

ABSTRACT
Using conditional knock-in mouse models, we and others have shown that despite the very high sequence identity between Nras and Kras proteins, oncogenic Kras displays a much stronger leukemogenic activity than oncogenic Nras in vivo. In this manuscript, we will summarize our recent work of characterizing wild-type Kras function in adult hematopoiesis and in oncogenic Kras-induced leukemogenesis. We attribute the strong leukemogenic activity of oncogenic Kras to 2 unique aspects of Kras signaling. First, Kras is required in mediating cell type- and cytokine-specific ERK1/2 signaling. Second, oncogenic Kras, but not oncogenic Nras, induces hyperactivation of wild-type Ras, which significantly enhances Ras signaling in vivo. We will also discuss a possible mechanism that mediates oncogenic Kras-evoked hyperactivation of wild-type Ras and a potential approach to down-regulate oncogenic Kras signaling.
KEYWORDS: leukemogenesis, oncogenic Kras, oncogenic Nras, SOS1, wild-type Kras
In mammals, there are 3 Ras genes (Hras, Nras, and Kras) encoding 4 homologous 21kD proteins: Hras, Nras, Kras4A and Kras4B.1 The first 85 amino-terminal residues are identical and the middle 78 amino acids share an 85% – 90% identity among all Ras isoforms, while the last 25 amino acids at the carboxyl-terminus are highly variable.2 In haematopoietic neoplasms, oncogenic mutations in the NRAS and KRAS genes are common, but rare in the HRAS gene.2 Despite the high similarity in protein sequences and largely overlapping expression patterns, accumulating evidence suggest that KrasG12D/+ expressed from its endogenous locus demonstrates much stronger leukemogenic activity than oncogenic Nras alleles (including NrasG12D alleles) (Fig. 1). Using conditional knock-in mouse models, we and others characterized multiple oncogenic Ras alleles in a pure C57BL/6 background, including the weak NrasG12D/+, the intermediate NrasQ61R/+ and NrasG12D/G12D, and the strong KrasG12D/+.3-8 All of these models involve a conditional knock-in oncogenic Ras allele and the interferon-inducible Mx1-Cre. Upon polyinosinic-polycytidylic acid (pI-pC) injections to induce the expression of Cre and subsequently the oncogenes, KrasG12D/+ mice die rapidly, while NrasG12D/G12D and NrasG12D/+ mice show incrementally prolonged survival (Fig. 1A). To eliminate the impact of acute interferon signaling on animal survival, we also took advantage of the leaky expression of Mx1-Cre over the time. Again, the non-pI-pC treated KrasG12D/+ mice display a significantly shorter survival than the corresponding NrasG12D/G12D mice (Fig. 1B). These results clearly demonstrate that oncogenic Kras is a much more potent oncogene than oncogenic Nras in leukemogenesis.
Figure 1.
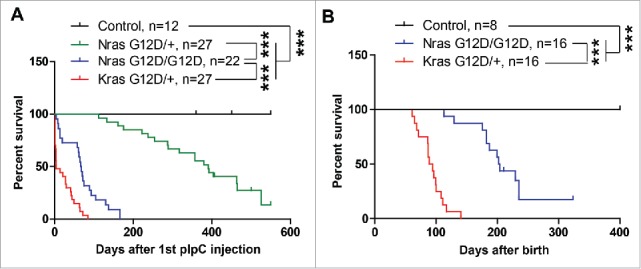
Oncogenic Kras displays more potent leukemogenic activity than oncogenic Nras. (A) Six-seven weeks old control, KrasG12D/+, NrasG12D/+, and NrasG12D/G12D mice were injected with pI-pC twice every other day. Kaplan-Meier survival curves were plotted against days after 1st pI-pC injection. (B) Kaplan-Meier survival curves of non-pI-pC treated control, KrasG12D/+, and NrasG12D/G12D mice. P values were determined using the Log-rank test. *** P < 0.001.
