

Abstract
In view of the diverse functional groups left on the MXene during the etching process, we computationally investigated the effects of surface-group types on the structural, electronic and optical properties of Sc2CT2 (T = -O, -OH, -F) MXenes. For all geometries of the Sc2CT2 MXenes, the geometry I of Sc2CT2, which has the functional groups locating above the opposite-side Sc atoms, are lowest-energy structure. Accordingly, the energetically favorable Sc2CF2-I, Sc2CO2-I and Sc2C(OH)2-I were selected for further evaluation of the photocatalytic activities. We found that the Sc2CO2-I is metallic, while Sc2CF2-I and Sc2C(OH)2 are semiconductors with visible-light absorptions and promising carrier mobilities. Compared with the Sc2C(OH)2-I, the Sc2CF2-I has not only more suitable band gap (1.91 eV), but also the higher redox capability of photo-activated carriers, which should have better photocatalytic performance.
Introduction
Since the discovery of graphene by Novoselov et al.1, two-dimensional (2D) materials have garnered tremendous interest in experimental and theoretical studies due to the unique properties of these 2D free-standing crystals. Several families of 2D materials have been recently identified and investigated, such as transition metal dichalcogenides (TMDs)2, hexagonal boron nitrides3, few-layer metal oxides4, metal chalcogenides (e.g. MoS2 5,WS2 6). Remarkably, the constellation of 2D materials has been augmented by a potentially quite large group of early transition metal carbides and carbonitrides (MXenes)7. MXenes was generally fabricated by selectively etching “A” layers from Mn+1AXn phases through acid solution etching8–12. Here, “M” is an early transition metal, “A” is mainly a group IIIA or IVA element (e.g. Al, Si), X is C and/or N, n = 1, 2, or 3.
During the etching process, “A” atoms were removed and functional groups (-O, -OH, or -F) were left on the surface of MXene to passivate the outer-layer metal atoms. MXene phases present a number of configurations which have variety of novel properties according to the species of functional groups. Many theoretical studies show that bare MXene are metallic and the majority of MXene are passivated by -OH, -F and -O groups still remain metallic. Exceptionally, some of the M2C (M = Ti, Zr, Hf, Sc,) MXenes turn from metallic to semiconducting by surface functionalization (-O, -OH, or -F), which have band gaps between 0.24 ∼ 1.8 eV13. The suitable band gaps of surface functionalized MXenes strongly indicate their visible-light absorption and possible photocatalytic applications.
Compared with other 2D materials, MXenes possess many exceptional properties for photocatalytic application. For example, the promising electric conductivity and carrier mobility could promote the migration of photo-induced carriers, resulting in more effective participation of e--h+ pairs during the photocatalytic process. The surface hydroxy group may serve as the anchor site for the reactants, resulting in more efficient adsorption and oxidation of the organic molecules. The band gaps of the functionalized MXenes are expected to be 0 ~ 2 eV, which reach the requirements of efficient visible-light absorption. Additionally, experimental studies of MXenes have mainly focused on their practical application of lithium-ion batteries, supercapacitors, and adsorbents, few results have been achieved on their photocatalytic utilization. In view of the above reasons, it is quite indispensable to evaluate the possibilities of photocatalytic application, in order to further explore the potential utilizations of these novel graphene-like materials.
In our previous work14, we theoretically investigated the electronic and optical properties of functionalized MXenes M2CT2 (M = Ti, Zr, Hf, T = -O, -OH, -F) with different functionalization geometries. Among the variously functionalized MXenes, only M2CO2 (M = Ti, Zr, Hf) with geometry I, which have the functional group located above the opposite-side metal atoms, have appropriate band gaps (0.92 ∼ 1.75 eV) for photocatalytic applications. Regarding to the Sc2CT2 (T = -O, -OH, -F) MXenes, first-principle calculations13 by using the Generalized Gradient Approximation (GGA) in the form of Perdew-Burke-Ernzerhof (PBE) functional found that the Sc2C(OH)2 has a direct band gap of 0.45 eV, while the Sc2CF2 and Sc2CO2 have indirect band gaps of 1.03 and 1.80 eV, respectively. As we known, the band gaps of these MXenes are underestimated due to the PBE functional. The hybrid functional of Heyd-Scuseria-Ernzerhof (HSE06) method was proven a reliable method in the calculation of electronic structures15,16. Moreover, the effects of surface group on the electronic and optical properties of Sc2CT2 (T = -O, -OH, -F) MXenes and the possibilities of their photoelectronic applications are yet to be addressed.
