

Abstract
Cancer stem cells have established mechanisms that contribute to tumor heterogeneity as well as resistance to therapy. Over 40% of hepatocellular carcinomas (HCCs) are considered to be clonal and arise from a stem‐like/cancer stem cell. Moreover, HCC is the second leading cause of cancer death worldwide, and an improved understanding of cancer stem cells and targeting these in this cancer are urgently needed. Multiple studies have revealed etiological patterns and multiple genes/pathways signifying initiation and progression of HCC; however, unlike the transforming growth factor β (TGF‐β) pathway, loss of p53 and/or activation of β‐catenin do not spontaneously drive HCC in animal models. Despite many advances in cancer genetics that include identifying the dominant role of TGF‐β signaling in gastrointestinal cancers, we have not reached an integrated view of genetic mutations, copy number changes, driver pathways, and animal models that support effective targeted therapies for these common and lethal cancers. Moreover, pathways involved in stem cell transformation into gastrointestinal cancers remain largely undefined. Identifying the key mechanisms and developing models that reflect the human disease can lead to effective new treatment strategies. In this review, we dissect the evidence obtained from mouse and human liver regeneration, and mouse genetics, to provide insight into the role of TGF‐β in regulating the cancer stem cell niche. (Hepatology Communications 2017;1:477–493)
Abbreviations
- β2SP
β2 spectrin
- BMP
bone morphogenetic protein
- BWS
Beckwith‐Weidemann syndrome
- Cdk
cyclin‐dependent kinase
- CSC
cancer stem cell
- CTCF
CCCTC‐binding factor
- ESC
embryonic stem cell
- HCC
hepatocellular carcinoma
- HGF
hepatocyte growth factor
- IL‐6
interleukin‐6
- LSC
liver stem cell
- NF‐κB
nuclear factor kappa B
- Oct3/4
octamer 3/4
- PHx
partial hepatectomy
- STAT3
signal transducer and activator of transcription 3
- TAK1
TGF‐β–activated kinase 1
- TBRII
TGF‐β type II receptor
- TBRI
TGF‐β type I receptor
- TGF‐β
transforming growth factor β
- TNF‐α
tumor necrosis factor α
Introduction
Cancer stem cells have established mechanisms that contribute to tumor heterogeneity as well as resistance to therapy.1, 2, 3, 4 Yet to date, the switches involved in stem cell transformation in the liver and the definitive role of key pathways involved in liver regeneration and cancer remain partially understood. Multiple studies have revealed etiological patterns and multiple genes/pathways signifying initiation and progression of HCC. These pathways include CTNNB1/WNT‐β‐catenin, TPp53, ARID1/2s, HGF/c‐Met, and vascular endothelial growth factor/angiogenic signaling.5, 6, 7, 8, 9, 10, 11, 12 However, unlike the transforming growth factor β (TGF‐β) pathway, loss of p53 and/or activation of β‐catenin do not spontaneously drive HCC in animal models.13, 14, 15
Primary cancer of the liver (HCC) currently remains among the most prevalent and lethal cancers, with ∼17% 5‐year survival rate (2007–2013).2, 16, 17, 18 Drug resistance is one of the causal factors for therapy failure and is associated with the existence of tumor‐like stem cells.15 Yet driving pathways and mechanistic insight into stem cell transformation, leading to targeted therapeutics, remain poorly understood for these cancers. We review insight into how TGF‐β drives these cancers and controls the switch from normal stem cells to cancer through mechanistic insight of mouse genetic models.13, 14 TGF‐β serves as an essential regulator of cell polarity, growth, differentiation, lineage specificity, tumor suppression, as well as tumor promotion in multiple cell types13, 19 (Figs. 1, 2, 3). Yet the significance of dichotomy in function remains unclear for the liver and gastrointestinal system. Defective TGF‐β signaling is implicated in multiple cancers due to frequent somatic mutations or deregulation of its components, such as Smad3, Smad4, and TGF‐β receptors 1 and 2 (TBRI and TBRII).15, 20, 21 Smads are the intracellular mediators of TGF‐β signaling,21, 22, 23, 24 and their function is modulated by adaptor proteins such as the Smad anchors and signal transducers, filamin, and microtubules,25, 26 as well as E3 ligases such as SMURFs, Ski, PRAJA, Sno, and others. Traditionally, TGF‐β has been considered to be mainly prominent during the termination phase of liver regeneration. However, several TGF‐β–associated genes have important roles throughout the three phases of liver regeneration and focusing on the temporal fluctuations of these TGF‐β–associated partners can provide an insight into their function. This review addresses the role of “core” TGF‐β pathway–related genes (based on a literature search) that are grouped into five different categories, including TGF‐β “receptors,” “ligands,” “receptor substrates,” “adaptors,” and “inhibitory SMADs.” These genes are TGF‐β1‐3, TBRIII, TGF‐βRAP1, BMP1‐7, BMP9,10, BMP15, BMPR1A, BMPR1B, BMPR2, SMAD1‐7, SMAD9, SPTBN1 (β2SP), ACVR1, ACVR1B, ACVR1C, ACVR2A, ACVR2B, ACVRL1, ZFYVE9, INHA, INHBA, INHBB, INHBC, INHBE, GDF1, GDF11, and NODAL. In addition, other molecules, including E3 ligases, are also associated upstream or downstream of the TGF‐β pathway and correlate their expression levels with TGF‐β pathway activity such as SKI, SMURF1‐2, ITCH, ITIH4, SARA, β2SP, ELF1‐5, PRAJA1‐2, MYC, TERT, RUNX, CTCF, ALDH2, IL‐6, STAT3, TWIST1‐2, ZEB1, CDK4, TGIF1‐2, STRAP, and SNAI1‐2.
Figure 1.
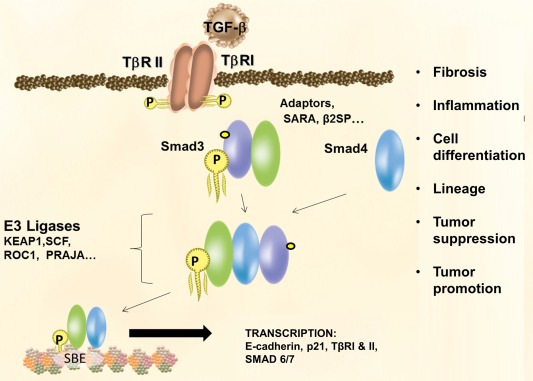
TGF‐β serves as an essential regulator of cell polarity, growth, differentiation, lineage specificity, tumor suppression, and tumor promotion in multiple cell types.
Figure 2.
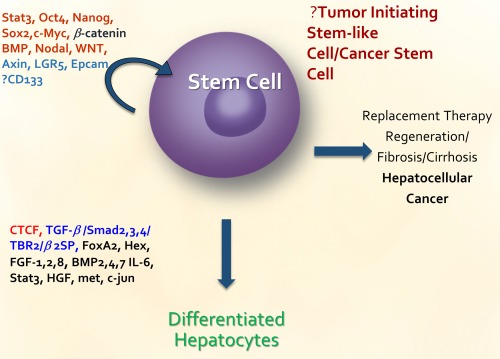
List of genes that has been identified to be important in liver cancer stem cell or normal stem cell function.
Figure 3.
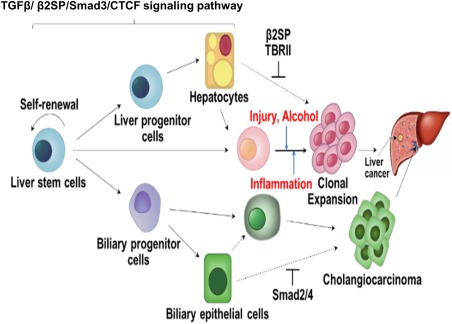
TGF‐β/SMAD/CTCF signaling pathway. The flow chart demonstrates roles of the TGF‐β/β2SP/Smad3/CTCF signaling pathway in liver stem cell homeostasis and response to alcohol‐induced injury and inflammation.
Liver Stem Cells and TGF‐β: Evidence From Mouse Knockout, Lineage Studies, and Human Liver Regeneration
Currently, at least three stem cell populations are known to exist in both mouse27 and human adult liver28: 1) pericentral Axin2+ hepatocytes that can regenerate liver in normal homeostasis29; 2) periportal cells positive for Lgr530; 3) Prom1+ liver stem cells that are located within or adjacent to the Krt19+ bile duct epithelium.31 Human periportal cells also label for octamer 3/4 (Oct3/4), β2 spectrin (β2SP), and TBRII in both human and mouse liver regeneration.27, 28, 32 Through previous studies in human liver donor and liver transplant specimens that represent human liver regeneration, as well as fulminant hepatic failure, putative liver progenitor/stem cell expansion has been observed during massive hepatic necrosis with fibrosis as well as submassive hepatic necrosis with hepatocytic lineage. The observation of fibrosis in the massive hepatic necrosis group may indicate aberrant TGF‐β signaling, which is not observed in the submassive necrosis group where a few hepatocytes are observed.33 In human liver regeneration, liver progenitor/stem cells are observed along the pericentral vein early in regeneration (before 6 weeks following post transplantation), and later at the portal tracts (≥6 weeks post transplantation)28 (Fig. 4). Anti‐Oct4 and Nanog are observed to label cells as early as the first 1‐3 weeks, representing early liver regeneration.28 These putative progenitor cells carry stem cell markers as well as TGF‐β markers, including TBRII and β2SP (TGF‐β component Smad3/4 adaptor). Although Smad3 is expressed ubiquitously, we found that the common mediator Smad4 is also expressed in this biliary region, perhaps signifying this population as “committed progenitor cells,” and suggesting that TGF‐β members play multiple and complex roles in liver stem cell function and in conferring the cell type. Future lineage tracing experiments will identify the cell populations responsible for liver regeneration following injury/damage in the context of TGF‐β signaling.
