

ABSTRACT
MP0250 is a multi-domain drug candidate currently being tested in clinical trials for the treatment of cancer. It comprises one anti-vascular endothelial growth factor-A (VEGF-A), one anti-hepatocyte growth factor (HGF), and two anti-human serum albumin (HSA) DARPin® domains within a single polypeptide chain. While there is first clinical validation of a single-domain DARPin® drug candidate, little is known about DARPin® drug candidates comprising multiple domains. Here, we show that MP0250 can be expressed at 15 g/L in soluble form in E. coli high cell-density fermentation, it is stable in soluble/frozen formulation for 2 years as assessed by reverse phase HPLC, it has picomolar potency in inhibiting VEGF-A and HGF in ELISA and cellular assays, and its domains are simultaneously active as shown by surface plasmon resonance. The inclusion of HSA-binding DARPin® domains leads to a favorable pharmacokinetic profile in mouse and cynomolgus monkey, with terminal half-lives of ∼ 30 hours in mouse and ∼ 5 days in cynomolgus monkey. MP0250 is thus a highly potent drug candidate that could be particularly useful in oncology. Beyond MP0250, the properties of MP0250 indicate that multi-domain DARPin® proteins can be valuable next-generation drug candidates.
KEYWORDS: DARPin®, HGF, multi-specificity, pharmacokinetics, serum albumin, VEGF
Introduction
The quest for more efficacious and safe cancer drug candidates has resulted in the establishment of non-immunoglobulin binding proteins as sources for novel drugs.1 Several drug candidates representing this class of molecules are either approved or at various stages of clinical development.2 The most advanced candidates are single-domain drugs clinically validating the diverse technology platforms. DARPin® domains are a promising class of non-immunoglobulin binding proteins.3 The most advanced DARPin® drug candidate is abicipar pegol, which is being evaluated in Phase 3 clinical trials as a treatment for macular degeneration (ClinicalTrials.gov Identifiers NCT02462486, NCT02462928). DARPin® domains are biophysically stable molecules that can be expressed at high levels in bacteria, and we have generated numerous high-affinity binding proteins with high target specificity. These findings and the fact that natural ankyrin repeat proteins are often found as multi-domain constructs suggest that the DARPin® platform is ideally suited for the generation of multi-functional molecules. Such approaches have the potential to overcome some of the technical limitations of antibodies and antibody fragments, enabling novel therapeutic strategies.
MP0250, a multi-domain DARPin® drug candidate3 with binding specificities for vascular endothelial growth factor A (VEGF-A),4 hepatocyte growth factor (HGF),5 and human serum albumin (HSA) is the first multi-functional DARPin® drug candidate in clinical studies. MP0250 is currently being evaluated in a Phase 1 study (ClinicalTrials.gov Identifier NCT02194426) in cancer patients. While the use of VEGF-A antagonists, especially the monoclonal antibody (mAb) bevacizumab, is successful in the clinic, the occurrence of resistance is a major drawback, which preclinical data suggests may be overcome by concomitant inhibition of additional pathways.6–8 With its target specificities, MP0250 may thus help to overcome this problem. Concomitant HGF inhibition appears to be an attractive option, since HGF/cMet signaling has been shown to trigger potent angiogenic signals promoting resistance to anti-angiogenic therapy in some cancer types.9 Cabozantinib, an orally dosed multi-specific tyrosine kinase inhibitor drug that interferes with both the VEGF-A and the HGF/cMet signaling pathways validates this approach.10–12 The drawback of broad specificity drugs (e.g., cabozantinib) and drugs with off-target toxicity (e.g., tivantinib)13,14 is a high incidence of adverse events.15,16 Interestingly, the anti-HGF mAb rilotumumab, which exhibits partial HGF inhibition,17 did not demonstrate efficacy as a single agent in Phase 2 trials in prostate cancer18,19 and in Phase 3 trials in gastric cancer combined with chemotherapy.20 Similarly, onartuzumab,21,22 an anti-cMet antibody, failed to show benefit when tested in combination with the EGFR inhibitor erlotinib in a Phase 3 non-small-cell lung cancer trial,23 which might be due to inappropriate patient selection.23, 24 Creating a treatment option that combines specific HGF/cMet pathway inhibition with concomitant VEGF/VEGF-R2 pathway inhibition thus remains desirable. MP0250 was designed to this end. Here, we describe the generation and characterization of MP0250.
Results
MP0250 consists of an N-terminal DARPin® domain binding HSA, followed by a DARPin® domain binding HGF, followed by a DARPin® domain binding VEGF-A, followed by a C-terminal DARPin® domain binding HSA, all linked by Pro/Thr-rich linkers (Figure 1). Here, we describe MP0250 in detail regarding recombinant expression, solubility, storage stability, potency, pre-clinical pharmacokinetic as well as pharmacological properties. Additional data, in particular data on efficacy in tumor animal models, are given elsewhere.25
Figure 1.
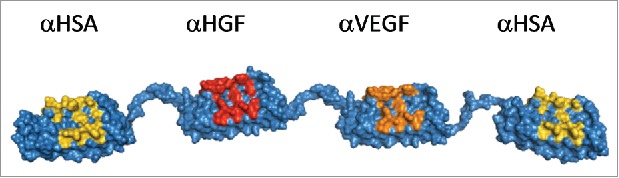
Molecular model of MP0250 consisting of four designed ankyrin repeat domains in a single polypeptide chain. The target specificity of every domain is indicated in the figure. For every designed ankyrin repeat domain, positions that were randomized in the DARPin® library and thus represent potential target interaction residues, are colored yellow (serum albumin), red (HGF), or orange (VEGF).