In this paper, the first-principle calculations were performed to explore the structural, electronic and optical properties of Sc2CT2 (T = -O, -OH, -F) and to further discuss their probability for photocatalytic applications. The crucial characteristics of these Sc2CT2 were summarized, including the thermodynamic stabilities, band structures, optical absorptions, redox potentials and carrier mobility. The computational results suggest that Sc2CF2-I with -F group locating at the opposite position of outer metal atoms have appropriate band gaps of 1.91 eV, visible-light absorption and strong redox capability of photo-induced excitons. Based on our calculation results, the fluorinated Sc2C MXene could be a promising candidate for high-performance photocatalysts.
Results
Thermodynamic stabilities of Sc2CT2 (T = -O, -OH, -F) MXenes
We firstly check the relative stabilities of the optimized MXenes with different geometries. The total energies (E tot) of the optimized MXenes are listed in Table 1. In general, our results are consistent with previous report13. In three geometries, geometry-I structures of Sc2CO2, Sc2C(OH)2, Sc2CF2 usually have the lowest total energy, which demonstrates that geometry I is energetically more favorable than geometry II and III.
Table 1.
Total energy (E tot in eV/unit cell) and Cohesive energies (E coh in eV/atom) of the functionalized Sc2CT2 (T = -O, -OH, -F) MXenes.
| MXenes | Total energy E tot | Cohesive energies E coh | ||||
|---|---|---|---|---|---|---|
| geometry | Sc2CO2 | Sc2C(OH)2 | Sc2CF2 | Sc2CO2 | Sc2C(OH)2 | Sc2CF2 |
| I | −40.58 | −49.13 | −37.88 | 6.38 | 5.46 | 6.25 |
| II | −39.24 | −48.36 | −36.67 | 6.11 | 5.35 | 6.01 |
| III | −39.90 | −48.77 | −37.33 | 6.24 | 5.41 | 6.14 |
The most stable geometry is highlighted in bold.
We further calculated the cohesive energy to evaluate the stabilities of the investigated MXenes. The cohesive energy (E coh) were defined as:
| 1 |
where E Sc, E C, are the total energies of a single Sc atom and C atom, E T is that of a single O, F atom or one O atom plus H atom, denotes the total energy of one unit cell of Sc2CT2 monolayer. N is the number of atoms in the supercell. The results of cohesive energy calculations are presented in Table 1. According to our results, the cohesive energies of the investigated Sc2CT2 MXene range from 5.35 to 6.38 eV/atom, which are comparable to those of FeB6 monolayer (5.56 ∼ 5.79 eV/atom)17, Al2C monolayer has (4.49 eV/atom)18, and Be2C monolayer (4.86 eV/atom)19. This indicates that the formation of strong chemical bonds between the Sc atom and T (T = -O, -OH, -F) groups. In view of the comparable E coh values of Sc2CT 2 MXenes (5.35 to 6.38 eV) and FeB6 monolayers (5.56 ~ 5.79 eV), it is proposed that the Sc2CT2 have the same type of chemical bonds with the FeB6 monolayer, in which there is obvious electron transition from the Fe atom to B framework, and thus the ionic bonding between the Fe and B atoms. Therefore, the high E coh values of Sc2CT2 MXenes also suggest the strong ionic bonds within them.
Generally, the structure with lower total energy usually imply the higher cohesive energy of the corresponding structure. In other word, the highest-cohesive-energy geometry should have the lowest total energy, and should be the energetically preferable structure. As shown in Table 1, The E coh values increase with functional group in the order of Sc2CO2 > Sc2CF2 > Sc2C(OH)2. The largest E coh values for O functionalized Sc2C may due to the stronger interaction between the O and Sc atom, which results from the shorter bond length of Sc-O (2.00 Å) than those of Sc-F (2.21 Å) and Sc-OH (2.27 Å). The same trend holds for most of other transition metal carbides (e.g Ti, V, Cr, Mo, Hf)20 Moreover, E coh values are also affected by the geometric structure. The cohesive energies of Sc2CT2 MXenes are in the decreasing order of Geometry-I > Geometry-III > Geometry-II. The highest cohesive energy of Geometry-I indicates that Geometry-I is the most beneficial to the formation of strong chemical bonds in the Sc2CT2. With a definite functional group, the preference of Geometry-I rather than Geometry-II and Geometry-III is affected by several factors such as the number of electrons demanded by the functional groups, the hybridization strength between the functional groups and transition metal13.