Figure 4.
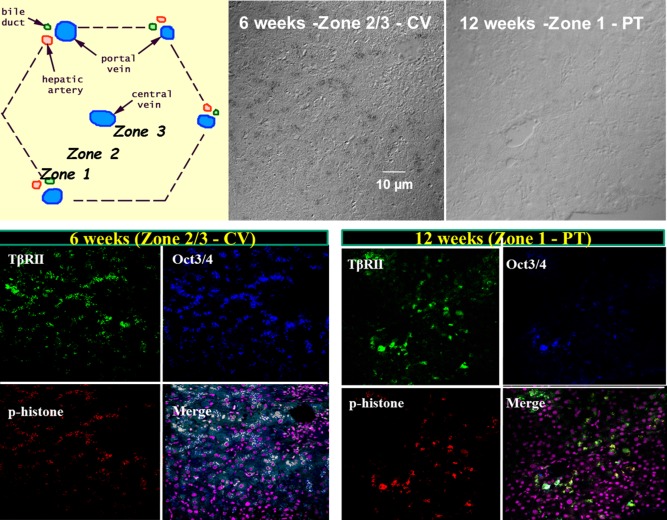
In human liver regeneration, liver progenitor/stem cells are observed along the pericentral vein early in regeneration (before 6 weeks), and later at the portal tracts (after 6 weeks). As demonstrated colocalization of Oct3/4 and p‐Histone, a marker of cell proliferation. Given that hepatocytes primarily drive liver regeneration after acute injury, it is likely that the Oct3/4 and AFP‐positive cells are proliferating hepatocytes and expression of these stem cell markers reflects their stem cell–like nature. More importantly, however, Oct3/4 and p‐Histone–positive cells also colocalize with β2SP and TBRII at all times, as evidenced in the merged images, where white represents colocalization.
TGF‐β, as a pleiotropic cytokine, has been proven to be differentially involved in the regulation of multilineage differentiation of stem cells (Fig. 5), through cross‐talk involving the Smad pathway, non‐Smad pathways including MAP kinase pathways, PI3K/AKT pathways, and Rho‐like GTPase signaling pathways. For instance, TGF‐β promotes the differentiation of stem cells into smooth muscle cells,34, 35, 36, 37 chondrocytes,38, 39, 40 neurocytes, hepatic stellate cells, Th17 cells, dendritic cells, and cardiomyocytes.41 However, TGF‐β inhibits the differentiation of stem cells into myotubes,42 adipocytes, endothelial cells, and natural killer cells.41 Additionally, TGF‐β plays a critical role in bone remodeling and can provide competence for early stages of osteoblastic differentiation, but at late stages, TGF‐β acts as an inhibitor.43, 44 In embryonic stem cells (ESCs), another TGF‐β family member, bone morphogenetic protein (BMP4) is required for ESC self‐renewal through a balanced inhibition of ESC lineage commitment. In mesenchymal stem cells, the BMP signal induces osteoblastic differentiation through Bmpr1b but inhibits osteoblastic differentiation through Bmpr1a.45 BMP signaling inhibits stem cell activation and expansion in intestinal stem cells.46 In hematopoietic stem cells, BMP signaling through Bmpr1a restricts stem cell number by controlling the niche size.47 In vitro and in vivo studies have demonstrated that Activin/Nodal signaling maintains pluripotency in human pluripotent stem cells48 and also in mouse epiblast stem cells.49 Absence of Nodal signaling results in the loss of pluripotency markers and the gain of ectopic neuroectoderm marker expression in the epiblast immediately after implantation.50, 51 BMP4 through Smad1/5/8 and Activin/Nodal through Smad2/3 compete to modulate the expression of key pluripotency markers such as Nanog.52 Activin B and several other genes that are known to be involved in enhancing Activin signaling, such as Wwp2, S100A4, Sulf2, and Inhbb, are also known to be involved in self‐renewal of hair follicle stem cells.53 Recently, TRIM33 has been discovered to act as a signal transducer and direct mediator of transcription in the TGF‐β pathway in ESCs. Ligand activation of nodal/Activin receptors induces the formation of TRIM33–Smad2/3 and Smad4–Smad2/3 protein complexes.54
Figure 5.
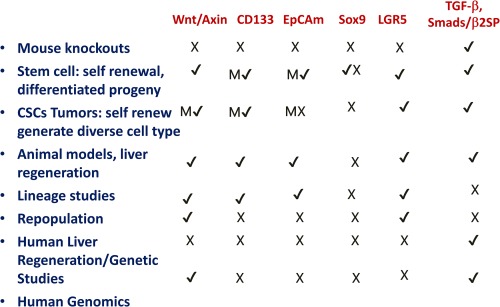
Summary of mouse knockout and lineage studies on candidate molecules in liver stem cells/cancer stem cells. Data were obtained from the Mouse Genome Informatics Database. Wnt/Axin: no liver phenotypes were observed, but 65 other phenotypes observed, including limb, kidney, and intestine. β‐catenin: 341 phenotypes were also observed, including bone and intestinal abnormalities. No liver phenotypes were observed. CD133: phenotypes observed included retinitis pigmentosa and retinal degeneration (137). EpCam: no liver abnormalities were observed, but small intestinal and trophoblast abnormalities were observed. Sox9: No liver phenotypes were observed, but 184 other phenotypes were observed, including bone, pancreas, and eye abnormalities. LGR5: 12 phenotypes were observed, including intestinal abnormalities, distended abdomen and neonatal death. In contrast, TGF‐β members display definitive foregut‐liver lineage and multiple liver cancer phenotypes. Smad4: 154 phenotypes were observed including ectoderm, mesoderm, no primitive foregut and multiple gastrointestinal cancers. Smad2/Smad3, β2SP: multiple liver and gastrointestinal abnormalities, and cancers were observed. (M = marker).
Several TGF‐β signaling components are tumor suppressors. Inactivation of at least one of these components occurs in almost all gastrointestinal tumors.17, 55 For instance, TBRII is mutated in up to 30% of colon cancers,56 TBRI is mutated in 15% of biliary cancers,57, 58, 59 and SMAD4 is deleted in 40%‐60% of pancreatic cancers and mutated in gastrointestinal cancer.60 Loss of β2SP is observed in human HCC.14, 61, 62, 63 Evidence from Smad4‐knockout mice, which develop head and neck cancers, demonstrates a significant role for Smad4 in promoting genomic stability.64 Another piece of evidence from studies of liver regeneration that implicates TGF‐β pathway in cancer involves vitamin D deficiency. Vitamin D supplementation is essential for TGF‐β pathway member expression levels. β‐catenin activation in fibrotic/cirrhotic human liver tissues and vitamin D deficiency promotes tumor growth in the context of Smad3 disruption, potentially through the regulation of TLR7 expression and β‐catenin activation. Whole genome and transcriptomic analyses of somatic mutations and alterations in genes involved in vitamin D metabolism, vitamin D–related genes, and the TGF‐β superfamily using The Cancer Genome Atlas database of 147 patients with liver cancer revealed positive correlation between inactivating somatic mutations for vitamin D–related genes and the TGF‐β pathway and plays a critical role in liver tumorigenesis.65 In addition, TGF‐β1 inhibits telomerase activity. Conversely, TGF‐β1–induced arrest of cell growth can be overcome by the activation of human TERT, the protein catalytic subunit of telomerase.66 Telomerase activation and maintenance is important for malignant transformation from normal cells.67 Keratinocytes cultured from TGF‐β1–null mice have marked genomic instability that could accelerate tumor progression.68 More recently, studies have been conducted in Smad4 conditional knockout mice that develop head and neck cancers, where Smad4 has been postulated as a “guardian of the genome” through regulation of the Fanconi anemia/Brca (Fanc/Brca) DNA repair pathway.64, 69 Interestingly, the development of HCCs in β2SP heterozygote mutants establishes β2SP as a nontraditional and functional tumor suppressor. Spectrins have been observed to associate with Fanconi proteins (G and D) as well as with DNA interstrand cross‐links.70, 71 Similarly, by virtue of its involvement in Smad3/4 localization and subsequent activation of Smad3/4, β2SP may enhance TGF‐β tumor suppressor function. Also, TGF‐β–deficient β2SP mutant mice are highly susceptible to alcohol injury, marked by an abnormal response to DNA cross‐linking repair. Taken together, these studies indicate TGF‐β as a potential processor of genomic stability through modulation of the Fanc pathway at interstrand crosslinks, yet clear mechanisms remain to be elucidated.