High-level recombinant expression
MP0250 can be expressed at high levels in soluble form in the cytoplasm of E. coli. Using standard shake-flask expression, 680 mg MP0250 can be expressed in E. coli BL21 per liter TB medium culture as judged by SDS-PAGE (Table 1). This yield is in the range of that seen for the individual DARPin® domains, whose expression varied between 320 mg/L and 700 mg/L in the same expression system. These findings indicate that there is no reduction of the expression level when going from a single DARPin® domain to a four-domain construct. Furthermore, the expression levels of the individual DARPin® proteins are significantly higher than the previously reported 200 mg/L for single DARPin® domains,26 which were obtained with E. coli XL-1 blue and LB medium, indicating significant improvement in yield can be achieved by choice of expression strain and medium. Indeed, when using E. coli BL21 and TB medium, the molecule E3_526 can be expressed at 370 mg/L. In fermenter fed-batch productions, titers in the range of 15 g MP0250 per liter culture were achieved as judged by SDS-PAGE, corresponding to ∼ 23% of the cell dry weight. These expression levels enable the efficient microbial production of MP0250 for clinical applications. MP0250 consistently can be purified to >99% homogeneity using standard chromatography methods such as anion-exchange chromatography, as assessed by size-exclusion chromatography (SEC) and SDS-PAGE.
Table 1.
Shake-flask expression levels of MP0250 and DARPin® domains (E. coli BL21, TB medium).
| Construct | MP0250 | αHSA | αHGF | αVEGF | E3_5* |
|---|---|---|---|---|---|
| Expression level [g/L] | 0.68 | 0.53 | 0.32 | 0.70 | 0.37 |
GenBank Accession AAO25689.126
Storage stability and solubility
MP0250, formulated in phosphate-buffered saline (PBS), was assessed at various concentrations with no visible alteration of the sample. In a preliminary long-term stability study at 15 mg/mL in PBS at 25°C, 5°C, −20°C, or −70°C, assessed by reverse-phase HPLC (Figure 2), MP0250 exhibited less than 1% loss of monomer peak over at least 18 months at 5°C (0.2% loss at 18 months), −20°C (0.0% loss at 18 months), or −70°C (0.0% loss at 18 months). Under accelerated conditions at 25°C, MP0250 exhibited 2.4% loss of monomer peak over a 6-month period. These results demonstrate the commercial feasibility of a liquid MP0250 formulation. Further analyses showed that MP0250 at 15 mg/mL in PBS sustains at least 5 freeze-thaw-cycles without visible changes in SEC, reverse-phase chromatography, and visual appearance (data not shown). The viscosity of MP0250 in PBS was determined to be 1.26 mPaS, 1.39 mPaS, 1.70 mPaS, and 4.16 mPaS at concentrations of 15 mg/mL, 30 mg/mL, 45 mg/mL, and 95 mg/mL, respectively (data not shown), indicating favorable viscosity properties suitable for liquid use of the drug candidate.
Figure 2.
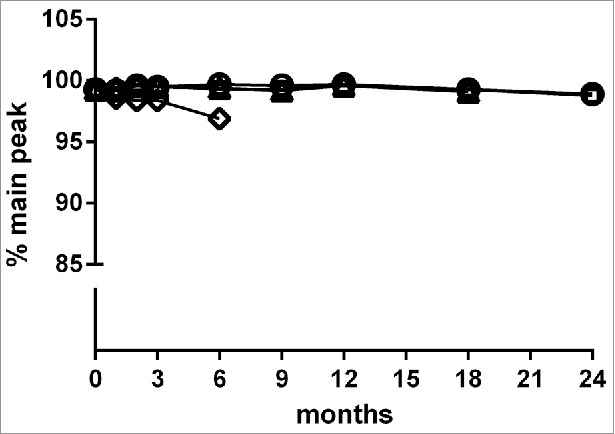
Storage stability of MP0250 assessed by reverse-phase HPLC analysis. MP0250 at ∼ 15 mg/mL in PBS was stored for up to 24 months at −70°C (open circles), −20°C (open squares), 5°C (open triangles), or 25°C (open diamonds) in glass vials. Reverse-phase HPLC (see Materials & Methods) was used to determine the % of main peak at 1, 2, 3, 6, 9, 12, 18, or 24 months of incubation. In frozen state at −70°C and −20°C, the % main peak value in reverse-phase HPLC is stable with only minor change. Similarly, the % main peak value for MP0250 stored at +5°C is stable with only minor change over the time measured. As expected, a decrease in % of main peak was observed when MP0250 was incubated at +25°C for several months.