Electronic structures and optical properties
According to the above results of cohesive energies, Geometry-I of Sc2CO2, Sc2C(OH)2, Sc2CF2 is the most stable structure. Therefore, we only calculated the electronic structures and optical properties of Sc2CO2-I, Sc2C(OH)2-I and Sc2CF2-I in order to find out whether they have appropriate characteristics for photocatalytic applications. Based on the HSE06 hybrid functional, the band structures of three Sc2CT2 (T = -O, -OH, -F) MXenes with geometry I are computed and presented in Fig. 1. We found that Sc2CO2-I is metallic, while the Sc2C(OH)2-I and Sc2CF2-I materials are semiconductors. Accordingly, the latter two MXene were selected for further investigation. The Sc2C(OH)2 presents a direct band gap of 0.81 eV with the valence band maximum (VBM) and the conduction band minimum (CBM) both located at Γ point, while Sc2CF2-I has an indirect band gap of 1.91 eV with the VBM located at Γ point and CBM at M point, which agree well with the previous computational results20,21. The indirect band gap is beneficial to the restraint of electron-hole recombination in the photocatalytic reaction22,23.
Figure 1.

Band structures of the functionalized MXenes with geometry I. The Fermi level is at 0 eV (the red dashed lines).
HSE06 hybrid functional combines one-quarter of the exact Hartree–Fock exchange energy with three-quarters of an approximate exchange-correlation energy. This combination in general produces more reliable band gaps than GGA-PBE. As expected, the values of band gaps obtained by our HSE06 computations are greater than those of Sc2C(OH)2 (0.45 eV) and Sc2CF2 (1.03 eV) in other calculation based on PBE functionals13. For Sc2CO2 materials, the Geometry-I structure with the lowest energy was selected for the comparison in our work, while the band structure of Geometry-III with the second lowest energy was computed in ref.13. Accordingly, the metallic property of Sc2CO2 in our work is not in accordance with the results in ref.13, in which the Sc2CO2 is semiconductor. The lowest energy of the Geometry-I indicates its best thermodynamic stability and the highest experimental feasibility, and accordingly, should be selected for the further evaluation of the photocatalytic activities.
We also calculated the dielectric constants of Sc2C(OH)2-I and Sc2CF2-I to further confirm their optical absorption properties. The imaginary parts of the dielectric constants consists of and , which are the components perpendicular and parallel to the z direction, respectively. We can see from Fig. 2 that the Sc2C(OH)2-I has three obvious absorption peaks at 1.04 eV, 2.34 eV and 2.99 eV in the visible-light region (<3.0 eV). The imaginary part of Sc2CF2-I has one peak at 2.76 eV. The Sc2CF2-I also has obvious absorption in the visible-light region (1.99 eV to 2.76 eV), implying the harvest of the major portion of visible light. These evidences of the absorption spectra indicate their potential application in visible light driven photocatalysis and other photoelectronic process.
Figure 2.

Imaginary parts of dielectric constants of the Sc2C(OH)2-I and Sc2CF2-I.
From the viewpoint of photoelectrochemistry, the photocatalytic activity of the materials is also affected by the band edge position. The band edge positions were obtained by calculating the CBM and VBM energies relative to the vacuum level at 0 eV in our computations. Additionally, in order to evaluate the oxidation/reduction ability of the investigated materials, a normal hydrogen electrode (NHE) is selected as a reference, which potential is equal to −4.5 V with respect to the vacuum level (Fig. 3 and Table S1). Generally, the photocatalytic material should have photo-induced electrons with the stronger reduction capabilities if the CBM level is more negative and photo-activated holes in it should have the greater oxidation capabilities if the VBM energy is more positive.
Figure 3.
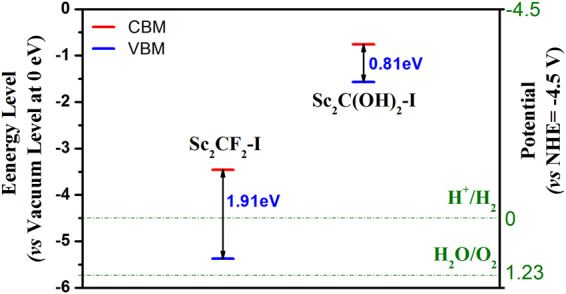
Band edge position of the Sc2C(OH)2-I and Sc2CF2-I. The redox potentials of H+/H2 and H2O/O2 at PH = 0 are also provided as a reference (the green dashed lines).
