

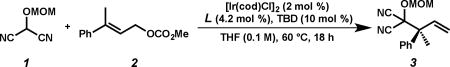
Abstract
The first highly enantioselective iridium-catalyzed allylic alkylation providing access to products bearing an allylic all-carbon quaternary stereogenic center has been developed. The reaction utilizes a masked acyl cyanide (MAC) reagent, which enables the one-pot preparation of α-quaternary carboxylic acids, esters, and amides with a high degree of enantioselectivity. The utility of these products is further explored via a series of diverse product transformations.
Keywords: Iridium, Allylic Alkylation, Umpolung, Quaternary Stereocenter, Carboxylic Acid Derivatives
COMMUNICATION

Ir-resistable: The first enantioselective iridium-catalyzed allylic alkylation providing access to products bearing an allylic all-carbon quaternary stereogenic center has been developed. The reaction utilizes a masked acyl cyanide (MAC) reagent, which enables a one-pot preparation of α-quaternary carboxylic acids, esters, and amides with a high degree of enantioselectivity. The utility of these products is further explored via a series of diverse product transformations.
The field of enantioselective iridium-catalyzed allylic alkylation has flourished in the 20 years since the seminal report by Helmchen.1 Over these two decades, the substrate scope with respect to the nucleophile has expanded significantly to encompass a vast array of both carbon2 and hetereoatom3 nucleophiles.4 Conversely, the scope of the electrophiles has remained largely unchanged, being limited to those that produce products bearing a tertiary allylic stereocenter (Figure 1a, left).5 Despite the longstanding interest in the synthesis of enantioenriched quaternary stereocenters within the synthetic community as well as the development of other transition metal-catalyzed processes to access all-carbon quaternary allylic stereocenters,6,7,8 iridium-catalyzed allylic alkylation reactions that furnish products possessing such a stereocenter remain conspicuously absent from the literature (Figure 1a, right).
Figure 1.

Synthesis of allylic all-carbon quaternary stereocenters via enantioselective iridium-catalyzed allylic alkylation.
As part of our ongoing program in developing iridium-catalyzed allylic alkylation technology and our continued interest in the catalytic, asymmetric synthesis of quaternary stereocenters,9 we were attracted to this unmet challenge. Moreover, we imagined that an umpolung strategy iridium-catalyzed allylic alkylation reaction of a trisubstituted allylic electrophile with a masked acyl cyanide (MAC) nucleophile would not only give rise to products containing an enantioenriched allylic all-carbon quaternary stereocenter, but also provide access to highly valuable acyclic α-quaternary carboxylic acid derivatives (i.e., acids, esters, amides) upon unmasking of the MAC functionality (Figure 1b).8,9e,10 However, success of this strategy hinged upon the implementation of a trisubstituted allylic electrophile, which were predicted to be unreactive in an enantioselective iridium-catalyzed allylic alkylation reaction. It is known that the reaction rates of these processes decrease with increasing substitution on the olefin of the electrophile.5,11,12 Herein, we unlock this heretofore unreactive class of electrophiles to achieve the first example of an enantioselective iridium-catalyzed allylic alkylation reaction forming a quaternary stereocenter at the allylic position.
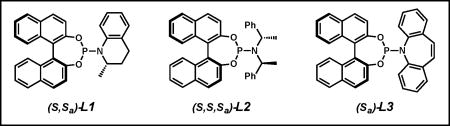
Preliminary studies focused on identifying a combination of ligand and additive to promote the reaction of MAC 1 and trisubstituted allylic electrophile 2 (Table 1). Application of our standard conditions for iridium-catalyzed allylic alkylation reactions of [Ir(cod)Cl]2, L1, and LiBr returned only starting material (Table 1, entry 1).9a,e A brief ligand screen revealed that while ligand L2 also resulted in no reaction (entry 2), the phosphoramidite L3 developed by Carreira provided desired product 3 in 13% yield with a moderate 79% ee (entry 3).3b Attempts to further increase yield and selectivity via an extensive evaluation of additives known to promote iridium-catalyzed allylic alkylations proved ineffective.2–4,9 As we hypothesized that the oxidative addition process is slow for trisubstituted allylic electrophiles, we reasoned that the inclusion of a strong Lewis acid would facilitate the ionization of the carbonate during the insertion event, leading to improved reactivity of these recalcitrant electrophiles. Toward this end, we substituted LiBr for triethylborane and were pleased to find that the yield nearly tripled and the enantioselectivity rose to 93% ee (entry 4).13 Upon varying the stoichiometry of nucleophile 1 to electrophile 2, we observed a dramatic increase in yield to 74% with no erosion of enantioselectivity (entry 5). Ultimately, we discovered that exposure of a mixture (1:2) of MAC 1 and trisubstituted allylic electrophile 2 afforded MAC adduct 3 in nearly quantitative yield and in 94% ee (entry 6).14
Table 1.