TGF‐β in Liver Cancer Stem Cells
Pathways involved in stem cell transformation into gastrointestinal cancers remain largely undefined and a black box. A recent discovery reports that TGF‐β–deficient mutant mice closely resemble a cancer stem cell disorder, including characteristic ear abnormalities and adrenal cytomegaly. Beckwith‐Wiedemann syndrome (BWS) is a human stem cell overgrowth disorder with an estimated prevalence of 1 in 14,00072 (Fig. 6). The syndrome includes heterogeneous features such as organomegaly and adrenal cytomegaly (a hallmark characteristic)73 (Fig. 7). BWS is associated with an 800‐fold increased risk of childhood neoplasms, and can develop multiple tumor types within the same organ simultaneously, an example including the co‐occurrence of a mesenchymal hamartoma, capillary hemangioma hepatoblastoma, and cholangiocarcinoma within the liver of one patient.74, 75 These events are suggestive of the multipotentiality of neoplastic transformation and imply dysfunctional processes as stem cells differentiate into mature adult cell types.76 Mechanistic insight into downstream effector pathways which lead to stem cell transformation and an integrated analysis from mouse models to human disease for BWS and associated cancers remain only partially defined.
Figure 6.
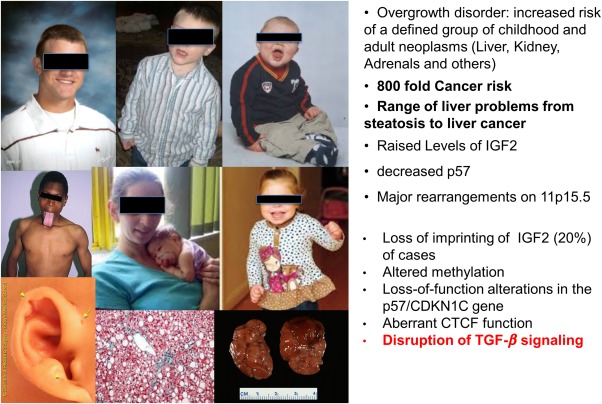
BWS is a human cancer stem cell overgrowth disorder with an estimated prevalence of 1 in 14,000. The syndrome includes heterogeneous features such as organomegaly and adrenal cytomegaly (a hallmark characteristic). Sptbn1+/−/Smad3+/− mice generated in the laboratory were phenocopies of BWS patients. Sptbn1+/−/Smad3+/− mice demonstrated microfacies and the classic metopic ridge frequently associated with human BWS and a high risk of cancers that include HCC.
Figure 7.
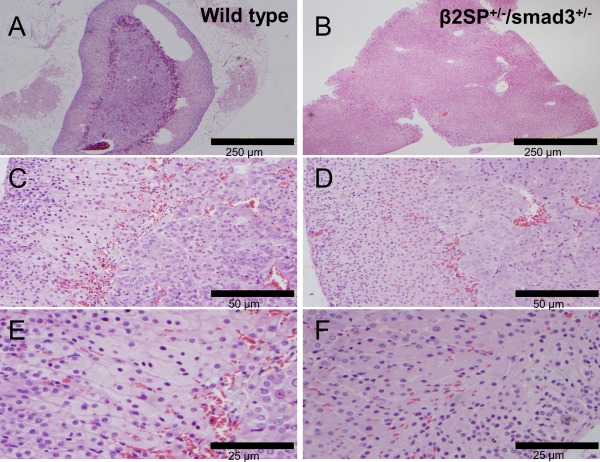
Identification of cytomegaly of the fetal cortex of the adrenal glands in a β2SP+/−/Smad3+/− mouse (B, D, F) compared with a wild‐type mouse (A, C, E).
These new studies demonstrate that TGF‐β induces chromatin insulator CCCTC‐binding factor (CTCF) which facilitates TGF‐β–mediated repression of TERT transcription via interactions with β2SP and SMAD3. This regulation is abrogated in TGF‐β–defective mice and BWS, resulting in TERT overexpression. Tert induction in Sptbn1+/−/Smad3+/− mouse embryonic fibroblasts suggests that dysregulated telomerase expression may be part of the molecular basis of tumor development in BWS patients. These results show that recruitment of the SMAD3/β2SP/CTCF complex at the TERT promoter resulting in tumorigenesis is dependent on the TGF‐β signaling pathway.14 Moreover, activation of the SMAD3/β2SP/CTCF complex on the TERT promoter region may cooperate with MYC activation. MYC activation and telomerase dysfunction have been shown to play prominent roles in early HCC initiation.77 Therefore, the TGF‐β–mediated β2SP/SMAD3/CTCF complex regulates telomerase activity and is part of a pathway that suppresses the switch to tumorigenesis in BWS‐associated cancers. Identifying similar key mechanisms through such mouse models that reflect the human disease could lead to effective new treatment strategies.
TGF‐β in an Invasive Cancer Stem Cell Model
How chronic inflammation modulates stem cells and cancer remains only partially defined, and is another black box. Chronic inflammation, often associated with liver injury, leads to secretion of cytokines, chemokines, free radicals, and other DNA‐damaging molecules, thereby changing the hepatic microenvironment.78 Persistent inflammation during this long‐term process leads to an expansion of hepatic stem and progenitor cells that accumulate genetic and epigenetic alterations. Thus, the highly inflamed liver immune microenvironment is a major driver of the transformation of normal liver stem cells (LSCs) to highly metastatic cancer stem cells (CSCs).
Molecular mechanisms that link chronic inflammatory responses with tumor initiation have been studied extensively.79, 80 Among numerous proinflammatory factors, interleukin‐6 (IL‐6) is the most prominently elevated in almost 40% of liver cancer patients, suggesting that IL‐6 is associated with HCC progression.81, 82, 83, 84, 85, 86, 87 The TGF‐β pathway induced IL‐6 secretion may confer chemotherapeutic resistance in HCC.32 IL‐6 is required for the priming of hepatocytes to leave their quiescent state (G0) and enter a prereplicative phase (G1), and transcriptionally up‐regulates an array of genes during liver growth.88 A recent study showed that signal transducer and activator of transcription 3 (STAT3), following its IL‐6–mediated activation, binds to the promoter element of CD133 to induce HCC progression.89 Liver cancer patients with high levels of CD133 expression have shorter overall survival and higher relapse rates than those with low levels of CD133 expression.90 IL‐6–mediated inflammation programs, constitutive activation of the TGF‐β–activated kinase 1 (TAK1)/nuclear factor kappa B (NF‐κB) signaling cascade in CD133+ LSCs, and this programming, interacts with deficient TGF‐β signaling, thereby accelerating the transformation of normal LSCs to metastatic CSCs.
CD133+ LSCs derived from preneoplastic livers of TGF‐β–deficient β2SP+/− mice treated with IL‐6 were highly tumorigenic and metastatic and exhibited nuclear localization of Twist and Slug (markers of epithelial‐mesenchymal transition) and constitutive activation of NF‐κB91 (Fig. 8). NF‐κB was activated by TAK1 (MAP3K7), which is associated with poor survival in HCC and IL‐6 expression. Hepatocyte‐specific deletion of TAK1 in mice activates the TGF‐β signaling pathway to induce spontaneous inflammation, fibrosis, and, eventually, hepatic tumorigenesis.92 Overall, this reciprocal regulation between β2SP and IL‐6 in the programming of liver CSCs provides insight into the mechanism by which normal stem cells transform into EMT‐positive CSCs.93 Therefore, these studies demonstrate that there exists a reciprocal cross‐talk between TGF‐β signaling and IL‐6–driven inflammation in preneoplastic liver tissues and defining the mechanisms by which the loss of TGF‐β/β2SP regulates the transition of hepatic stem cells to cells with EMT phenotypes in an inflammation‐driven hepatic immune environment. Discovery of this mechanistic insight will improve modern therapeutic approaches to eliminate metastatic tumor stem cells at an early stage before tumor initiation and will shed new light on the black box of mechanisms of stem cell transformation. Furthermore, this discovery will provide a scientific basis for a specific targeted therapy of metastatic CSCs for preventing invasion, resistance, and relapse.
Figure 8.