As represented in Fig. 3, Sc2C(OH)2-I has a narrow band gap of 0.81 eV, which has absorption peak at the energies of 1.04 eV, 2.34 eV and 2.99 eV. However, the tiny band gap of it may result in the easier transition of the activated electrons from CBM to VBM, which lead to the fast recombination of photo-induced e- -h+ pairs16. The Sc2C(OH)2-I with narrow bandgap may be utilized to form composites with other wide gap semiconductors, just like the graphene/TiO2 24 and graphene/ZnO nanocomposites25. These composites with well-matched band structures can broaden light absorption and enhance the separation of photo-generated electrons and holes. Compared to other composite structure26–28, in which the great achievements have been obtained, the Sc2C(OH)2-I MXene may have its unique superiority. For instance, the -OH group at the surface of Sc2C(OH)2 MXene may be promising active sites for the effective adsorption of reactants, such as the organic molecules and heavy metal ions. The high carrier mobility and good electric conductivity of Sc2C(OH)2-I could lead to the efficient migration of the photo-activated electron and holes, which would effectively participate in the photoreaction on the surface of composite-structure photocatalysts.
The CBM position of the Sc2CF2-I is around −1.038 V (vs. NHE at PH = 0), which is more negative than the reduction potential of carbon dioxide (CO2) into hydrocarbons, such as methane (CH4), methanol (CH3OH), formic acid (HCOOH). It is also more negative than the potential of H2 evolution (0 V vs. NHE at PH = 0)16. Therefore, the Sc2CF2-I can be used for photocatalytic reduction of CO2 or photocatalytic generation of hydrogen.
In semiconductors, the valence-band holes that have chemical potential of +1.0 to +3.5 V (vs. NHE) are powerful oxidants, while the conduction-band electrons with chemical potential of +0.5 to −1.5 V (vs. NHE) are good reductants29. The VBM (vs. NHE) of the Sc2CF2-I MXene is 0.87 V. So the Sc2CF2-I MXene also has oxidation capacity at a certain extent. Furthermore, the band edge position can be effectively modified by approaches of Doping30,31, tensile strain32 and heterojunctions33 to reach the requirement of the photocatalytic process.
Migration mechanism of the carrier
It is necessary to calculate the spatial charge distribution because most photo-induced electrons and holes will transfer to spatial locations of CBM and VBM, respectively. Figure 4a shows the spatial charge distribution of the Sc2C(OH)2-I. the CBM predominantly distribute around OH layer, while the VBM are located at the carbon layer. Regarding to the spatial charge distribution of the Sc2CF2-I in Fig. 4b, CBM mostly distribute around Sc atoms, the distribution of VBM is similar to that of the Sc2C(OH)2-I. These different locations of CBM and VBM could lead to spatial separation of photo-activated electron and holes and inhibit the recombination of electron-hole pairs.
Figure 4.

Spatial charge distributions for (a) Sc2C(OH)2-I (b) Sc2CF2-I.
We also computed the electron localization function (ELF)34 of the Sc2C(OH)2-I and Sc2CF2-I to analyze their electron distributions. Since the electron localization function (ELF)34 of the Sc2C(OH)2-I and Sc2CF2-I is similar, we only list the result of the Sc2CF2-I for analysis. From ELF map in Fig. S2, the red region around the C atoms indicates that the electrons are mainly located around the C atoms and there is remarkable electron transfer from Sc to C atoms. As a consequence, C atoms can act as the negative charge center and efficiently capture the photo-activated holes in Sc2CF2-I material, which results in the efficient separation of photo-generated e- -h+ pairs.
The carrier mobility is another important factor affecting photocatalytic activity. We calculated the carrier mobility of the Sc2C(OH)2-I and Sc2CF2-I along x-direction and y-direction on the basis of the hexagonal cell (Fig. S3). The results of the carrier mobility are shown in Fig. 5 and the related calculation details are presented in Fig. S4 and Table S2. As shown in Fig. 5, we found that the electron mobilities of Sc2C(OH)2-I and Sc2CF2-I are 1558 cm2 V−1 s−1 and 1288 cm2 V−1 s−1 along x direction, as well as 1230 cm2 V−1 s−1, 520 cm2 V−1 s−1 along y direction, respectively, which are comparable to those of monolayer phosphorene (1140 cm2 V−1 s−1 along x direction and 80 cm2 V−1 s−1 along y direction)27, Ti2CO2 monolayer MXene (611 cm2 V−1 s−1)35, and MoS2 (about 200 cm2 V−1 s−1)36. These results reveal the photo-induced electron can quickly migrate to the active site of photocatalytic reaction on the investigated MXenes.