Optimization of reaction parameters.a
| |||||
|---|---|---|---|---|---|
| Entry | L | 1:2 | Additive (200 mol %) |
Yield (%)b |
ee (%)c |
| 1 | L1 | 2:1 | LiBr | 0 | – |
| 2 | L2 | 2:1 | LiBr | 0 | – |
| 3 | L3 | 2:1 | LiBr | 13 | 79 |
| 4 | L3 | 2:1 | BEt3 | 34 | 93 |
| 5 | L3 | 1:1.2 | BEt3 | 74 | 92 |
|
| |||||
| 6 | L3 | 1:2 | BEt3 | 99 | 94 |
Reactions performed on 0.1 mmol scale.
1H NMR yield based on internal standard.
Determined by chiral HPLC analysis.
TBD = 1,3,5-triazabicyclo[4.4.0]dec-5-ene.
During the course of our optimization studies, we discovered both the surprising necessity of the guanidine base TBD as well as the importance of electrophile stereochemistry in our newly developed reaction. Though previously reported conditions for the use of L3 in iridium-catalyzed allylic alkylations do not require a base additive,15 we found the inclusion of TBD during the catalyst prestir to be critical to the success of the reaction. We hypothesize that TBD may be serving as either a placeholder ligand to prevent the formation of an inactive catalyst16 or as a base to promote the formation of an active iridicycle.2a,c Additionally, we noted that use of the E-trisubstituted allylic electrophile was required, as Z-olefin isomer 4 led to markedly decreased yield and selectivity (Table 2).17 Moreover, neither a kinetic nor a dynamic kinetic resolution occurs under the reaction conditions with the use of terminal olefin rac-5 (Table 2).
Table 2.
Electrophile Isomers.a

|
Reactions performed with 1 (0.1 mmol) and 2, 4, or 5 (0.2 mmol).
1H NMR yield based on internal standard.
Determined by chiral HPLC analysis.
Before substrate scope exploration commenced, we identified an additional opportunity for innovation. We imagined that hydrolysis of the MAC functionality of product 3 could be performed in the same reaction vessel as the iridium-catalyzed allylic alkylation reaction to provide direct access to the corresponding carboxylic acid in a one-pot, two-step procedure.10 Moreover, we envisioned that these carboxylic acid products would be amenable to purification by a simple acid/base extraction. To this end, we subjected the crude allylic alkylation mixture to hydrolysis with 6M HCl at 80 °C and were pleased to find that pure carboxylic acid 7a was obtained after an aqueous work-up with no need for column chromatography (Table 3).
Table 3.
Aryl Substituent Substrate Scope.a

|
Reactions performed on 0.2 mmol scale.
Isolated yield.
Determined by chiral HPLC or SFC analysis.
Electrophile 6f used as the bis-carbonate which was deprotected during hydrolysis.
Reaction run for 48 h.
Absolute stereochemistry determined via single crystal X-ray analysis, the absolute stereochemistry of all other compounds has been assigned by analogy.
Reaction performed with double catalyst loading.
With the optimized protocol in hand, we first explored the effect of substitution on the aryl moiety of electrophile 6 (Table 3). We were pleased to find that para-substitution was well tolerated to provide acids 7b–f in consistently high enantioselectivities, though electron-rich substrates provided decreased yields. Meta-substituted products 7g and 7h were obtained in similarly high enantioselectivites (92% and 87% ee, respectively), and bulky naphthyl-substituted acid 7i was furnished in 92% ee, albeit in a moderate 66% yield. Further exploration of steric effects using methyl-substituted derivatives demonstrated that while a single meta-substituent is tolerated to access 7j in 68% yield with 93% ee, the bis-meta-substituted derivative 7k was afforded in a drastically lower 32% yield but with good enantioselectivity (85% ee). Finally, we discovered that ortho-substitution was not tolerated and only starting material was recovered from the reaction.18
With the general trends in reactivity corresponding to aryl substitution elucidated, we next turned our attention to the scope of the reaction with respect to the aliphatic moiety of the electrophile (Table 4). We found that extension of the alkyl chain led to decreased yields with ethyl-substituted 9a and n-butyl-substituted 9b isolated in 61% and 14% yield, respectively, though both were obtained in similarly excellent enantioselectivities. Furthermore, branched-substituted electrophiles were unreactive and only starting material was recovered in attempts to prepare isopropyl-substituted 9c.