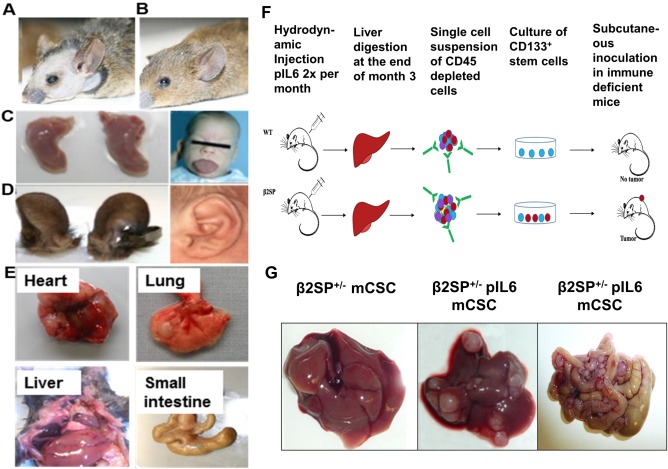
Cancer stem cell models of HCC‐TGF‐β, IL‐6. (A) β2SP+/−/Smad3+/− mouse phenocopy of BWS compared with (B) wild‐type mouse. (C) Macroglossia and (D) abnormal anterior ear creases in β2SP+/−/Smad3+/− mice and BWS patients. (E) β2SP+/−/Smad3+/− mice spontaneously develop liver and gastrointestinal cancers by 12 months of age. (F) Schematic representation of chronic treatment with IL‐6–induced cancer stem cells in β2SP+/− mice. (G) Orthotopic invasive tumor development after stem cell inoculation shown in panel F.
The Phases of Liver Regeneration
In the adult liver, mature hepatocytes seldom proliferate and have a life span of over a year.94 After partial hepatectomy, however, proliferation of the normally quiescent hepatocytes and cholangiocytes, followed by proliferation of the hepatic stellate cells and endothelial cells quickly restores the liver to its original mass. In the rodent model, DNA synthesis starts 12 to 16 hours after the standard partial hepatectomy (PHx) and peaks at 24‐48 hours. The original organ mass is almost restored 3‐7 days postresection, and by 3‐4 months in humans.95, 96 Liver regeneration therefore represents an example of precisely controlled initiation and synchronized cell proliferation in vivo, in which normally quiescent hepatocytes exit G0, reenter the cell cycle, and undergo one or two rounds of replication, with restoration of liver mass and function.94, 97, 98 The initiation step is characterized by priming of quiescent hepatocytes by factors such as tumor necrosis factor α (TNF‐α), IL‐6, and nitric oxide. These cytokines are released within minutes of partial hepatectomy from nonparenchymal liver cells and induce hepatocytes to synthesize further acute phase proteins—mainly protease inhibitors—through activation of hepatocyte DNA‐binding proteins.99, 100 Therefore, more than 100 immediate early genes are activated by latent transcription factors at the transition between G0 and G1. For instance, within minutes, specific transcription factors such as NF‐κB, STAT3, and AP1 are rapidly activated in remnant hepatocytes, as are intracellular signaling pathways such as mitogen‐activated protein kinase, phosphorylated extracellular signal‐regulated kinases, and Jun amino‐terminal kinase.101, 102, 103 The result is an induction of hepatocytes to become sensitive to growth factors and competent for replication.
The proliferation step arises when hepatocytes enter the cell cycle as G1 phase and are stimulated by complete mitogens including hepatocyte growth factor (HGF). HGF increases 10‐ to 20‐fold in the plasma within the first 3 hours after PHx and activates the HGF receptor cMet within 30‐60 minutes.104, 105 Similarly, plasma concentration of TNF‐α, IL‐6, epidermal growth factor, and TGF‐β1 increase within 1‐2 hours after PHx. These hepatomitogens, together with co‐mitogens such as norepinephrine and potentiating factors such as insulin, induce hepatocytes to override the mitogen restriction point at two‐thirds of the G1 phase and progress into DNA synthesis. These factors induce cyclins and cyclin‐dependent kinases that play critical roles in cell cycle progression.106, 107, 108 Intracellularly, β‐catenin and the Notch1 intracellular domain translocate to hepatocyte nuclei within 15‐30 minutes, and enhanced activation of STAT3 and NF‐κB within 1 hour contributes to activation of signaling pathways leading to cell cycle progression.
Cell cycle progression, regulated by the sequential formation, activation, and inactivation of complexes composed of cyclin‐dependent kinases (Cdks) and cyclins proceeds in a synchronized pattern following PHx. In mid‐ to late G1, phosphorylation of the retinoblastoma protein by Cdk4/6‐cyclin D complexes initiates the cell cycle and mediates the G1/S‐phase transition.109 Cdk2 then successively associates with cyclins E and A, completes phosphorylation of retinoblastoma protein, promotes activation of the DNA replication machinery, and regulates centrosome duplication, completing transition into S‐phase. Cdk1, in association with cyclins A and B, is then essential for entry and exit from mitosis. Cyclin D1 has been shown to be activated by 6 hours and maximal levels of Cdk4 are present at 24 hours after PHx in rats.110 Meanwhile, Cdk1 is sharply induced between 18 and 24 hours, followed by a transient decrease, before another increase at 30 hours post‐PHx in rats.111 Regeneration is complete, when an appropriate functional size is reached. TGF‐β plays a prominent role in this phase through inhibition of DNA synthesis in regenerating hepatocytes.
Temporal and Spatial Fluctuations of TGF‐β Associated Members in Liver Regeneration: An Insight Into Their Function
TGF‐β signaling has been shown to reversibly inhibit the proliferative response following partial hepatectomy.112 TGF‐β1 levels are raised in the first 2 hours after PHx, and expression levels of downstream Smads, phospho‐Smad2, Smad2, and Smad4 are similarly elevated.95, 113 Concomitant up‐regulation of TGF‐β inhibitory proteins, SnoN and Ski, and a down‐regulation of the TGF‐β receptors, allows hepatocytes to transition from G1 to S phase.114 TBRII‐conditional knockout mice demonstrate accelerated proliferation and an increased liver mass to body weight ratio after PHx.115
TGF‐β is primarily synthesized by stellate cells, a cell type that resides within the space of Disse (perisinusoidal space) in recesses between hepatocytes. There are approximately 2‐20 stellate cells per 100 hepatocytes, and these are activated during liver injury and become myofibroblasts that produce extracellular matrix, leading to progression of fibrosis and liver disease in the aberrant state. TGF‐β released from these stellate cells has a paracrine effect on hepatocytes. TGF‐β perturbations occur primarily in hepatocytes or stellate cells, depending on which cell type is dominantly undergoing proliferative changes after liver injury. Each isoform of TGF‐β has its own characteristic pattern of messenger RNA (mRNA) expression (which is assumed to have a high degree of correlation to secreted protein levels116) during a 72‐hour period after PHx (Figs. 9 and 10).116 The early phase of TGF‐β1 expression (2‐ to 6‐hour time point) precedes the major peak of hepatocyte growth, while the 48‐ to 72‐hour time point coincides with the decrease in liver cell turnover.116 The β1 isoform has an early peak at 2 hours, but then decreases and spikes again between 48 and 72 hours in hepatocytes. This suggests that the β1 isoform probably plays two different roles in liver regeneration: a major role both during the initial phase as liver regeneration progresses to DNA synthesis, as well as role in the later, termination phase of liver regeneration after injury. The β2 isoform is increased in all liver cell types at hour 6, but at the later time point, the hepatocyte fraction shows a further increase, whereas the nonparenchymal cell fractions decline. The β3 isoform shows major increases in all cell fractions at the early time point, but only the hepatocyte fraction maintains this increase at the later time point.116, 117
Figure 9.
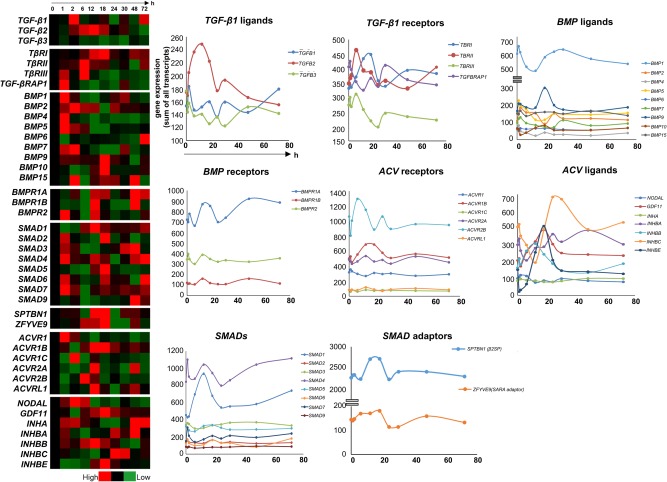
TGF‐β–associated genes had a characteristic pattern of mRNA expression during a 72‐hour period after PHx. Core TGF‐β pathway–related genes were evident in liver regeneration.
Figure 10.
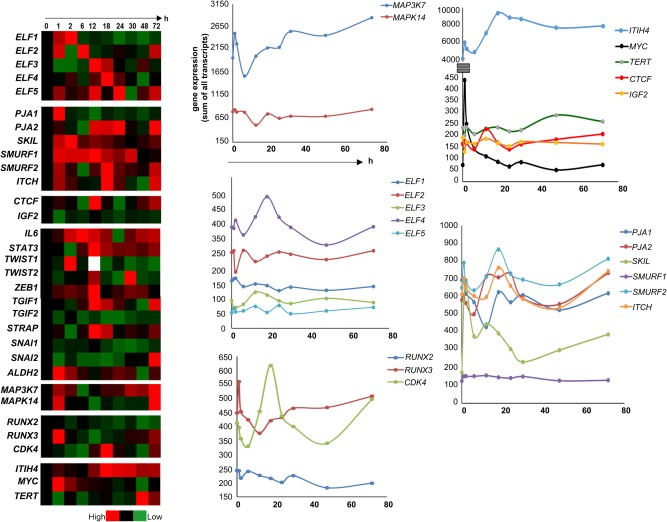
mRNA expression of other TGF‐β pathway–related genes was evident in liver regeneration during a 72‐hour period after PHx.