Figure 5.

The carrier mobility of the Sc2C(OH)2-I and Sc2CF2-I along x-direction and y-direction.
Furthermore, in both the Sc2C(OH)2-I and Sc2CF2-I, the electron mobility is much higher than the hole mobility in x or y direction, which can be ascribed to the faster migration of electrons within the Sc2C(OH)2-I and Sc2CF2-I. The great difference between the mobility of electron and holes should result in different transport efficiency of photo-activated electron and holes within these materials. For this reason, when the photo-induced electrons reach the surface, the holes should still stay in the inner layer. This property should be valuable for the separation of electrons and holes during the photocatalytic process.
Discussion
By using first-principle computation, we have systematically investigated the functional-group-dependent structural, electronic and optical properties of Sc2CT2 (T = -O, -OH, -F) MXenes. We found that geometry-I of Sc2CT2 is the most stable one in all three geometries, which is selected to the further studies on the evaluation of their photocatalytic activity. The Sc2CO2-I is metallic, while the Sc2C(OH)2-I and Sc2CF2-I are semiconductors with band gaps of 0.81 eV and 1.91 eV, respectively. Both the Sc2C(OH)2-I and Sc2CF2-I exhibit very good optical absorption in the visible-light region. The presence of absorption peaks in the visible light region indicates their potential application as photocatalysts.
Additionally, the Sc2CF2-I MXene has an excellent reduction potential of −1.038 V (vs. NHE) and could be used for photocatalytic reduction of CO2 or for photocatalytic hydrogen generation. The photo-induced holes within it is also a good oxidant with redox potential of 0.87 V (vs. NHE). Through further studying the migration mechanism of the carrier, we confirmed that both Sc2C(OH)2-I and Sc2CF2-I have the efficiently separated electron-hole pairs and good carrier mobility. Relatively, the Sc2CF2-I can be more conducive to the visible-light photocatalysis due to its more suitable band gap and band edge alignment. Our results provide some fundamental data on the possible MXene photocatalysts, which is helpful in further exploring the practical applications of MXenes.
Computational Method
For the first-principles calculations, the Vienna Ab-initio Simulation Package (VASP) were employed37. The GGA with the PBE38 functional was adopted for the exchange-correlation functional. The ion-electron interaction was treated by the projector augmented wave (PAW) method39. The energy cutoff was set to 600 eV and the energy precision of the computations was 10−5 eV. The atomic positions were fully relaxed until the maximum force on each atom was less than 10−3 eV/Å. In the geometry optimization and self-consistent computations, a 9 × 9 × 1 Monkhorst-Pack k-point grid centered on the à point was used for the Brillouin zone. All of the band structures were calculated with the HSE06 hybrid functional40. The electronic structures were visualized using the VESTA code41.
The optical absorption properties of the Sc2CT2 were investigated by computing the complex dielectric constants (ε) at a given frequency (ω) by using the HSE06 hybrid functional with a 21 × 21 × 1 k-point mesh. The dielectric constants can be defined as . The absorption coefficient I(ω) can be calculated through following equation42:
| 2 |
As shown in the equation (1), only if the imaginary part , will be above zero. Therefore, the imaginary part reflects the light absorption at a given frequency. The imaginary part can be determined as43:
| 3 |
here the indices c and í are restricted to the conduction and valence band states and is the cell periodic part of the wavefunctions at the k-point.
The carrier mobility of the Sc2CT2 was calculated which is based on deformation potential theory44 with the following Equation45–47:
| 4 |
here the temperature of T = 300 K was adopted in this study; e, and k B are electron charge, the reduced Planck constant and the Boltzmann constant, respectively. C is the elastic modulus under the uniaxial strain along the transport direction, given by , where is the total energy of a unit cell under different uniaxial strain å and S 0 is the area of the unit cell in the xy plane; m * is the effective mass of the carrier along the transport direction, which is determined by ; E 1 is the deformation potential (DP) constant of VBM for holes and CBM for electrons, calculated by using . E edge is the energy of VBM or CBM along the transport direction.
