS Nano. 2015;9(10):10268–10277. doi: 10.1021/acsnano.5b04243. [DOI] [PubMed] [Google Scholar]; (i) Chen JX, Wang HY, Li C, Han K, Zhang XZ, Zhuo RX. Biomaterials. 2011;32(6):1678–1684. doi: 10.1016/j.biomaterials.2010.10.047. [DOI] [PubMed] [Google Scholar]; (j) Wang C, Tao H, Cheng L, Liu Z. Biomaterials. 2011;32(26):6145–6154. doi: 10.1016/j.biomaterials.2011.05.007. [DOI] [PubMed] [Google Scholar]; (k) Idris NM, Gnanasammandhan MK, Zhang J, Ho PC, Mahendran R, Zhang Y. Nat Med. 2012;18(10):1580–1585. doi: 10.1038/nm.2933. [DOI] [PubMed] [Google Scholar]; (l) Huang P, Lin J, Wang S, Zhou Z, Li Z, Wang Z, Zhang C, Yue X, Niu G, Yang M, et al. Biomaterials. 2013;34(19):4643–4654. doi: 10.1016/j.biomaterials.2013.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Park YI, Kim HM, Kim JH, Moon KC, Yoo B, Lee KT, Lee N, Choi Y, Park W, Ling D, et al. Adv Mater. 2012;24(42):5755–5761. doi: 10.1002/adma.201202433. [DOI] [PubMed] [Google Scholar]; (n) Levine DH, Ghoroghchian PP, Freudenberg J, Zhang G, Therien MJ, Greene MI, Hammer DA, Murali R. Methods. 2008;46(1):25–32. doi: 10.1016/j.ymeth.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Cheng Y, Samia AC, Meyers JD, Panagopoulos I, Fei B, Burda C. J Am Chem Soc. 2008;130(32):10643–10647. doi: 10.1021/ja801631c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Rieffel J, Chen F, Kim J, Chen G, Shao W, Shao S, Chitgupi U, Hernandez R, Graves SA, Nickles RJ, et al. Adv Mater. 2015;27(10):1785–1790. doi: 10.1002/adma.201404739. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Luo D, Carter KA, Miranda D, Lovell JF. Adv Sci. 2016 doi: 10.1002/advs.201600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Lu K, He C, Lin W. J Am Chem Soc. 2014;136(48):16712–16715. doi: 10.1021/ja508679h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu K, He C, Lin W. J Am Chem Soc. 2015;137(24):7600–7603. doi: 10.1021/jacs.5b04069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castano AP, Mroz P, Hamblin MR. Nat Rev Cancer. 2006;6(7):535–545. doi: 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Couzin-Frankel J. Science. 2013;342(6165):1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]; (b) Pardoll DM. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma P, Allison JP. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 8.Zou W. Nat Rev Cancer. 2005;5(4):263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 9.(a) Gilboa E. Nat Rev Cancer. 2004;4(5):401–411. doi: 10.1038/nrc1359. [DOI] [PubMed] [Google Scholar]; (b) Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. J Exp Med. 1999;189(9):1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Science. 1998;281(5380):1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 11.Yue EW, Douty B, Wayland B, Bower M, Liu X, Leffet L, Wang Q, Bowman KJ, Hansbury MJ, Liu C, et al. J Med Chem. 2009;52(23):7364–7367. doi: 10.1021/jm900518f. [DOI] [PubMed] [Google Scholar]
- 12.Khleif S, Munn D, Nyak-Kapoor A, Mautino MR, Kennedy E, Vahanian NN, Link CJ. ASCO Annual Meeting Proceedings. 2014:TPS3121. [Google Scholar]
- 13.Morris W, Volosskiy B, Demir S, Gándara F, McGrier PL, Furukawa H, Cascio D, Stoddart JF, Yaghi OM. Inorg Chem. 2012;51(12):6443–6445. doi: 10.1021/ic300825s. [DOI] [PubMed] [Google Scholar]
- 14.(a) Park J, Jiang Q, Feng D, Mao L, Zhou HC. J Am Chem Soc. 2016;138(10):3518–3525. doi: 10.1021/jacs.6b00007. [DOI] [PubMed] [Google Scholar]; (b) Liu J, Yang Y, Zhu W, Yi X, Dong Z, Xu X, Chen M, Yang K, Lu G, Jiang L, et al. Biomaterials. 2016;97:1–9. doi: 10.1016/j.biomaterials.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 15.Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Nat Med. 2003;9(10):1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 16.Newton RC, Scherle PA, Bowman K, Liu X, Beatty GL, O’Dwyer PJ, Gajewski T, Bowman J, Schaub R, Leopold L. ASCO Annual Meeting Proceedings. 2012:2500. [Google Scholar]
- 17.Janeway CA, Travers P, Walport M, Capra JD. Immunobiology: the immune system in health and disease. Taylor & Francis; 2005. [Google Scholar]
- 18.Siegel R, DeSantis C, Jemal A. Cancer J Clin. 2014;64(2):104–117. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 19.(a) Huxford RC, Della Rocca J, Lin W. Curr Opin Chem Biol. 2010;14(2):262–268. doi: 10.1016/j.cbpa.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lin W, Rieter WJ, Taylor KM. Angew Chem, Int Ed. 2009;48(4):650–658. doi: 10.1002/anie.200803387. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Della Rocca J, Liu D, Lin W. Acc Chem Res. 2011;44(10):957–968. doi: 10.1021/ar200028a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) He C, Liu D, Lin W. Chem Rev. 2015;115(19):11079–11108. doi: 10.1021/acs.chemrev.5b00125. [DOI] [PubMed] [Google Scholar]
- 20.(a) Cui Y, Yue Y, Qian G, Chen B. Chem Rev. 2012;112(2):1126–1162. doi: 10.1021/cr200101d. [DOI] [PubMed] [Google Scholar]; (b) Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM. Science. 2013;341(6149):1230444. doi: 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]; (c) Horcajada P, Gref R, Baati T, Allan PK, Maurin G, Couvreur P, Ferey G, Morris RE, Serre C. Chem Rev. 2011;112(2):1232–1268. doi: 10.1021/cr200256v. [DOI] [PubMed] [Google Scholar]
- 21.Palucka K, Banchereau J. Nat Rev Cancer. 2012;12(4):265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kousis PC, Henderson BW, Maier PG, Gollnick SO. Cancer Res. 2007;67(21):10501–10510. doi: 10.1158/0008-5472.CAN-07-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.