Levels of TBRI and TBRII involved in TGF‐β signaling are decreased in the early liver regeneration phase while TBRIII and TGF‐βRAP1/TRAP1 which are known to be inhibitors of TGF‐β signaling118, 119 are elevated. These expression levels are reversed in correlation to TGF‐β peaks in the 6‐ and 72‐hour phases. Bone morphogenetic proteins (BMPs) are members of the TGF‐β family and act as constitutively expressed repressors of regeneration. Consistent with this hypothesis, BMPs are expressed in the initial phase within 2 hours and then down‐regulated as TGF‐β spikes.116, 120 In the normal liver, strong BMP2 expression is observed around the central and portal veins. The observed down‐regulation of BMP2 in rat liver following partial hepatectomy suggests that such down‐regulation may be necessary for hepatocyte proliferation.121 Mice driven to maintain BMP4 expression in the liver, display inhibited hepatocyte proliferation and restoration of liver mass after hepatectomy, suggesting that reduced BMP4 is necessary for normal regeneration.120 Consistent with this finding, the BMP receptors also follow an inverse pattern of up‐regulation compared with TGF‐β activation and are mostly up‐regulated in the anti‐proliferative/termination phase.116
Hepatocyte‐specific deletion of the BMP receptor Activin receptor‐like kinase 3 enhances regeneration.120 The BMP4 antagonist Noggin has also been reported to enhance regeneration. BMP7 expression is absent in liver; however, neutralization of circulating endogenous BMP7 results in significantly impaired regeneration of the liver after partial hepatectomy, whereas therapeutic administration of recombinant human BMP7 significantly enhances liver regeneration. BMP9 stimulation of cultured hepatocytes inhibited proliferation. Constitutive expression of low levels of BMP9 stabilizes hepatocyte function in the healthy liver. Acute liver injury caused by partial hepatectomy results in transient down‐regulation of hepatic BMP9 mRNA expression and following HSC activation, endogenous BMP9 levels again increase.122 Regulation of Activin signaling through receptors is another major factor determining liver regeneration after liver injury. Activins inhibit DNA synthesis in hepatocytes and Activin and their receptors are initially down‐regulated and later induced between 24 and 72 hours after the proliferation slows down.123 Likewise, Inhibins which have biological effects directly opposite to those of Activins are induced in the initial phase, down‐regulated when Activins are up‐regulated, and elevated again in the later phase when activin expression is reduced.116
Smad proteins are intracellular effectors of TGF‐β signaling and transduce signals from TGF‐β superfamily ligands that regulate cell proliferation, differentiation and death through activation of receptor serine/threonine kinases. Partial hepatectomy stimulates a strong regenerative response with elevated expression of Smad2/3 phosphorylation in the first 2 hours followed by IL‐6, TNF‐α, and STAT3 induction 24 hours post‐PHx in both hepatocytes and nonparenchymal cells.116, 124 Smad3 deficiency leads to reduced hepatocyte proliferation 42 hours post‐PHx, a process that correlated with and was preceded by significant reductions in IL‐6 expression and STAT3 phosphorylation. STAT3, upon activation by a number of factors including IL‐6, regulates cell survival and proliferation and liver regeneration and loss of STAT3 in hepatocytes reduces their proliferation early during regeneration after PHx.125, 126
Among the other SMADs, suppression of SMAD1, SMAD5, and SMAD9 is known to repress liver regeneration.127 SMAD6 inhibits Wnt/β‐catenin signaling and suppresses the growth and self‐renewal of hepatic progenitor cells.128 Adaptors such as β2 spectrin (β2SP) are involved in hepatocyte proliferation through the interaction of TGF‐β/Smad and PI3K/AKT signaling.129 β2SP deficiency results in dysfunctional hepatocyte cell cycle progression and delayed liver regeneration at 48 hours after PHx. This defect is mediated by dysfunctional expression of cell cycle proteins and by increased DNA damage.27 Spatial and temporal expansion of TBRI and β2SP expression occurs as regeneration proceeds. TBRI and β2SP expression increases gradually in the initial phase, until approximately 18 hours after hepatic injury, and then decreases.116 The spatial expansion of TBRI and β2SP proceeds from periportal to pericentral areas of lobules, suggesting an important role for the TGF‐β signaling molecules in liver regeneration in response to liver injury.28 An interesting transition between PRAJA (PJA1), an E3‐dependent ubiquitin ligase of β2SP, and β2SP proteins, occurs at 6 hours postinjury, with expression of PJA1, and β2SP predictably inversely proportional to each other. At 6 hours, PJA1 levels begin to decrease, allowing the up‐regulation of β2SP on a background of TGF‐β expression, which is already high compared with expression in normal liver. As PJA1 expression continues to decrease over 6‐12 hours postinjury, β2SP remains accumulated in the cells.130 Potentially targeting similar E3 ligases that are activated in the setting of loss of TGF‐β tumor suppressor activity, could provide attractive new therapeutics for HCC.
Serial transplantation experiments have shown that hepatocytes have a near infinite capacity to proliferate.94, 131, 132 When mature hepatocytes and cholangiocytes are damaged or inhibited in their replication, however, a reserve compartment of hepatic progenitor cells are activated.133 In human liver donor transplant recipients, early on within the first 6 weeks, expansion of cells expressing stem cell markers Oct3/4, AFP, and TGF‐β members is observed in zone 1, zone 2,28 and surprisingly also zone 3 (central vein)134, 135, 136 and appears to give rise to hepatocytes. These studies suggest that in submassive hepatic necrosis with intact zones 1 and 2, cells express stem cell markers and potentially lead the regenerative process.28, 33 The activation of the stem cell compartment, originally referred to as a “ductular reaction” in humans and “oval cell reaction” in rodents, is observed in circumstances of prolonged necrosis, cirrhosis, and chronic inflammatory liver diseases. In summary, rodent studies have led to the identification of the three stem cell compartments displayed by markers of the Wnt/Axin/Lgr5 family, EpCam, and CD133. Yet mouse mutants of the single gene knockouts do not reveal significant liver pathology (Fig. 5). Mouse knockouts, human liver regeneration, and functional studies thus reveal a pivotal role for TGF‐β in suppressing cancer stem cells, as well as modulating processes of liver regeneration and fibrosis.27, 28 However, exactly how it is defined temporally with existing stem cell markers such as Axin1, Lgr5, Fancd2, and CD133 remains to be explored. Importantly, the studies provide a key role for the TGF‐β pathway in suppressing cancer stem cells, and the loss of TGF‐β signaling in these cells could lead to better identification of these cells, as well as prediction of tumor behavior and ultimately lead to precise targeting of cancer stem cells in this lethal cancer.
Potential conflict of interest: Nothing to report.
REFERENCES
- 1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014;64:9‐29. [DOI] [PubMed] [Google Scholar]
- 2. White DL, Thrift AP, Kanwal F, Davila J, El‐Serag HB. Incidence of hepatocellular carcinoma in all 50 United States, from 2000 through 2012. Gastroenterology 2017;152:812‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hepatocellular carcinoma. Nat Rev Dis Primers 2016;2:16019. [DOI] [PubMed] [Google Scholar]
- 4. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359‐E86. [DOI] [PubMed] [Google Scholar]
- 5. Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, et al. Trans‐ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 2014;46:1267‐1273. [DOI] [PubMed] [Google Scholar]
- 6. Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol 2015;12:408‐424. [DOI] [PubMed] [Google Scholar]
- 7. Li S, Mao M. Next generation sequencing reveals genetic landscape of hepatocellular carcinomas. Cancer Lett 2013;340:247‐253. [DOI] [PubMed] [Google Scholar]
- 8. Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, Pawlik TM, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet 2011;43:828‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, et al. Integrated analysis of somatic mutations and focal copy‐number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012;44:694‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, et al. Whole‐genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet 2012;44:760‐764. [DOI] [PubMed] [Google Scholar]
- 11. Clevers H, Nusse R. Wnt/beta‐catenin signaling and disease. Cell 2012;149:1192‐1205. [DOI] [PubMed] [Google Scholar]
- 12. Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, LeBlanc AC, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 2008;68:6779‐6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mishra L, Derynck R, Mishra B. Transforming growth factor‐beta signaling in stem cells and cancer. Science 2005;310:68‐71. [DOI] [PubMed] [Google Scholar]
- 14. Chen J, Yao ZX, Chen JS, Gi YJ, Munoz NM, Kundra S, Herlong HF, et al. TGF‐beta/beta2‐spectrin/CTCF‐regulated tumor suppression in human stem cell disorder Beckwith‐Wiedemann syndrome. J Clin Invest 2016;126:527‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katz LH, Likhter M, Jogunoori W, Belkin M, Ohshiro K, Mishra L. TGF‐beta signaling in liver and gastrointestinal cancers. Cancer Lett 2016;379:166‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. National Cancer Institute Surveillance, Epidimiology and End Results (SEER) Program. Cancer stat facts: liver and intrahepatic bile duct cancer. https://seer.cancer.gov/statfacts/html/livibd.html. 2016. Accessed June 20, 2017.