It was well identified in previous works13,48 that each functionalized Sc2C MXene has three different geometries on its surface. We take the Sc2CO2 as an example to illustrate them. The top view and side view of Sc2CO2 structure are shown in Fig. 6, Sc2C(OH)2 and Sc2CF2 are shown in Fig. S1 (Supporting Information). As seen in Fig. 1a, Bare Sc2C MXene has one carbon layer sandwiched by two scandium layers. The functionalized Sc2C MXenes have three configurations: the geometry I with surface O atoms located above the opposite-side Sc atoms (Fig. 6b, Sc2CO2-I), the geometry II with two terminated O atoms located on the middle C atoms (Fig. 6c, Sc2CO2-II), as well as the geometry III with one terminated O atom lies on the top of the opposite Sc atom and another one lies on the top of the middle C atom (Fig. 6d, Sc2CO2-III). A vacuum space of 20 Å was inserted to avoid any interaction between MXene layers along the z axis.
Figure 6.

Top view (upper) and side view (lower) of the geometries for (a) bare Sc2C; (b) Sc2CO2-I; (c) Sc2CO2-II; (d) Sc2CO2-III.
Electronic supplementary material
Acknowledgements
This work was supported by the Natural Science Foundation of China (21403001, 11304001, 61671017), National Key R&D Program of China (Grant No. 2017YFA0403600), Science and technology research project of Jiangxi Provincial Department of Education, China (No. GJJ160483) and the Science Foundation of East China Jiaotong University (No. 15LX04).
Author Contributions
Haijun Zhang conceived the idea, designed the study and improved the manuscript. Kuangwei Xiong, Zhongfei Liu and Xin Xu performed the calculation process and wrote the paper. Guang Yang provided and compiled the Vienna Ab-initio Simulation Package. Shaowei Jin and Peihong Wang prepared and improved the Figures and Tables. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-15233-8.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Haijun Zhang, Email: hjzhang_ahu@163.com.
Shaowei Jin, Email: jinsw@mail.ustc.edu.cn.
References
- 1.Novoselov KS, et al. Electric field effect in atomically thin carbon films. Science. 2004;306:666–669. doi: 10.1126/science.1102896. [DOI] [PubMed] [Google Scholar]
- 2.Coleman JN, et al. Two-dimensional nanosheets produced by liquid exfoliation of layered materials. Science. 2011;331:568–571. doi: 10.1126/science.1194975. [DOI] [PubMed] [Google Scholar]
- 3.Novoselov KS, et al. Two-dimensional atomic crystals. P. Natl. Acad. Sci. USA. 2005;102:10451–10453. doi: 10.1073/pnas.0502848102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sreedhara MB, Matte HSSR, Govindaraj A, Rao CNR. Synthesis, characterization, and properties of few-layer MoO3. Chem-Asian. J. 2013;8:2430–2435. doi: 10.1002/asia.201300470. [DOI] [PubMed] [Google Scholar]
- 5.Joensen P, Frindt RF, Morrison SR. Single-layer MoS2. Mater. Res. Bull. 1986;21:457–461. doi: 10.1016/0025-5408(86)90011-5. [DOI] [Google Scholar]
- 6.Seo JW, et al. Two-dimensional nanosheet crystals. Angew. Chem. Int. Edit. 2007;46:8828–8831. doi: 10.1002/anie.200703175. [DOI] [PubMed] [Google Scholar]
- 7.Naguib M, Gogotsi Y, Barsoum MW. Two-dimensional nanocrystals produced by exfoliation of Ti3AlC2. Adv. mater. 2011;23:4248–4253. doi: 10.1002/adma.201102306. [DOI] [PubMed] [Google Scholar]
- 8.Naguib M, Gogotsi Y, Barsoum MW. Two-dimensional transition metal carbides. Acs Nano. 2012;6:1322–1331. doi: 10.1021/nn204153h. [DOI] [PubMed] [Google Scholar]
- 9.Naguib M, et al. New Two-dimensional niobium and vanadium carbides as promising materials for Li-ion batteries. J. Am. Chem. Soc. 2013;135:15966–15969. doi: 10.1021/ja405735d. [DOI] [PubMed] [Google Scholar]
- 10.Lukatskaya MR, Mashtalir O, Chang ER, Gogotsi Y. Cation intercalation and high volumetric capacitance of two-dimensional titanium carbide. Science. 2013;341:1502–1505. doi: 10.1126/science.1241488. [DOI] [PubMed] [Google Scholar]
- 11.Mashtalir O, et al. Intercalation and delamination of layered carbides and carbonitrides. Nat. Commun. 2013;4:1716. doi: 10.1038/ncomms2664. [DOI] [PubMed] [Google Scholar]