Table 4.
Non-Aryl Substituent Substrate Scope.a

|
Reactions performed on 0.2 mmol scale.
Isolated yield.
Determined by chiral HPLC or SFC analysis.
We then moved to explore the necessity of the aryl functionality. We hypothesized that cyclohexyl- and cyclohexenyl-substituted electrophiles 8d and 8e would mimic the sterics of the phenyl moiety of 6a when interacting with the chiral catalyst, but we found that only trace products 9d and 9e were observed under our reaction conditions (Table 4). Use of bis-n-alkyl-substituted electrophile 8f provided the corresponding acid 9f in moderate yield, though no enantioselectivity was observed. Finally, we were pleased to find that prenyl methyl carbonate (8g) was a competent electrophile furnishing acid 9g in 63% yield.
As MAC adducts can be transformed to essentially any carboxylic acid derivative,10 we endeavored to develop additional one-pot transformations to access both α-quaternary esters and amides. Gratifyingly, we found that alkanolysis of the crude MAC alkylation product with either methanol or allyl alcohol provided methyl ester 10 and allyl ester 11 in 88% and 74% yield, respectively (Figure 2). Similarly, aminolysis provided access to both tertiary amide 12 in 61% yield and secondary amide 13 in 63% yield.
Figure 2.

(a) i. CSA, AcOH/DME, 60 °C, 6 h, ii. MeOH, Et3N, −40 °C → 23 °C, 18 h, 88% yield; (b) i. CSA, AcOH/DME, 60 °C, 6 h, ii. allyl alcohol, Et3N, 0 °C → 23 °C, 18 h, 74% yield; (c) CSA, AcOH/DME, 60 °C, 6 h, ii. pyrrolidine, Et3N, −40 °C → 23 °C, 18 h, 61% yield; (d) i. CSA, AcOH/DME, 60 °C, 6 h, ii. allyl amine, Et3N, 0 °C → 23 °C, 18 h, 63% yield.
In order to demonstrate the synthetic utility of the enantioenriched α-quaternary carboxylic acid derivatives, a series of transformations were performed to access a diverse array of chiral building blocks starting from ester derivative 10 (Figure 3). Hydrogenation of olefin 10 delivered ethyl-substituted 14 in 97% yield. Alcohol 15 was accessed via reduction of the ester moiety in 73% yield. Dihydroxylation of the pendant olefin proceeded with concomitant lactonization to furnish γ-butyrolactone 16 in 82% yield as a mixture (1:1) of diastereomers. Finally, ozonolysis furnished aldehyde 17 in moderate yield.
Figure 3.

(a) Pd/C, H2 (balloon), EtOAc, 23 °C, 18 h, 97% yield; (b) DIBAL, Et2O, 0 °C, 2 h, 73% yield; (c) K2OsO4, NMO, THF/H2O (4:1), 23 °C, 18 h, 82% yield (1:1 dr); (d) i. O3, NaHCO3, MeOH/CH2Cl2 (1:5), −78 °C, 0.5 h, ii. DMS, −78 °C→ 23 °C, 18 h, 50% yield.
In conclusion, we have developed the first synthesis of all-carbon quaternary allylic stereocenters via enantioselective iridium-catalyzed allylic alkylation. The unprecedented combination of triethylborane and a catalyst prepared from [Ir(cod)Cl]2, L3, and TBD was used to coerce reactivity from a once poorly reactive class of trisubstituted allylic electrophiles. Furthermore, the use of a single masked acyl cyanide nucleophile facilitated the one-pot syntheses of enantioenriched α-quaternary acids, esters, and amides. The protocol is tolerant of a wide range of substitution on the aryl moiety to provide the corresponding products with good yields and excellent enantioselectivites. This methodology is critical in laying the groundwork for the future development of technology to access vicinal quaternary stereocenters via iridium-catalyzed allylic alkylation of prochiral nucleophiles. Work to elucidate the nature of this catalyst system and further expand the substrate scope will be reported in due course.