- 17. Majumdar A, Curley SA, Wu X, Brown P, Hwang JP, Shetty K, Yao ZX, et al. Hepatic stem cells and transforming growth factor beta in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2012;9:530‐538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bruix J, Reig M, Sherman M. Evidence‐based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology 2016;150:835‐853. [DOI] [PubMed] [Google Scholar]
- 19. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000;103:295‐309. [DOI] [PubMed] [Google Scholar]
- 20. Chen J, Raju GS, Jogunoori W, Menon V, Majumdar A, Chen JS, Gi YJ, et al. Mutational profiles reveal an aberrant TGF‐beta‐CEA regulated pathway in colon adenomas. PLoS One 2016;11:e0153933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi Y, Massague J. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 2003;113:685‐700. [DOI] [PubMed] [Google Scholar]
- 22. Moses HL, Serra R. Regulation of differentiation by TGF‐beta. Curr Opin Genet Dev 1996;6:581‐586. [DOI] [PubMed] [Google Scholar]
- 23. Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence‐specific transcription activators. Mol Cell 1998;1:611‐617. [DOI] [PubMed] [Google Scholar]
- 24. Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF‐beta signaling. Cell 1998;94:585‐594. [DOI] [PubMed] [Google Scholar]
- 25. Wu, G , Chen YG, Ozdamar B, Gyuricza CA, Chong PA, Wrana JL, Massague J, et al. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science 2000;287:92‐97. [DOI] [PubMed] [Google Scholar]
- 26. Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 1998;95:779‐791. [DOI] [PubMed] [Google Scholar]
- 27. Thenappan A, Shukla V, Abdul Khalek FJ, Li Y, Shetty K, Liu P, Li L, et al. Loss of transforming growth factor beta adaptor protein beta‐2 spectrin leads to delayed liver regeneration in mice. Hepatology 2011;53:1641‐1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thenappan A, Li Y, Kitisin K, Rashid A, Shetty K, Johnson L, Mishra L. Role of transforming growth factor beta signaling and expansion of progenitor cells in regenerating liver. Hepatology 2010;51:1373‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang B, Zhao L, Fish M, Logan CY, Nusse R. Self‐renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature 2015;524:180‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, Sato T, et al. In vitro expansion of single Lgr5 + liver stem cells induced by Wnt‐driven regeneration. Nature 2013;494:247‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu L, Finkelstein D, Gao C, Shi L, Wang Y, Lopez‐Terrada D, Wang K, et al. Multi‐organ mapping of cancer risk. Cell 2016;166:1132‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, Mueller SC, Ressom HW, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF‐beta and IL‐6 signaling. Proc Natl Acad Sci U S A 2008;105:2445‐2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nissim O, Melis M, Diaz G, Kleiner DE, Tice A, Fantola G, Zamboni F, et al. Liver regeneration signature in hepatitis B virus (HBV)‐associated acute liver failure identified by gene expression profiling. PLoS One 2012;7:e49611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kinner B, Zaleskas JM, Spector M. Regulation of smooth muscle actin expression and contraction in adult human mesenchymal stem cells. Exp Cell Res 2002;278:72‐83. [DOI] [PubMed] [Google Scholar]
- 35. Gojo S, Gojo N, Takeda Y, Mori T, Abe H, Kyo S, Hata J, et al. In vivo cardiovasculogenesis by direct injection of isolated adult mesenchymal stem cells. Exp Cell Res 2003;288:51‐59. [DOI] [PubMed] [Google Scholar]
- 36. Tamama K, Sen CK, Wells A. Differentiation of bone marrow mesenchymal stem cells into the smooth muscle lineage by blocking ERK/MAPK signaling pathway. Stem Cells Dev 2008;17:897‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vo E, Hanjaya‐Putra D, Zha Y, Kusuma S, Gerecht S. Smooth‐muscle‐like cells derived from human embryonic stem cells support and augment cord‐like structures in vitro. Stem Cell Rev 2010;6:237‐247. [DOI] [PubMed] [Google Scholar]
- 38. Kim HJ, Kim YJ, Im GI. Is continuous treatment with transforming growth factor‐beta necessary to induce chondrogenic differentiation in mesenchymal stem cells? Cells Tissues Organs 2009;190:1‐10. [DOI] [PubMed] [Google Scholar]
- 39. Kawamura K, Chu CR, Sobajima S, Robbins PD, Fu FH, Izzo NJ, Niyibizi C. Adenoviral‐mediated transfer of TGF‐beta1 but not IGF‐1 induces chondrogenic differentiation of human mesenchymal stem cells in pellet cultures. Exp Hematol 2005;33:865‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Solorio LD, Fu AS, Hernandez‐Irizarry R, Alsberg E. Chondrogenic differentiation of human mesenchymal stem cell aggregates via controlled release of TGF‐beta1 from incorporated polymer microspheres. J Biomed Mater Res A, 2010;92:1139‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang MK, Sun HQ, Xiang YC, Jiang F, Su YP, Zou ZM. Different roles of TGF‐beta in the multi‐lineage differentiation of stem cells. World J Stem Cells 2012;4:28‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Massague J. The transforming growth factor‐beta family. Annu Rev Cell Biol 1990;6:597‐641. [DOI] [PubMed] [Google Scholar]
- 43. Bonewald LF, Mundy GR. Role of transforming growth factor‐beta in bone remodeling. Clin Orthop Relat Res 1990:261‐276. [PubMed] [Google Scholar]
- 44. Karsdal MA, Hjorth P, Henriksen K, Kirkegaard T, Nielsen KL, Lou H, Delaisse JM, et al. Transforming growth factor‐beta controls human osteoclastogenesis through the p38 MAPK and regulation of RANK expression. J Biol Chem 2003;278:44975‐44987. [DOI] [PubMed] [Google Scholar]
- 45. Chen D, Ji X, Harris MA, Feng JQ, Karsenty G, Celeste AJ, Rosen V, et al. Differential roles for bone morphogenetic protein (BMP) receptor type IB and IA in differentiation and specification of mesenchymal precursor cells to osteoblast and adipocyte lineages. J Cell Biol 1998;142:295‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, et al. BMP signaling inhibits intestinal stem cell self‐renewal through suppression of Wnt‐beta‐catenin signaling. Nat Genet 2004;36:1117‐1121. [DOI] [PubMed] [Google Scholar]
- 47. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003;425:836‐841. [DOI] [PubMed] [Google Scholar]
- 48. Vallier L, Reynolds D, Pedersen RA. Nodal inhibits differentiation of human embryonic stem cells along the neuroectodermal default pathway. Dev Biol 2004;275:403‐421. [DOI] [PubMed] [Google Scholar]
- 49. Brons IG, Smithers LE, Trotter MW, Rugg‐Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007;448:191‐195. [DOI] [PubMed] [Google Scholar]
- 50. Camus A, Perea‐Gomez A, Moreau A, Collignon J. Absence of Nodal signaling promotes precocious neural differentiation in the mouse embryo. Dev Biol 2006;295:743‐755. [DOI] [PubMed] [Google Scholar]
- 51. Mesnard D, Guzman‐Ayala M, Constam DB. Nodal specifies embryonic visceral endoderm and sustains pluripotent cells in the epiblast before overt axial patterning. Development 2006;133:2497‐2505. [DOI] [PubMed] [Google Scholar]
- 52. Xu RH, Sampsell‐Barron TL, Gu F, Root S, Peck RM, Pan G, Yu J, et al. NANOG is a direct target of TGFbeta/activin‐mediated SMAD signaling in human ESCs. Cell Stem Cell 2008;3:196‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kadaja M, Keyes BE, Lin M, Pasolli HA, Genander M, Polak L, Stokes N, et al. SOX9: a stem cell transcriptional regulator of secreted niche signaling factors. Genes Dev 2014;28:328‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xi Q, Wang Z, Zaromytidou AI, Zhang XH, Chow‐Tsang LF, Liu JX, Kim H, et al. A poised chromatin platform for TGF‐beta access to master regulators. Cell 2011;147:1511‐1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, et al. TGF‐beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004;303:848‐851. [DOI] [PubMed] [Google Scholar]
- 56. Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, et al. Inactivation of the type II TGF‐beta receptor in colon cancer cells with microsatellite instability. Science 1995;268:1336‐1338. [DOI] [PubMed] [Google Scholar]
- 57. Schlitter AM, Born D, Bettstetter M, Specht K, Kim‐Fuchs C, Riener MO, Jeliazkova P, et al. Intraductal papillary neoplasms of the bile duct: stepwise progression to carcinoma involves common molecular pathways. Mod Pathol 2014;27:73‐86. [DOI] [PubMed] [Google Scholar]