MP0250 is highly potent in inhibiting VEGF-A and HGF
By embedding an individual domain in a multi-domain molecule, its activity may be altered. It is thus important that MP0250 exhibits sufficient affinity and potency for each of its functionalities. MP0250 is able to bind VEGF-A with an apparent EC50 of 24 pM (Figure 3 and Table 2). It binds VEGF-A of human, mouse, and cynomolgus monkey, whereas it does not bind to the related VEGF-C or PDGF-AB (Figure 3a). In sandwich ELISA, MP0250 showed an IC50 of 4.5 pM in inhibiting human VEGF-A, which is comparable to the individual VEGF-A-binding DARPin® domain (IC50 of 13 pM) and the value reported for other high-affinity single domain DARPin® proteins27 (data not shown). In cell assays, MP0250 was able to inhibit VEGF-A-induced human umbilical vein endothelial cell (HUVEC) proliferation with an IC50 of 231 pM, comparable to the individual VEGF-A-binding DARPin® domain (IC50 of 208 pM; Figure 4), which was statistically not significant (p = 0.79, Student's t-test). These IC50 values are most likely underestimates because they were limited by the amount of VEGF-A (190 pM) needed in this cell assay. In a VEGF/VEGF-receptor competition fluorescence resonance energy transfer assay, MP0250 was shown to inhibit the interaction between VEGF-A and VEGF-R2 with an apparent IC50 of 0.7 nM (data not shown). MP0250 binds human HGF with an apparent EC50 of 24 pM (Figure 3 and Table 2). HGF of mouse and cynomolgus monkey are likewise bound with high affinity by MP0250 (range of 40 to 45 pM; Table 2). HGF inhibition was additionally shown in an A549 cMet phosphorylation cell assay, where MP0250 exhibits an IC50 of 310 pM (1 nM HGF used for stimulation; Figure 4), again comparable to the HGF-inhibiting individual DARPin® domain (200 pM, difference statistically non-significant; Figure 4). In summary, these data indicate that individual DARPin® domains can be included in a multi-domain construct without compromising functionality. The apparent EC50 of the HSA binding of MP0250 is significantly improved (13 pM; Figure 3c and Table 2) compared to that observed for the individual HSA-binding DARPin® domain (437 pM; Table 2), which is expected due to the presence of two HSA-binding domains in MP0250 (avidity effect). Species serum-binding ELISA indicate that MP0250 is able to bind serum albumin of human, mouse, and monkey (Figure 3c), as well as rat and dog (data not shown). Overall, these measurements indicate that MP0250 has the potency required for therapeutic use, and that it can validly be used in preclinical mouse and cynomolgus monkey models.
Figure 3.
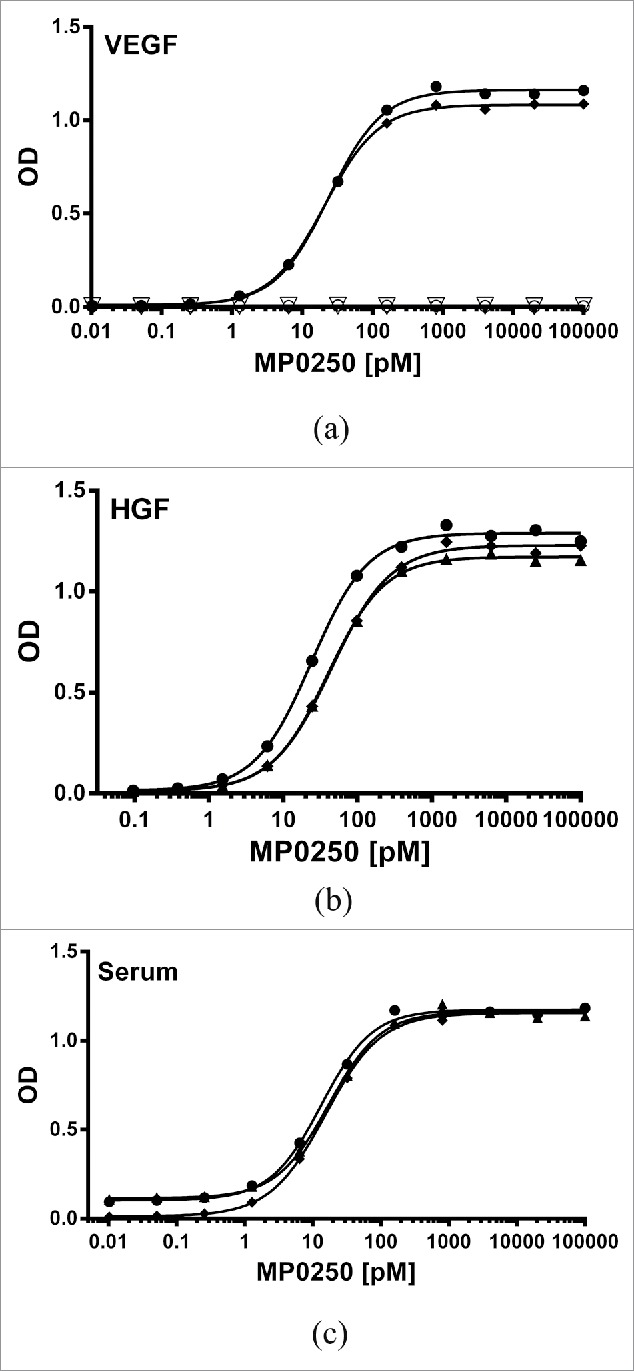
Potent binding of MP0250 in ELISA. (a) VEGF-A binding ELISA. The binding signal of various concentrations of MP0250 to immobilized VEGF-A of human (filled circles; 100% sequence identity to cynomolgus monkey VEGF-A), and mouse (filled diamonds), as well as human VEGF-C (open inverse triangles), and human PDGF-AB (open circles), and the corresponding fitting inhibition curves are shown. (b) HGF binding ELISA. The binding signal of various concentrations of MP0250 to immobilized HGF of human (filled circles), cynomolgus monkey (filled triangles), and mouse (filled diamonds), and the corresponding fitting inhibition curves are shown. (c) Serum binding ELISA. The binding signal of various concentrations of MP0250 to immobilized serum of human (filled circles), cynomolgus monkey (filled triangles), and mouse (filled rhombus) and the corresponding fitting inhibition curves are shown. For all graphs, the ELISA signal (OD 450 nm – OD 620 nm) is plotted as a function of the MP0250 concentration in pM. Experimental details are described in the Materials & Methods section. Fitted EC50 values are shown in Table 2.