Supplementary Material
Acknowledgments
The NIH-NIGMS (R01GM080269) and Caltech are thanked for support of our research program. J.C.H. thanks the Camille and Henry Dreyfus postdoctoral program, and S.E.S. thanks the NIH-NIGMS for a predoctoral fellowship (F31GM120804). Dr. Michael Takase, Dr. Lawrence Henling, and Niklas Thompson are acknowledged for assistance with X-ray analysis. Dr. Mona Shahgholi and Naseem Torian are thanked for mass spectrometry assistance. Dr. Scott Virgil is thanked for assistance with instrumentation.
References
- 1.Janssen JP, Helmchen G. Tetrahedron Lett. 1997;38:8025–8026. [Google Scholar]
- 2.For selected recent examples, see: Jiang X, Beiger JJ, Hartwig JF. J. Am. Chem. Soc. 2017;139:87–90. doi: 10.1021/jacs.6b11692.Sandmeier T, Krautwald S, Zipfel HF, Carreira EM. Angew. Chem. Int. Ed. 2015;54:14363–14367. doi: 10.1002/anie.201506933.Angew. Chem. 2015;127:14571–14575.Liu WB, Zheng C, Zhuo C-X, Dai L-X, You S-L. J. Am. Chem. Soc. 2012;134:4812–4821. doi: 10.1021/ja210923k.
- 3.For selected recent examples, see: Seehafer K, Malakar CC, Bender M, Qu J, Liang C, Helmchen G. Eur. J. Org. Chem. 2016:493–501.Defieber C, Ariger MA, Moriel P, Carreira EM. Angew. Chem. Int. Ed. 2007;46:3139–3143. doi: 10.1002/anie.200700159.Angew. Chem. 2007;119:3200–3204.Ueda M, Hartwig JF. Org. Lett. 2010;12:92–94. doi: 10.1021/ol9023248.
- 4.For selected reviews, see: Helmchen G, Dahnz A, Dübon P, Schelwies M, Weihofen R. Chem. Commun. 2007:675–691. doi: 10.1039/b614169b.Hartwig JF, Pouy MJ. Top. Organomet. Chem. 2011;34:169–208.Liu W-B, Xia J-B, You S-L. Top. Organomet. Chem. 2012;38:155–208.Hethcox JC, Shockley SE, Stoltz BM. ACS Catal. 2016;6:6207–6213. doi: 10.1021/acscatal.6b01886.
- 5.A singular example of an iridium-catalyzed allylic alkylation reaction producing a product bearing an allylic all-carbon stereocenter has been reported with 11% yield and 21% ee: Onodera G, Watabe K, Matsubara M, Oda K, Kezuka S, Takeuchi R. Adv. Synth. Catal. 2008;350:2725–2732.
- 6.a) Douglas CJ, Overman LE. Proc. Natl. Acad. Sci. USA. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Das JP, Marek I. Chem. Commun. 2011;47:4593–4623. doi: 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]; c) Quasdorf KW, Overman LE. Nature. 2014;516:181–191. doi: 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Corey EJ, Guzman-Perez A. Angew. Chem. Int. Ed. 1998;37:388–401. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998;110:402–415. [Google Scholar]; e) Christoffers J, Mann A. Angew. Chem. Int. Ed. 2001;40:4591–4597. doi: 10.1002/1521-3773(20011217)40:24<4591::aid-anie4591>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001;113:4725–4732. [Google Scholar]; f) Trost BM, Jiang C. Synthesis. 2006:369–396. [Google Scholar]; g) Liu Y, Han S-J, Liu W-B, Stoltz BM. Acc. Chem. Res. 2015;48:740–751. doi: 10.1021/ar5004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For selected recent examples of the enantioselective transition metal-catalyzed synthesis of allylic all-carbon quaternary stereocenters, see: Zhang A, RajanBabu TV. J. Am. Chem. Soc. 2006;128:5620–5621. doi: 10.1021/ja060999b.Falciola CA, Alexakis A. Chem. Eur. J. 2008;14:10615–10627. doi: 10.1002/chem.200801309.Xiong Y, Zhang G. Org. Lett. 2016;18:5094–5097. doi: 10.1021/acs.orglett.6b02540.Guduguntla S, Gualtierotti J-B, Goh SS, Feringa BL. ACS Catal. 2016;6:6591–6595.Trost BM, Jiang C. J. Am. Chem. Soc. 2001;123:12907–12908. doi: 10.1021/ja012104v.Hou X-L, Sun N. Org. Lett. 2004;6:4399–4401. doi: 10.1021/ol048729q.Zhang P, Le H, Kyne RE, Morken JP. J. Am. Chem. Soc. 2011;133:9716–9719. doi: 10.1021/ja2039248.