- 12.Tang Y, Zhu J, Yang C, Wang F. Enhanced capacitive performance based on diverse layered structure of two-dimensional Ti3C2 MXene with long etching time. J. Electrochem. Soc. 2016;163:A1975–A1982. doi: 10.1149/2.0921609jes. [DOI] [Google Scholar]
- 13.Khazaei M, Arai M, Sasaki T, Chung CY. Novel electronic and magnetic properties of two-dimensional transition metal carbides and nitrides. Adv. Funct. Mater. 2013;23:2185–2192. doi: 10.1002/adfm.201202502. [DOI] [Google Scholar]
- 14.Zhang H, et al. Computational studies on the structural, electronic and optical properties of graphene-like MXenes (M2CT2, M = Ti, Zr, Hf; T = O, F, OH) and their potential applications as visible-light driven photocatalysts. J. Mater. Chem. A. 2016;4:12913–12920. doi: 10.1039/C6TA04628B. [DOI] [Google Scholar]
- 15.Zhang H, Wu D, Tang Q, Liu L, Zhou Z. ZnO–GaN heterostructured nanosheets for solar energy harvesting: computational studies based on hybrid density functional theory. J. Mater. Chem. A. 2013;1:2231–2237. doi: 10.1039/C2TA00706A. [DOI] [Google Scholar]
- 16.Zhang H, Zuo X, Tang H, Li G, Zhou Z. Origin of photoactivity in graphitic carbon nitride and strategies for enhancement of photocatalytic efficiency: insights from first-principles computations. Phys. Chem. Chem. Phys. 2015;17:6280–6288. doi: 10.1039/C4CP05288A. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Li Y, Hou J, Tu K, Chen Z. FeB6 monolayers: the graphene-like material with hypercoordinate transition metal. J. Am. Chem. Soc. 2016;138:5644–5651. doi: 10.1021/jacs.6b01769. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Liao Y, Schleyer PVR, Chen Z. Al2C monolayer: the planar tetracoordinate carbon global minimum. Nanoscale. 2014;6:10784–10791. doi: 10.1039/C4NR01972E. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Liao Y, Chen Z. Be2C monolayer with quasi-Planar hexacoordinate carbons: A Global Minimum Structure. Angew. Chem. Int. Edit. 2014;53:7248–7252. doi: 10.1002/anie.201403833. [DOI] [PubMed] [Google Scholar]
- 20.Zha XH, Luo K, Li QW. Role of the surface effect on the structural, electronic and mechanical properties of the carbide MXenes. EPL. 2015;111:26007. doi: 10.1209/0295-5075/111/26007. [DOI] [Google Scholar]
- 21.Yang JH, Luo XP, Zhang SZ, Chen L. Investigation of magnetic and electronic properties of transition metal doped Sc2CT2 (T = O, OH or F) using a first principles study. Phys. Chem. Chem. Phys. 2016;18:12914. doi: 10.1039/C6CP00138F. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Liu L, Zhou Z. Towards better photocatalysts: first-principles studies of the alloying effects on the photocatalytic activities of bismuth oxyhalides under visible light. Phys. Chem. Chem. Phys. 2012;14:1286–1292. doi: 10.1039/C1CP23516H. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, Liu L, Zhou Z. First-principles studies on facet-dependent photocatalytic properties of bismuth oxyhalides (BiOXs) Rsc. Adv. 2012;2:9224–9229. doi: 10.1039/c2ra20881d. [DOI] [Google Scholar]
- 24.Akhavan O, Abdolahad M, Esfandiar A, Mohatashamifar M. Photodegradation of graphene oxide sheets by TiO2 nanoparticles after a photocatalytic reduction. J. Phys. Chem. C. 2010;114:12955–12959. doi: 10.1021/jp103472c. [DOI] [Google Scholar]
- 25.Williams G, Kamat PV. Graphene-semiconductor nanocomposites: excited-state interactions between ZnO nanoparticles and graphene oxide. Langmuir. 2009;25:13869–13873. doi: 10.1021/la900905h. [DOI] [PubMed] [Google Scholar]
- 26.Sun ZQ, Kim JH, Zhao Y, Attard D, Dou SX. Morphology-controllable 1D/3D nanostructured TiO2 bilayer photoanodes for dye-sensitized solar cells. Chem. Commun. 2013;49:966–968. doi: 10.1039/C2CC37212F. [DOI] [PubMed] [Google Scholar]
- 27.Sun ZQ, et al. Rational design of 3D Dendritic TiO2 nanostructures with favorable architectures. J. Am. Chem. Soc. 2011;133:19314–19317. doi: 10.1021/ja208468d. [DOI] [PubMed] [Google Scholar]