Table 2.
Apparent EC50 values (and 95% confidence interval) of MP0250 for binding VEGF-A, HGF and serum albumin of different species.
| Species | EC50 [pM] VEGF-A | EC50 [pM] HGF | EC50 [pM] SA |
|---|---|---|---|
| Human | 24 (20–27) | 24 (20–29) | 13 (10–16)* |
| Mouse | 21 (19–23) | 45 (38–52) | 15 (13–17) |
| Cynomolgus monkey | n.a. (24 (20–27))† | 40 (36–44) | 17 (13–21) |
n.a. not analyzed; 100% sequence identity to human VEGF-A, thus human VEGF-A value listed.
A single HSA-binding DARPin® domain exhibits an EC50 of 437 pM in the same assay for HSA.
Figure 4.
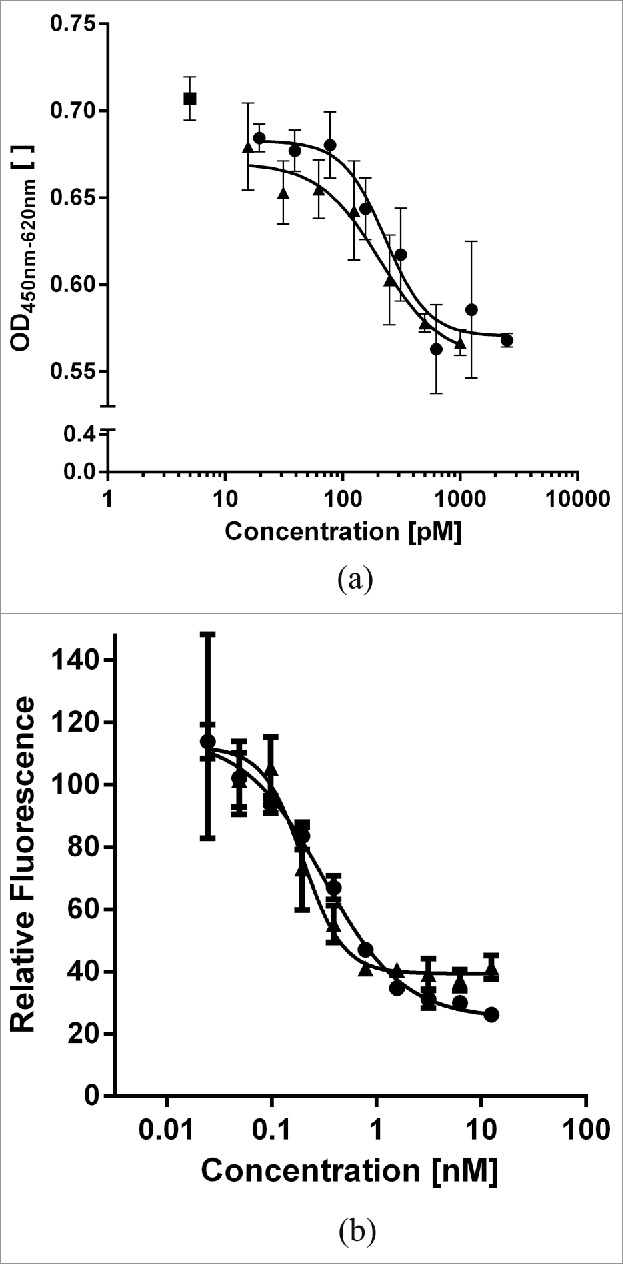
Comparison of His-tagged MP0250 (circles) in comparison to individual DARPin® domains (triangles) in cellular assays. (a) HUVEC proliferation inhibition assay. MP0250 is equivalently potent as the individual VEGF-inhibiting DARPin® domain of MP0250 in inhibiting HUVEC proliferation. Reference (square): sample with no inhibitor present. (b) c-Met phosphorylation inhibition assay. MP0250 is equivalently potent as the individual HGF-inhibiting DARPin® domain of MP0250 in inhibiting c-Met phosphorylation. The cellular assays were performed as described in the Materials & Methods section.
MP0250 simultaneously binds all targets
While it might not be required for clinical efficacy, it is of interest whether MP0250 can simultaneously bind to all of its targets. SPR analyses indicate that HGF, VEGF-A, and HSA can be bound simultaneously by MP0250 (Figure 5). Judging from the serum albumin signal, it is likely that MP0250 binds two HSA molecules and thus that all four DARPin® domains simultaneously bind their targets (Figure 5, trace #1). In SEC coupled with multi-angle static light scattering experiments, the ability of MP0250 to simultaneously bind two HSA molecules could indeed be confirmed (data not shown). These findings support the notion that individual domains can be included in a multi-domain molecule with little compromise of functionality.
Figure 5.