- 8.For the only enantioselective reports to access acyclic quaternary α-vinyl, α-aryl carbonyl derivatives, see: Murphy KE, Hoveyda AH. Org. Lett. 2005;7:1255–1258. doi: 10.1021/ol047331r.Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2006;128:15604–15605. doi: 10.1021/ja067456m.Gao F, Lee Y, Mandai K, Hoveyda AH. Angew. Chem. Int. Ed. 2010;49:8370–8374. doi: 10.1002/anie.201005124.Angew. Chem. 2010;122:8548–8552.Hojoh K, Ohmiya H, Sawamura M. J. Am. Chem. Soc. 2017;139:2184–2187. doi: 10.1021/jacs.6b12881.
- 9.a) Liu W-B, Reeves CM, Stoltz BM. J. Am. Chem. Soc. 2013;135:17298–17301. doi: 10.1021/ja4097829. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu W-B, Reeves CM, Virgil SC, Stoltz BM. J. Am. Chem. Soc. 2013;135:10626–10629. doi: 10.1021/ja4052075. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu W-B, Okamoto N, Alexy EJ, Hong AY, Tran K, Stoltz BM. J. Am. Chem. Soc. 2016;138:5234–5237. doi: 10.1021/jacs.6b02153. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hethcox JC, Shockley SE, Stoltz BM. Angew. Chem. Int. Ed. 2016;55:16092–16095. doi: 10.1002/anie.201609960. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016;128:16326–16329. [Google Scholar]; e) Hethcox JC, Shockley SE, Stoltz BM. Org. Lett. 2017;19:1527–1529. doi: 10.1021/acs.orglett.7b00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Nemoto H, Kubota Y, Yamamoto Y. J. Org. Chem. 1990;55:4515–4516. [Google Scholar]; b) Yang KS, Nibbs AE, Türkmen YE, Rawal VH. J. Am. Chem. Soc. 2013;135:16050–16053. doi: 10.1021/ja409012q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Madrahimov ST, Hartwig JF. J. Am. Chem. Soc. 2012;134:8136–8147. doi: 10.1021/ja212217j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Madrahimov ST, Li Q, Sharma A, Hartwig JF. J. Am. Chem. Soc. 2015;137:14968–14981. doi: 10.1021/jacs.5b08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.One report of a trisubstituted allylic electrophile in an enantioselective iridium-catalyzed allylic alkylation has been disclosed, but the products bear a tertiary allylic stereocenter: Chen M, Hartwig JF. Angew. Chem. Int. Ed. 2016;55:11651–11655. doi: 10.1002/anie.201607053.Angew. Chem. 2016;128:11823–11827.
- 13.To the best of our knowledge, only one report of a borane additive (Ph3B) in iridium-catalyzed allylic alkylation reactions has been disclosed: Yamashita Y, Gopalarathnam A, Hartwig JF. J. Am. Chem. Soc. 2007;129:7508–7509. doi: 10.1021/ja0730718.
- 14.It should be noted that excess electrophile 2 is not consumed and can be recovered following the iridium-catalyzed allylic alkylation reaction.
- 15.Rössler SL, Krautwald S, Carreira EM. J. Am. Chem. Soc. 2017;139:3603–3606. doi: 10.1021/jacs.6b12421. [DOI] [PubMed] [Google Scholar]
- 16.TBD is included with ligands L1 and L2 to form an active iridicycle catalyst; however, Carreira has demonstrated that ligand L3 does not form an iridicycle, see ref. 15.
- 17.We rationalize this difference in reactivity via the preferred conformation of the reactants. Whereas 2 may exist in a planar conformation, the phenyl group of 4 likely prefers to rotate out of plane to alleviate A1,3 strain. In adopting this perpendicular conformation, the phenyl ring has now increased the sterics above and below the olefin as well as become σ-withdrawing rather than π-donating.
- 18.Thiophene- and furan-substituted allylic electrophiles were well tolerated in the iridium-catalyzed allylic alkylation reaction but were not amenable to the hydrolysis conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.