- 28.Ferrighi L, Fazio G, Valentin CD. Charege carriers separation at the graphene/(101) anatase TiO2interface. Adv. Mater. Interfaces. 2016;3:1500624. doi: 10.1002/admi.201500624. [DOI] [Google Scholar]
- 29.Hoffmann MR, Martin ST, Choi WY, Bahnemann DW. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995;95:69–96. doi: 10.1021/cr00033a004. [DOI] [Google Scholar]
- 30.Asahi R, Morikawa T, Irie H, Ohwaki T. Nitrogen-doped titanium dioxide as visible-light-sensitive photocatalyst: designs, developments, and prospects. Chem. Rev. 2014;114:9824–9852. doi: 10.1021/cr5000738. [DOI] [PubMed] [Google Scholar]
- 31.Yang K, Dai Y, Huang B, Whangbo MH. Density functional characterization of the band edges, the band gap states, and the preferred doping sites of halogen-doped TiO2. Chem. Mater. 2008;20:6528–6534. doi: 10.1021/cm801741m. [DOI] [Google Scholar]
- 32.Xing SL, Wei W, Qi LS, Feng PL, Bai BH. Ying, Dai., Two-dimensional germanium monochalcogenides for photocatalytic water splitting with high carrier mobility. Appl. Catal. B: Environ. 2017;217:275–284. doi: 10.1016/j.apcatb.2017.05.087. [DOI] [Google Scholar]
- 33.Wang M, et al. 2D-2D MnO2/g-C3N4 heterojunction photocatalyst: In-situ synthesisand enhanced CO2 reduction activity. Carbon. 2017;120:23–31. doi: 10.1016/j.carbon.2017.05.024. [DOI] [Google Scholar]
- 34.Savin A, Nesper R, Wengert S, Thomas EF. ELF: The electron localization function. Angew. Chem., Int. Ed. Engl. 1997;36:1808–1832. doi: 10.1002/anie.199718081. [DOI] [Google Scholar]
- 35.Zhang X, Zhao XD, Wu DH, Jing Y, Zhou Z. High and anisotropic carrier mobility inexperimentally possible Ti2CO2 (MXene) monolayers and nanoribbons. Nanoscale. 2015;7:16020. doi: 10.1039/C5NR04717J. [DOI] [PubMed] [Google Scholar]
- 36.disavljevic B, Radenovic A, Brivio J, Giacometti V, Kis A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011;6:147–150. doi: 10.1038/nnano.2010.279. [DOI] [PubMed] [Google Scholar]
- 37.Kresse G, Furthmuller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996;54:169–186. doi: 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- 38.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 39.Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999;59:1758–1775. doi: 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- 40.Heyd J, Scuseria GE, Ernzerhof M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2006;124:219906. doi: 10.1063/1.2204597. [DOI] [Google Scholar]
- 41.Momma K, Izumi F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011;44:1272–1276. doi: 10.1107/S0021889811038970. [DOI] [Google Scholar]
- 42.Sonali S, Sinha TP. Electronic structure, chemical bonding, and optical properties of paraelectric BaTiO3. Phys. Rev. B: Condens. Matter. Mater. Phys. 2000;73:045112. [Google Scholar]
- 43.Gajdos M, Hummer K, Kresse G, Furthmuller J, Bechstedt F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B. 2006;73:045112. doi: 10.1103/PhysRevB.73.045112. [DOI] [Google Scholar]
- 44.Bardeen J, Shockley W. Scattering of electrons in crystals in the presence of large electric fields. Phys. Rev. 1950;80:72–80. doi: 10.1103/PhysRev.80.72. [DOI] [Google Scholar]
- 45.Qiao J, Kong X, Hu ZX, Yang F, Ji W. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat. Commun. 2014;5:4475. doi: 10.1038/ncomms5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fei R, Yang L. Strain-engineering the anisotropic electrical conductance of few-layer black phosphorus. Nano. Lett. 2014;14:2884–2889. doi: 10.1021/nl500935z. [DOI] [PubMed] [Google Scholar]
- 47.Bruzzone S, Fiori G. Ab-initio simulations of deformation potentials and electron mobility in chemically modified graphene and two-dimensional hexagonal boron-nitride. Appl. Phys. Lett. 2011;99:222108. doi: 10.1063/1.3665183. [DOI] [Google Scholar]
- 48.Xie Y, Kent PRC. Hybrid density functional study of structural and electronic properties of functionalized Tin+1Xn (X = C, N) monolayers. Phys. Rev. B. 2013;87:235441. doi: 10.1103/PhysRevB.87.235441. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.