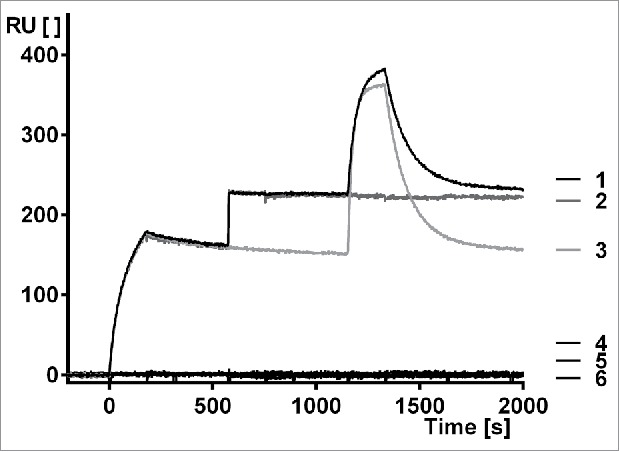
Surface plasmon resonance measurement to show simultaneous binding of MP0250 to HGF, VEGF-A, and human serum albumin. Setup: Human HGF is immobilized, MP0250 is injected (time 0 to 180 seconds) followed by a short buffer injection, human VEGF-A is then injected (time 600 to 780 seconds) followed by a short buffer injection, and finally HSA is injected (time 1200 to 1380 seconds), followed by a longer buffer injection. Trace 1 is the full measurement, traces 2–6 are control measurements (2: No HSA injected; 3: no human VEGF-A injected; 4: No MP0250, no human VEGF-A, and no HSA injected; 5: No MP0250 and no HSA injected; 6: No MP0250 and no human VEGF injected). RU: resonance units [ ]; t: time [s]. The measurement indicates that MP0250 binds to the immobilized HGF and can then bind VEGF-A to full saturation, indicating complete HGF/VEGF/MP0250 complex formation. This complex is then able to bind HSA, indicating that MP0250 can bind all targets simultaneously. The maximum signal of trace 1 being higher than trace 3 indicates that at least a fraction of the complexes consists of the full MP0250/HGF/VEGF-A/HSA complex. The controls indicate that no unspecific binding occurs in the measurement.
The HSA-binding technology leads to a favorable pharmacokinetic profile of MP0250
DARPin® domains without a serum albumin-binding moiety or polyethylene glycol are known to have very fast clearance.28 While serum albumin binding for pharmacokinetic engineering is a long-standing approach to improve the pharmacokinetic properties of small proteins,29 no such drug is on the market currently. The background on serum albumin-binding DARPin® domains is described in detail elsewhere.30 We applied the approach to MP0250, using two HSA-binding DARPin® domains flanking the other two domains. We analyzed the pharmacokinetic properties of MP0250 in single dose experiments in both mouse and cynomolgus monkey. Concentration-time profiles of MP0250 are shown in Figure 6 and the pharmacokinetic parameters calculated by non-compartmental analyses are given in Table 3. In cynomolgus monkey, the pharmacokinetic properties were analyzed in a broad dose range (1 to 100 mg/kg). While Cmax values increased in a dose-proportional manner, exposure (AUC) in the dose range from 1 to 10 mg/kg increased slightly more than dose-proportional, leading to a decrease in clearance between the low and higher dose levels. Since time points for serum sampling was limited to 168 h (one week), a substantial part of the overall exposure (AUC_inf) was extrapolated, which might have contributed to the observation of a slight non-dose linear behavior. The absence of obvious concentration outlier measurements in both mouse and monkey indicates the absence of anti-drug antibodies, which is in line with expectations for one-week studies. The pharmacokinetics both in mouse and cynomolgus monkey indicate the feasibility of preclinical testing in mouse and cynomolgus monkey. Furthermore, a potential terminal half-life of 11 days in human is extrapolated for MP0250 from mouse and cynomolgus monkey data by allometric scaling.31, 32 This value is in a similar range as the value predicted for bevacizumab by allometric scaling.33 These results indicate a pharmacokinetic profile of MP0250 suitable for therapeutic use as a multi-functional antagonist.
Figure 6.
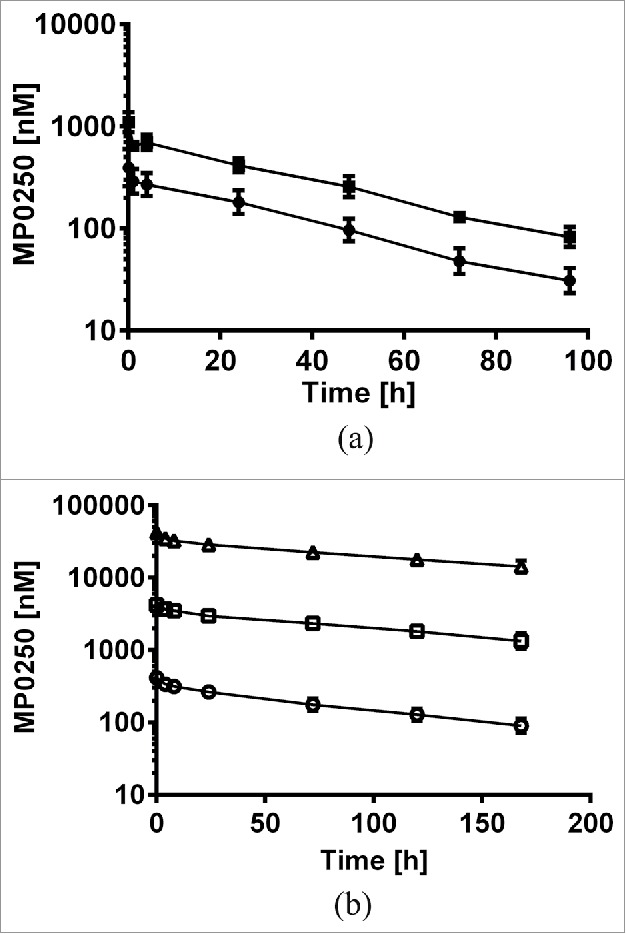
Concentration-time profiles of MP0250. (a) Mouse pharmacokinetic profile of MP0250 at 1 mg/kg (filled circles) and 2 mg/kg (filled squares). (b) Cynomolgus monkey pharmacokinetic profile of MP0250 at 1 mg/kg (open circles), 10 mg/kg (open squares), and 100 mg/kg (open triangles). Mean concentrations and standard deviations are plotted over time. Pharmacokinetic parameters derived from the experiments are given in Table 3.