- 58. Schwetz V, Uhrig S, Spuller E, Deutschmann A, Hogenauer C. Manifestations of juvenile polyposis syndrome in SMAD4 mutation carriers of a kindred. Eur J Gastroenterol Hepatol 2012;24:988‐994. [DOI] [PubMed] [Google Scholar]
- 59. Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, McPherson JR, et al. Exome sequencing of liver fluke‐associated cholangiocarcinoma. Nat Genet 2012;44:690‐693. [DOI] [PubMed] [Google Scholar]
- 60. Takeda H, Rust AG, Ward JM, Yew CC, Jenkins NA, Copeland NG. Sleeping Beauty transposon mutagenesis identifies genes that cooperate with mutant Smad4 in gastric cancer development. Proc Natl Acad Sci U S A. 2016;113:E2057‐E2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yao ZX, Jogunoori W, Choufani S, Rashid A, Blake T, Yao W, Kreishman P, et al. Epigenetic silencing of beta‐spectrin, a TGF‐beta signaling/scaffolding protein in a human cancer stem cell disorder: Beckwith‐Wiedemann syndrome. J Biol Chem 2010;285:36112‐36120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor‐beta signaling in ELF beta‐spectrin‐deficient mice. Science 2003;299:574‐577. [DOI] [PubMed] [Google Scholar]
- 63. Kitisin K, Ganesan N, Tang Y, Jogunoori W, Volpe EA, Kim SS, Katuri V, et al. Disruption of transforming growth factor‐beta signaling through beta‐spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene 2007;26:7103‐7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bornstein S, White R, Malkoski S, Oka M, Han G, Cleaver T, Reh D, et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest 2009;119:3408‐3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen J, Katz LH, Munoz NM, Gu S, Shin JH, Jogunoori WS, Lee MH, et al. Vitamin D deficiency promotes liver tumor growth in transforming growth factor‐beta/Smad3‐deficient mice through Wnt and Toll‐like receptor 7 pathway modulation. Sci Rep 2016;6:30217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stampfer MR, Garbe J, Levine G, Lichtsteiner S, Vasserot AP, Yaswen P. Expression of the telomerase catalytic subunit, hTERT, induces resistance to transforming growth factor beta growth inhibition in p16INK4A(‐) human mammary epithelial cells. Proc Natl Acad Sci U S A 2001;98:4498‐4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cong Y, Shay JW. Actions of human telomerase beyond telomeres. Cell Res 2008;18:725‐732. [DOI] [PubMed] [Google Scholar]
- 68. Glick A, Popescu N, Alexander V, Ueno H, Bottinger E, Yuspa SH. Defects in transforming growth factor‐beta signaling cooperate with a Ras oncogene to cause rapid aneuploidy and malignant transformation of mouse keratinocytes. Proc Natl Acad Sci U S A, 1999;96:14949‐14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Korc M. Smad4: gatekeeper gene in head and neck squamous cell carcinoma. J Clin Invest 2009;119:3208‐3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Oberbeck N, Langevin F, King G, de Wind N, Crossan GP, Patel KJ. Maternal aldehyde elimination during pregnancy preserves the fetal genome. Mol Cell 2014;55:807‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011;475:53‐58. [DOI] [PubMed] [Google Scholar]
- 72. Pettenati MJ, Haines JL, Higgins RR, Wappner RS, Palmer CG, Weaver DD. Wiedemann‐Beckwith syndrome: presentation of clinical and cytogenetic data on 22 new cases and review of the literature. Hum Genet 1986;74:143‐154. [DOI] [PubMed] [Google Scholar]
- 73. Weksberg R, Shuman C, Beckwith JB. Beckwith‐Wiedemann syndrome. Eur J Hum Genet 2010;18:8‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hadzic N, Finegold MJ. Liver neoplasia in children. Clin Liver Dis 2011;15:443‐462. [DOI] [PubMed] [Google Scholar]
- 75. Keller RB, Demellawy DE, Quaglia A, Finegold M, Kapur RP. Methylation status of the chromosome arm 19q MicroRNA cluster in sporadic and androgenetic‐biparental mosaicism‐associated hepatic mesenchymal hamartoma. Pediatr Dev Pathol 2015;18:218‐227. [DOI] [PubMed] [Google Scholar]
- 76. Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet 2006;7:21‐33. [DOI] [PubMed] [Google Scholar]
- 77. Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest 2004;113:160‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest 2013;123:1911‐1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schneider C, Teufel A, Yevsa T, Staib F, Hohmeyer A, Walenda G, Zimmermann HW, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine‐induced liver cancer. Gut 2012;61:1733‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, et al. Metabolic activation of intrahepatic CD8 + T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross‐talk with hepatocytes. Cancer Cell 2014;26:549‐564. [DOI] [PubMed] [Google Scholar]
- 81. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, Simeone DM, et al. Tumor‐associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014;147:1393‐1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chang TS, Wu YC, Chi CC, Su WC, Chang PJ, Lee KF, Tung TH, et al. Activation of IL6/IGFIR confers poor prognosis of HBV‐related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin Cancer Res 2015;21:201‐210. [DOI] [PubMed] [Google Scholar]
- 83. Hatting M, Spannbauer M, Peng J, Al Masaoudi M, Sellge G, Nevzorova YA, Gassler N, et al. Lack of gp130 expression in hepatocytes attenuates tumor progression in the DEN model. Cell Death Dis 2015;6:e1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. He G, Dhar D, Nakagawa H, Font‐Burgada J, Ogata H, Jiang Y, Shalapour S, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL‐6 signaling. Cell 2013;155:384‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Porta C, De Amici M, Quaglini S, Paglino C, Tagliani F, Boncimino A, Moratti R, et al. Circulating interleukin‐6 as a tumor marker for hepatocellular carcinoma. Ann Oncol 2008;19:353‐358. [DOI] [PubMed] [Google Scholar]
- 86. van der Zee M, Sacchetti A, Cansoy M, Joosten R, Teeuwssen M, Heijmans‐Antonissen C, Ewing‐Graham PC, et al. IL6/JAK1/STAT3 Signaling blockade in endometrial cancer affects the ALDHhi/CD126 + stem‐like component and reduces tumor burden. Cancer Res 2015;75:3608‐3622. [DOI] [PubMed] [Google Scholar]
- 87. Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy supports breast cancer stem cell maintenance by regulating IL6 secretion. Mol Cancer Res 2015;13:651‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Streetz KL, Luedde T, Manns MP, Trautwein C. Interleukin 6 and liver regeneration. Gut 2000;47:309‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Won C, Kim BH, Yi EH, Choi KJ, Kim EK, Jeong JM, Lee JH, et al. Signal transducer and activator of transcription 3‐mediated CD133 up‐regulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015;62:1160‐1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Song W, Li H, Tao K, Li R, Song Z, Zhao Q, Zhang F, et al. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int J Clin Pract 2008;62:1212‐1218. [DOI] [PubMed] [Google Scholar]
- 91. Mitra A, Yan J, Xia X, Zhou S, Chen J, Mishra L, Li S. IL6‐mediated inflammatory loop reprograms normal to epithelial‐mesenchymal transition + metastatic cancer stem cells in preneoplastic liver of transforming growth factor beta‐deficient beta2‐spectrin+/‐ mice. Hepatology 2017;65:1222‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yang L, Inokuchi S, Roh YS, Song J, Loomba R, Park EJ, Seki E. Transforming growth factor‐beta signaling in hepatocytes promotes hepatic fibrosis and carcinogenesis in mice with hepatocyte‐specific deletion of TAK1. Gastroenterology 2013;144:1042‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mitra A, Satelli A, Xia X, Cutrera J, Mishra L, Li S. Cell‐surface Vimentin: a mislocalized protein for isolating csVimentin(+) CD133(‐) novel stem‐like hepatocellular carcinoma cells expressing EMT markers. Int J Cancer 2015;137:491‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Michalopoulos GK, DeFrances MC. Liver regeneration. Science 1997;276:60‐66. [DOI] [PubMed] [Google Scholar]
- 95. Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol 2004;5:836‐847. [DOI] [PubMed] [Google Scholar]
- 96. Sell S. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatology 2001;33:738‐750. [DOI] [PubMed] [Google Scholar]
- 97. Michalopoulos GK. Liver regeneration. J Cell Physiol 2007;213:286‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology 2006;43(2 Suppl 1):S45‐S53. [DOI] [PubMed] [Google Scholar]
- 99. Diehl AM, Rai RM. Liver regeneration 3: regulation of signal transduction during liver regeneration. FASEB J 1996;10:215‐227. [DOI] [PubMed] [Google Scholar]