To decipher the unique function of oncogenic Kras signaling in leukemogenesis, we first investigated how loss of wild-type (WT) Kras impacts on adult hematopoiesis.9 We found that loss of Kras leads to greatly reduced TPO signaling in haematopoietic stem cells (HSCs), while SCF-evoked ERK1/2 activation is not affected. The compromised TPO signaling is associated with reduced long term- and intermediate-term HSC compartments and their reduced self-renewal capability (Fig. 2). Although GM-CSF-evoked ERK1/2 activation is only moderately decreased in Kras−/− myeloid progenitors, it is blunted in neutrophils and neutrophil survival is significantly reduced in vitro. Similarly, the ERK1/2 pathway is down-regulated in Kras−/− B cells, which is associated with B cell differentiation defects at both pre-B cell and mature B cell stages.10 These results reveal a surprising, indispensible role of Kras in mediating cell type- and cykokine-specific ERK1/2 activation, especially in differentiated cells (e.g. neutrophils). This conclusion is further supported by the finding that in Kras−/−; NrasQ61R/+ neutrophils, GM-CSF-evoked ERK1/2 activation remains blunted (our unpublished result).
Figure 2.
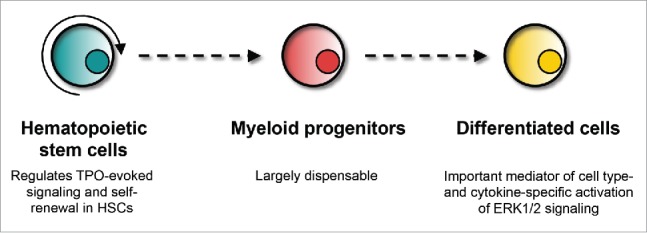
Summary of wild-type Kras function in adult hematopoiesis.
Interestingly, at 9–12 months old, a fraction of Kras conditional knockout mice develop profound haematopoietic defects (e.g., splenomegaly and an expanded neutrophil compartment), suggesting that they are highly prone to myeloid diseases. Indeed, we recently found that loss of WT Kras promotes oncogenic Kras-induced myeloproliferative neoplasm in a cell autonomous manner.11 The most striking result from this study is that oncogenic Kras but not oncogenic Nras leads to cross-activation of WT Ras in total bone marrow cells, while loss of WT Kras further promotes the activation of all Ras isoforms. Previous crystallographic analyses of Ras bound to the catalytic module of Sos indicate that Ras-GTP binds to the catalytic domain of Sos at a site distal to the active site of Sos and stabilizes this active site allosterically, significantly increasing the rate of Sos-stimulated GDP release from Ras.12 Subsequently, Bar-Sagi's group reported that cross-activation of WT Ras by oncogenic Ras is mediated by Sos1 and is essential for oncogenic Ras-induced tumorigenesis in cancer cell lines13 (Fig. 3). Our recent in vivo work is also in agreement with this conclusion (our unpublished results). Of note, the earlier studies imply that cross-activation of WT Ras is applicable to all Ras isoforms, while our in vivo study demonstrates that this phenomenon is specific to oncogenic Kras in total bone marrow cells. We speculate that this specificity might be related to the differential membrane trafficking and/or localization of different Ras isoforms, which is mediated by their highly variable C-termini.14-19
Figure 3.
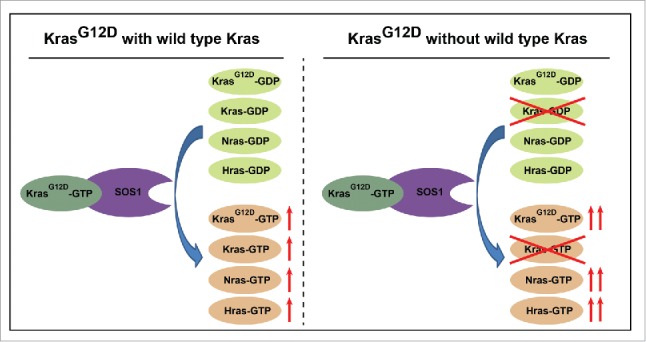
Schematic illustration of a potential mechanism how loss of WT Kras promotes activation of all Ras isoforms.