Table 3.
Pharmacokinetic properties* of MP0250 i.v. administered in mouse and cynomolgus monkey.
| Parameter | Unit | Balb/c mice 1 mg/kg | Balb/c mice 2 mg/kg | Cynomolgus monkey | Cynomolgus monkey | Cynomolgus monkey |
|---|---|---|---|---|---|---|
| 1 mg/kg | 10 mg/kg | 100 mg/kg | ||||
| Number of animals | 10f | 10f | 5f/5m | 5f/5m | 5f/5m | |
| AUCINF_pred | hr*(nmol/L) | 12901 | 32727 | 43217 | 643593 | 6698389 |
| AUClast | hr*(nmol/L) | 11693 | 29309 | 30633 | 384761 | 3755348 |
| Cmax | nmol/L | 395 | 1097 | 416 | 4212 | 42230 |
| Tmax | hr | 0.083 | 0.083 | 0.583 | 1.375 | 0.583 |
| Cl_pred | mL/(hr*kg) | 1.2 | 0.98 | 0.388 | 0.266 | 0.250 |
| Vz | mL/kg | 51.1 | 41.9 | 52.1 | 45.9 | 48.3 |
| t1/2 | hr | 28.5 | 29.7 | 94.6 | 127.0 | 138.9 |
Averaged all animals per group.
Discussion
Having well-behaved individual domains is an important starting point for achieving a developable multi-domain drug candidate. Here, we show that the advantageous properties seen in single DARPin® domains are retained in the multi-domain DARPin® drug candidate MP0250. For example, it has a high recombinant expression yield, high solubility and high storage stability, enabling successful manufacturing of the drug. Its pharmacokinetic profile and pharmacologic activity make MP0250 a promising candidate for cancer therapy. Fiedler et al.25 have shown efficacy of MP0250 in a number of mouse tumor xenograft models, indicating that the drug candidate indeed has the potential to improve patient benefit beyond single agent anti-VEGF therapy. Based on the results shown here, the preclinical in vivo data, as well as safety and tolerability assessments, clinical testing of MP0250 has been initiated (ClinicalTrials.gov Identifier NCT02194426).
Materials and methods
Ribosome display and cloning
Individual DARPin® domains binding VEGF-A, HGF, and HSA, respectively, were selected by ribosome display as described in patent applications WO2010060748, WO2014191574, and WO2012069654, respectively. The DARPin® domain amino acid sequences used to build MP0250 and the amino acid sequence of MP0250 are detailed in patent application PCT/EP2016/057272. MP0250 and proteins comprising one or more DARPin® domains were generated using DNA synthesis or by standard cloning methods.
Protein expression and purification
Single domain proteins and multi-domain proteins were expressed and purified as described previously26, 27 or using standard chromatography methods. For the expression tests, E. coli BL21 transformed with a standard T5 expression plasmid34 harboring MP0250 or individual DARPin® domains was used with 50 mL TB growth medium in 300 mL shake-flasks at 220 rpm in a Novotron (Infors, Switzerland) shaker at 37°C. Cultures were induced by adding 0.5 mM isopropyl-β-D-thiogalactopyranosid (IPTG) at an OD600 of 1.0 and the cultures were incubated for five more hours. Cells were harvested by centrifugation. Equivalent amounts of cells were analyzed by SDS-PAGE. Proteins were quantified by graphical methods in comparison to defined amounts of individual DARPin® proteins or MP0250. For high cell-density fermentation, E. coli HMS174 transformed with the expression plasmid harboring MP0250 was used with minimal medium using a 5 L bioreactor (Infors Labfors 5), in a fed-batch mode controlling pH at 6.8, temperature and at 37°C, pO2 level above 20%. Product expression was induced by adding 1 mM IPTG per liter at a cell density of 50 g/L dry cell weight. Amounts of product were judged using SDS-PAGE and cell dry weight was determined using standard methods.
Standard protein buffer for the experiments shown here was PBS unless stated otherwise.
Storage stability measurement
For storage stability analyses, proteins were concentrated to 15 mg/mL followed by incubation in glass vials at −70°C, −20°C, 5°C, and 25°C for 0, 1, 2, 3, 6, 9, 12, 18, and 24 months, respectively. The 5°C incubation and the 25°C incubation were performed up to 18 months or 6 months, respectively. Reverse-phase HPLC analysis was performed on an Agilent 1200 system (Agilent Technologies) using a Jupiter 5 µ C4 300A column (Phenomenex) and a standard water/0.12% trifluoroacetic acid (TFA) to acetonitrile/0.12% TFA gradient. Sample dilution buffer and reference was 100 mM Tris (Merck 1.08382). The percentage of the main peak compared to the total area under the curve was determined using the Agilent software.