- 100. Hui TT, Mizuguchi T, Sugiyama N, Avital I, Rozga J, Demetriou AA. Immediate early genes and p21 regulation in liver of rats with acute hepatic failure. Am J Surg 2002;183:457‐463. [DOI] [PubMed] [Google Scholar]
- 101. Haber BA, Mohn KL, Diamond RH, Taub R. Induction patterns of 70 genes during nine days after hepatectomy define the temporal course of liver regeneration. J Clin Invest 1993;91:1319‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kelley‐Loughnane N, Sabla GE, Ley‐Ebert C, Aronow BJ, Bezerra JA. Independent and overlapping transcriptional activation during liver development and regeneration in mice. Hepatology 2002;35:525‐534. [DOI] [PubMed] [Google Scholar]
- 103. Arai M, Yokosuka O, Chiba T, Imazeki F, Kato M, Hashida J, Ueda Y, et al. Gene expression profiling reveals the mechanism and pathophysiology of mouse liver regeneration. J Biol Chem 2003;278:29813‐29818. [DOI] [PubMed] [Google Scholar]
- 104. Pediaditakis P, Lopez‐Talavera JC, Petersen B, Monga SP, Michalopoulos GK. The processing and utilization of hepatocyte growth factor/scatter factor following partial hepatectomy in the rat. Hepatology 2001;34:688‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Stolz DB, Mars WM, Petersen BE, Kim TH, Michalopoulos GK. Growth factor signal transduction immediately after two‐thirds partial hepatectomy in the rat. Cancer Res 1999;59:3954‐3960. [PubMed] [Google Scholar]
- 106. Fausto N. Liver regeneration. J Hepatol 2000;32(1 Suppl):19‐31. [DOI] [PubMed] [Google Scholar]
- 107. Serandour AL, Loyer P, Garnier D, Courselaud B, Theret N, Glaise D, Guguen‐Guillouzo C, et al. TNFalpha‐mediated extracellular matrix remodeling is required for multiple division cycles in rat hepatocytes. Hepatology 2005;41:478‐486. [DOI] [PubMed] [Google Scholar]
- 108. Oosthuizen MM, Ndaba N, Myburgh JA. Rat hepatoproliferin revealed the status of a complete hepatomitogen in human hepatoma cells. Transplant Proc 2005;37:89‐92. [DOI] [PubMed] [Google Scholar]
- 109. Morgan DO. Cyclin‐dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 1997;13:261‐291. [DOI] [PubMed] [Google Scholar]
- 110. Jaumot M, Estanyol JM, Serratosa J, Agell N, Bachs O. Activation of cdk4 and cdk2 during rat liver regeneration is associated with intranuclear rearrangements of cyclin‐cdk complexes. Hepatology 1999;29:385‐395. [DOI] [PubMed] [Google Scholar]
- 111. Garnier D, Loyer P, Ribault C, Guguen‐Guillouzo C, Corlu A. Cyclin‐dependent kinase 1 plays a critical role in DNA replication control during rat liver regeneration. Hepatology 2009;50:1946‐1956. [DOI] [PubMed] [Google Scholar]
- 112. Russell WE, Coffey RJ, Jr ., Ouellette AJ, Moses HL. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc Natl Acad Sci U S A 1988;85:5126‐5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Braun L, Mead JE, Panzica M, Mikumo R, Bell GI, Fausto N. Transforming growth factor beta mRNA increases during liver regeneration: a possible paracrine mechanism of growth regulation. Proc Natl Acad Sci U S A 1988;85:1539‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Macias‐Silva M, Li W, Leu JI, Crissey MA, Taub R. Up‐regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor‐beta signals during liver regeneration. J Biol Chem 2002;277:28483‐28490. [DOI] [PubMed] [Google Scholar]
- 115. Romero‐Gallo J, Sozmen EG, Chytil A, Russell WE, Whitehead R, Parks WT, Holdren MS, et al. Inactivation of TGF‐beta signaling in hepatocytes results in an increased proliferative response after partial hepatectomy. Oncogene 2005;24:3028‐3041. [DOI] [PubMed] [Google Scholar]
- 116. Otu HH, Naxerova K, Ho K, Can H, Nesbitt N, Libermann TA, Karp SJ. Restoration of liver mass after injury requires proliferative and not embryonic transcriptional patterns. J Biol Chem 2007;282:11197‐11204. [DOI] [PubMed] [Google Scholar]
- 117. Bissell DM, Wang SS, Jarnagin WR, Roll FJ. Cell‐specific expression of transforming growth factor‐beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest 1995;96:447‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wurthner JU, Frank DB, Felici A, Green HM, Cao Z, Schneider MD, McNally JG, et al. Transforming growth factor‐beta receptor‐associated protein 1 is a Smad4 chaperone. J Biol Chem 2001;276:19495‐19502. [DOI] [PubMed] [Google Scholar]
- 119. Eickelberg O, Centrella M, Reiss M, Kashgarian M, Wells RG. Betaglycan inhibits TGF‐beta signaling by preventing type I‐type II receptor complex formation. Glycosaminoglycan modifications alter betaglycan function. J Biol Chem 2002;277:823‐829. [DOI] [PubMed] [Google Scholar]
- 120. Do N, Zhao R, Ray K, Ho K, Dib M, Ren X, Kuzontkoski P, et al. BMP4 is a novel paracrine inhibitor of liver regeneration. Am J Physiol Gastrointest Liver Physiol 2012;303:G1220‐G1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Xu CP, Ji WM, van den Brink GR, Peppelenbosch MP. Bone morphogenetic protein‐2 is a negative regulator of hepatocyte proliferation downregulated in the regenerating liver. World J Gastroenterol 2006;12:7621‐7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Breitkopf‐Heinlein K, Meyer C, Konig C, Gaitantzi H, Addante A, Thomas M, Wiercinska E, et al. BMP‐9 interferes with liver regeneration and promotes liver fibrosis. Gut 2017;66:939‐954. [DOI] [PubMed] [Google Scholar]
- 123. Takamura K, Tsuchida K, Miyake H, Tashiro S, Sugino H. Activin and activin receptor expression changes in liver regeneration in rat. J Surg Res 2005;126:3‐11. [DOI] [PubMed] [Google Scholar]
- 124. Kremer M, Son G, Zhang K, Moore SM, Norris A, Manzini G, Wheeler MD, et al. Smad3 signaling in the regenerating liver: implications for the regulation of IL‐6 expression. Transpl Int 2014;27:748‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Moh A, Iwamoto Y, Chai GX, Zhang SS, Kano A, Yang DD, Zhang W, et al. Role of STAT3 in liver regeneration: survival, DNA synthesis, inflammatory reaction and liver mass recovery. Lab Invest 2007;87:1018‐1028. [DOI] [PubMed] [Google Scholar]
- 126. Li W, Liang X, Kellendonk C, Poli V, Taub R. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem 2002;277:28411‐28417. [DOI] [PubMed] [Google Scholar]
- 127. Ho KJ, Do NL, Otu HH, Dib MJ, Ren X, Enjyoji K, Robson SC, et al. Tob1 is a constitutively expressed repressor of liver regeneration. J Exp Med 2010;207:1197‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ding ZY, Liang HF, Jin GN, Chen WX, Wang W, Datta PK, Zhang MZ, et al. Smad6 suppresses the growth and self‐renewal of hepatic progenitor cells. J Cell Physiol 2014;229:651‐660. [DOI] [PubMed] [Google Scholar]
- 129. Wang Z, Song Y, Tu W, He X, Lin J, Liu F. beta‐2 spectrin is involved in hepatocyte proliferation through the interaction of TGFbeta/Smad and PI3K/AKT signalling. Liver Int 2012;32:1103‐1111. [DOI] [PubMed] [Google Scholar]
- 130. Saha T, Vardhini D, Tang Y, Katuri V, Jogunoori W, Volpe EA, Haines D, et al. RING finger‐dependent ubiquitination by PRAJA is dependent on TGF‐beta and potentially defines the functional status of the tumor suppressor ELF. Oncogene 2006;25:693‐705. [DOI] [PubMed] [Google Scholar]
- 131. Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology 2004;39:1477‐1487. [DOI] [PubMed] [Google Scholar]
- 132. Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene 2006;25:3818‐3822. [DOI] [PubMed] [Google Scholar]
- 133. Roskams T, Yang SQ, Koteish A, Durnez A, DeVos R, Huang X, Achten R, et al. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am J Pathol 2003;163:1301‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Bhanumathy CD, Tang Y, Monga SP, Katuri V, Cox JA, Mishra B, Mishra L. Itih‐4, a serine protease inhibitor regulated in interleukin‐6‐dependent liver formation: role in liver development and regeneration. Dev Dyn 2002;223:59‐69. [DOI] [PubMed] [Google Scholar]
- 135. Mishra L, Tully RE, Monga SP, Yu P, Cai T, Makalowski W, Mezey E, et al. Praja1, a novel gene encoding a RING‐H2 motif in mouse development. Oncogene 1997;15:2361‐2368. [DOI] [PubMed] [Google Scholar]
- 136. Mishra L, Cai T, Yu P, Monga SP, Mishra B. Elf3 encodes a novel 200‐kD beta‐spectrin: role in liver development. Oncogene 1999;18:353‐364. [DOI] [PubMed] [Google Scholar]
- 137. Morita H, Mazerbourg S, Bouley DM, Luo CW, Kawamura K, Kuwabara Y, Baribault H, et al. Neonatal lethality of LGR5 null mice is associated with ankyloglossia and gastrointestinal distension. Mol Cell Biol 2004;24:9736‐9743. [DOI] [PMC free article] [PubMed] [Google Scholar]