To our surprise, loss of WT Kras not only further promoted activation of WT Hras and Nras but also significantly enhanced oncogenic Kras-GTP levels. It is known that RasG12D mutation affects the intrinsic GTPase activity of Ras proteins by preventing proper position of the Ras-GAP (GTPase activating protein) arginine finger within its catalytic site.20-22 Conceivably, although majority of KrasG12D protein binds to GTP, a fraction of KrasG12D protein binds to GDP and is inactive. One possibility is that all GDP-bound forms of Ras, including WT Ras and a small proportion of KrasG12D, compete for the binding to the activate site of Sos catalytic domain to release GDP. Without WT Kras, inactive WT Hras and Nras as well as inactive KrasG12D have a higher chance to be activated by Sos (Fig. 3). Alternatively, it could be that expression level of oncogenic Kras from a single copy of the oncogene is upregulated to a level comparable to that from 2 copies of the oncogene, as suggested by our quantitative Western blot analysis.11 The mechanism underlying this upregulation is unknown, but perhaps mediated through an epigenetic regulation. We would like to point out that these 2 possibilities are not mutually exclusive. In fact, it is highly likely that both of them contribute to the higher Kras-GTP level we observed.
Our work demonstrates that in bone marrow cells, expression of oncogenic Kras is equivalent to the activation of all Ras isoforms, including WT Nras and Hras, which could significantly contribute to the potent leukemogenic activity of oncogenic Kras. The question is “Can we target the interaction between oncogenic Kras and Sos1 to ameliorate oncogenic Kras-induced leukemias?” The answer to this question might be complex. In one hand, inhibition of this interaction could block activation of WT Ras and alleviate at least some of the leukemia phenotypes. On the other hand, due to the unique function of oncogenic Kras signaling in certain cell types as described above, inhibition of the interaction between oncogenic Kras and Sos1 would have none or minimal effects in these cells. Therefore, our data suggest that targeting this interaction should be combined with other therapies, such as inhibitors targeting Ras down stream MEK1/2, to achieve more potent anti-leukemia effects.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Barbacid M. ras genes. Annu Rev Biochem 1987; 56:779-827; PMID:3304147; https://doi.org/ 10.1146/annurev.bi.56.070187.004023 [DOI] [PubMed] [Google Scholar]
- [2].Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012; 120:3397-406; PMID:22898602; https://doi.org/ 10.1182/blood-2012-05-378596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang J, Wang J, Liu Y, Sidik H, Young KH, Lodish HF, Fleming MD. Oncogenic Kras-induced leukemogeneis: hematopoietic stem cells as the initial target and lineage-specific progenitors as the potential targets for final leukemic transformation. Blood 2009; 113:1304-14; PMID:19066392; https://doi.org/ 10.1182/blood-2008-01-134262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang JY, Liu YG, Li ZY, Du J, Ryu MJ, Taylor PR, Fleming MD, Young KH, Pitot H, Zhang J. Endogenous oncogenic Nras mutation leads to aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood 2010; 116:5991-6002; PMID:20921338; https://doi.org/ 10.1182/blood-2010-04-281527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li Q, Haigis KM, McDaniel A, Harding-Theobald E, Kogan SC, Akagi K, Wong JC, Braun BS, Wolff L, Jacks T, et al.. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood 2011; 117:2022-32; PMID:21163920; https://doi.org/ 10.1182/blood-2010-04-280750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang JY, Liu YG, Li ZY, Wang ZD, Tan LX, Ryu MJ, Meline B, Du J, Young KH, Ranheim E, et al.. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood 2011; 118:368-79; PMID:21586752; https://doi.org/ 10.1182/blood-2010-12-326058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xu J, Haigis KM, Firestone AJ, McNerney ME, Li Q, Davis E, Chen SC, Nakitandwe J, Downing J, Jacks T, et al.. Dominant role of oncogene dosage and absence of tumor suppressor activity in Nras-driven hematopoietic transformation. Cancer Discov 2013; 3:993-1001; PMID:23733505; https://doi.org/ 10.1158/2159-8290.CD-13-0096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kong G, Chang YI, You X, Ranheim EA, Zhou Y, Burd CE, Zhang J. The ability of endogenous Nras oncogenes to initiate leukemia is codon-dependent. Leukemia 2016; https://doi.org/ 10.1038/leu.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Damnernsawad A, Kong G, Wen Z, Liu Y, Rajagopalan A, You X, Wang J, Zhou Y, Ranheim EA, Luo HR, et al.. Kras is required for adult hematopoiesis. Stem Cells 2016; 34(7):1859-71. Epub on March 28; PMID:26972179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen Y, Zheng Y, You X, Yu M, Fu G, Su X, Zhou F, Zhu W, Wu Z, Zhang J, et al.. Kras is critical for B cell lymphopoiesis. J Immunol 2016; 196:1678-85; https://doi.org/ 10.4049/jimmunol.1502112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kong G, Chang YI, Damnernsawad A, You X, Du J, Ranheim RA, Lee W, Ryu MJ, Zhou Y, Xing Y, et al.. Loss of wild-type Kras promotes activation of all Ras isoforms in oncogenic Kras-induced leukemogenesis. Leukemia 2016; 30(7):1542-51. Epub on April 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras GTP of the Ras-specific nucleotide exchange factor SOS. Cell 2003; 112:685-95 [DOI] [PubMed] [Google Scholar]
- [13].Jeng HH, Taylor LJ, Bar-Sagi D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat Commun 2012; 3:1168; PMID:23132018; https://doi.org/ 10.1038/ncomms2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Roy S, Luetterforst R, Harding A, Apolloni A, Etheridge M, Stang E, Rolls B, Hancock JF, Parton RG. Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nat Cell Biol 1999; 1:98-105; PMID:10559881; https://doi.org/ 10.1038/15687 [DOI] [PubMed] [Google Scholar]
- [15].Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 1999; 98:69-80; PMID:10412982; https://doi.org/ 10.1016/S0092-8674(00)80607-8 [DOI] [PubMed] [Google Scholar]
- [16].Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol 2000; 20:2475-87; PMID:10713171; https://doi.org/ 10.1128/MCB.20.7.2475-2487.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prior IA, Harding A, Yan J, Sluimer J, Parton RG, Hancock JF. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol 2001; 3:368-75; PMID:11283610; https://doi.org/ 10.1038/35070050 [DOI] [PubMed] [Google Scholar]
- [18].Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL 2nd, Cox AD, Philips MR. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol 2002; 4:343-50; PMID:11988737 [DOI] [PubMed] [Google Scholar]
- [19].Wang Y, Velho S, Vakiani E, Peng S, Bass AJ, Chu GC, Gierut J, Bugni JM, Der CJ, Philips M, et al.. Mutant N-RAS protects colorectal cancer cells from stress-induced apoptosis and contributes to cancer development and progression. Cancer Discov 2013; 3:294-307; PMID:23274911; https://doi.org/ 10.1158/2159-8290.CD-12-0198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krengel U, Schlichting I, Scherer A, Schumann R, Frech M, John J, Kabsch W, Pai EF, Wittinghofer A. Three-dimensional structures of H-ras p21 mutants: molecular basis for their inability to function as signal switch molecules. Cell 1990; 62:539-48; PMID:2199064; https://doi.org/ 10.1016/0092-8674(90)90018-A [DOI] [PubMed] [Google Scholar]
- [21].Tong LA, de Vos AM, Milburn MV, Kim SH. Crystal structures at 2.2 A resolution of the catalytic domains of normal ras protein and an oncogenic mutant complexed with GDP. J Mol Biol 1991; 217:503-16; PMID:1899707; https://doi.org/ 10.1016/0022-2836(91)90753-S [DOI] [PubMed] [Google Scholar]
- [22].Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997; 277:333-8; PMID:9219684; https://doi.org/ 10.1126/science.277.5324.333 [DOI] [PubMed] [Google Scholar]