ELISA, competition ELISA and homogeneous time-resolved fluorescence
For EC50 measurements, 50 µL or 100 µL of 20 nM target (human VEGF-A, mouse VEGF-A, human VEGF-C, human PDGF-AB (all R&D Systems); human (Peprotech), cynomolgus monkey (Sino Biological), and mouse (R&D Systems) HGF) or 100 µL of 1/30′000 diluted serum (human (Sigma), mouse (Innovative research), cynomolgus monkey (collected using standard methods) serum; corresponding to 20 nM serum albumin assuming 600 µM serum albumin present in serum) in PBS per well were immobilized in Maxisorp plates (Nunc, Denmark) overnight at 4°C. After washing 5 times with 300 µL PBST (PBS supplemented with 0.1% Tween 20), the wells were blocked with 300 µL PBST-C (PBST supplemented with 0.25% casein) for 1 h or 2 h at room temperature with shaking (Titramax 1000 shaker, 450 rpm to 600 rpm; Heidolph, Germany). After washing 5 times as described above, 100 µL/well or 50 µL/well MP0250 or individual DARPin® proteins (concentrations ranging from 100 nM to 0.01 pM) in PBST-C were applied and incubated for 1 h to 2 h at room temperature with shaking. After washing 5 times as described above, binding of MP0250 or individual DARPin® proteins was detected using 100 µL or 50 µL/well rabbit anti-DARPin® domain mAb (generated using standard rabbit immunization methods) in PBST-C for 1 h at room temperature with shaking. After washing 5 times as described above, bound anti-DARPin® domain mAb was detected using 100 µL or 50 µL/well goat anti-rabbit IgG-HRP conjugate (Thermo Fisher 31460; 1:5000 dilution) in PBST-C for 1 h at room temperature with shaking at 450 rpm. After washing 5 times as described above, the ELISA was then developed using 100 µL or 50 µL BM soluble blue POD substrate (Roche, Switzerland), diluted 1:4 in water. The reaction was stopped after 5 min using 100 µL or 50 µL 1 M H2SO4 and the OD (OD 450 nm – OD 620 nm) was recorded. The serum albumin specificity was further verified using purified serum albumin ELISA using the same setup (data not shown). VEGF inhibition potency was further assessed using VEGF-A competition ELISA according to the manufacturer (VEGF Quantikine, R&D Systems).27
For the homogeneous time-resolved fluorescence (HTRF) VEGF-A/VEGF-R interaction analyses, purified MP0250 at varying concentration was incubated with biotinylated VEGF-A (40 nM; ReliaTech) for 1 hour at room temperature in PBS supplemented with 0.2% BSA and 0.01% Tween 20. During incubation, 5 µL of 80 nM hVEGF-R2 (ReliaTech) and 10 µL of a mixture of 1:100 dilutions each of streptavidin-Tb (Cisbio) and polyclonal anti-human-IgG-d2 (Cisbio) were added to each well of a 384-well HTRF white plate (Proxiplate 384, PerkinElmer). Five µL of the incubation mixtures were then added to the wells of the plate and the plate was incubated for 1 hour protected from light at room temperature. The ratio of fluorescence at 665 nm and 620 nm was measured.
Surface plasmon resonance
SPR measurements were performed using a ProteOn XPR36 instrument (BioRad). Running buffer was PBS pH 7.4 containing 0.005% Tween 20. 3000 RU of HGF were immobilized on a GLC chip using 100 nM HGF (Peprotech, 100–39) in 10 mM sodium acetate buffer pH 5.3. For the analysis, 100 nM MP0250 (180 s association, 60 s dissociation) were first injected, followed by 100 nM VEGF (180 s association, 60 s dissociation; Relia Tech, 300-036-L), and 100 nM serum albumin (180 s association, 300 s dissociation; CSL Behring 100 mL, 200 g/L). Different controls were made by omitting one each of the injected components (see Figure 5). All signals were referenced to the interspots.
Cellular assays
Purified MP0250 and individual DARPin® domains were tested in cellular assays, including a HUVEC proliferation assay to assess VEGF-A inhibition, and a cMet phosphorylation assay to assess HGF inhibition. For the HUVEC proliferation assay, human VEGF-A was used at a concentration of 8 ng/mL (corresponding to EC80 as determined in a proliferation assay; ∼ 190 pM), and MP0250 or the individual VEGF-A-inhibiting DARPin® domain of MP0250 were titrated in a concentration range between 15 pM and 2.5 nM. About 3000 cells were seeded in 50 µL EBM-2 medium (Lonza) supplemented with penicillin/streptomycin and 5% fetal calf serum (assay medium) in CellBind plates (Sigma-Aldrich) in a CO2 incubator at 37°C. Four-fold MP0250/individual DARPin® domain dilutions (in assay medium) were made by serial dilution 1:2 fold in a dilution plate. The four-fold protein concentration dilutions were mixed with four-fold VEGF-A stock (32 ng/mL) in a ratio 1:1, yielding a mixture comprising a mixture of two-fold concentrated MP0250/individual DARPin® domain and VEGF-A each. 50 µL of the two-fold mixtures were added to the cells for 72 h (1:1, yielding final concentration of MP0250/individual DARPin® domain and VEGF-A). Cell proliferation was determined by monitoring metabolic activity using WST-1 (Sigma Aldrich) and an optical reader (450 nm; 620 nm reference).
Inhibition of cMet phosphorylation by MP0250 was measured using A549 cells and a DuoSet P-cMet-ELISA (RnD Systems). Cells were seeded in complete medium with 10% fetal calf serum in 12-well plates at 200′000 cells per well in complete medium. After 24 h, medium was replaced by serum-free medium. Cells were incubated for another 24 h and stimulated by 1 nM human HGF (or PBS for negative control) in the presence and absence of MP0250 or the individual HGF-inhibiting DARPin® domain. HGF and MP0250 or the individual DARPin® domain were preincubated for at least 30 minutes at room temperature prior to addition to cells. Cells were stimulated for 10 minutes at room temperature. Stimulation was terminated by removing the cell supernatant, cell washing with PBS, and addition of ice-cold cell lysis buffer (Sample diluent concentrate 2x, DYC002 (RnD Systems) plus phosphatase inhibitor (Roche) plus protease inhibitor (Roche)). Cell lysates were kept at −20°C until the ELISA experiment in a 96-well plate.
Pharmacokinetic measurements
Single-dose tail vein intravenously administered dose pharmacokinetic measurements in female Balb/c mice (n = 10 per group) were performed at target doses of 1 mg/kg and 2 mg/kg MP0250. Blood samples were collected pre-dose and again at 5 min, 1, 4, 24, 48, 72, and 96 hours post-injection. Concentrations were determined as described below. The pharmacokinetic profile of MP0250 in cynomolgus monkey was assessed by administering single doses of MP0250 via intravenous infusion for 30 min at target dose levels of 1 mg/kg, 10 mg/kg, and 100 mg/kg to 5 each male and female animals (n = 10 per group). Blood samples were collected pre-dose and again at 5 min and 4, 8, 24, 72, 120 and 168 hours post-end of infusion. Serum concentrations of MP0250 were determined by sandwich ELISA using a rabbit monoclonal anti-DARPin® domain antibody as capture reagent and murine monoclonal anti-DARPin® domain antibody as detection reagent (both generated using standard immunization and hybridoma techniques), and using a standard curve. Pharmacokinetic parameters were determined using the software Phoenix WinNonLin (Certara, Princeton, USA) or GraphPadPrism (GraphPad Software, La Jolla, USA) and non-compartmental analyses. Results are shown in Table 3. Cynomolgus monkey anti-drug antibodies were assessed by ELISA coating MP0250 in Maxisorp plates, applying serum sample (or control antibodies), and detecting bound antibody with cynomolgus monkey cross-reactive anti-human-IgG/IgE/IgM-antibody-HRP conjugates (Sigma A0170; AbDSerotec STAR147P and STAR145P). Allometric scaling31, 32 to human (80 kg) was calculated using mouse (18 g; 1.19 d terminal half-life at 1 mg/mL) and cynomolgus monkey (5 kg; 5.29 d terminal half-life at 10 mg/kg).
Disclosure of potential conflicts of interest
All authors hold options or shares in Molecular Partners AG.
Acknowledgments
Drs. Keith M. Dawson, Jörg Herbst, Patrik Forrer, Peter Hornung, Ivo Sonderegger, and Frieder Merz, as well as Mirela Matzner, Ralph Bessey, Feyza Sacarcelik and Julie Lobstein are acknowledged for experimental input and support. Dr. Johannes Schilling is acknowledged for the preparation of the schematic MP0250 model.
References
- 1.Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–68. doi: 10.1038/nbt1127. PMID:16211069 [DOI] [PubMed] [Google Scholar]
- 2.Vazquez-Lombardi R, Phan TG, Zimmermann C, Lowe D, Jermutus L, Christ D. Challenges and opportunities for non-antibody scaffold drugs. Drug Discov Today. 2015;20:1271–83. doi: 10.1016/j.drudis.2015.09.004. PMID:26360055 [DOI] [PubMed] [Google Scholar]
- 3.Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, Grütter MG, Plückthun A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22:575–82. doi: 10.1038/nbt962. PMID:15097997 [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–74. doi: 10.1038/nature04483. PMID:16355214 [DOI] [PubMed] [Google Scholar]
- 5.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–16. doi: 10.1038/nrd2530. PMID:18511928 [DOI] [PubMed] [Google Scholar]
- 6.Escudier B. Anti-VEGF therapy for renal cell carcinoma. Clin Adv Hematol Oncol. 2007;5:530–1. PMID:17679926 [PubMed] [Google Scholar]
- 7.Hurwitz H. Integrating the anti-VEGF-A humanized monoclonal antibody bevacizumab with chemotherapy in advanced colorectal cancer. Clin Colorectal Cancer. 2004;4(Suppl 2):S62–8. doi: 10.3816/CCC.2004.s.010. PMID:15479481 [DOI] [PubMed] [Google Scholar]
- 8.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–49. doi: 10.1056/NEJMra0706596. PMID:18463380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K, Tom MW, Paquette J, Tokuyasu TA, Tsao S, et al.. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin Cancer Res. 2013;19:1773–83. doi: 10.1158/1078-0432.CCR-12-1281. PMID:23307858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castellone MD, Carlomagno F, Salvatore G, Santoro M. Receptor tyrosine kinase inhibitors in thyroid cancer. Best Pract Res Clin Endocrinol Metab. 2008;22:1023–38. doi: 10.1016/j.beem.2008.09.012. PMID:19041829 [DOI] [PubMed] [Google Scholar]
- 11.Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al.. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10:2298–308. doi: 10.1158/1535-7163.MCT-11-0264. PMID:21926191 [DOI] [PubMed] [Google Scholar]
- 12.Smith DC, Smith MR, Sweeney C, Elfiky AA, Logothetis C, Corn PG, Vogelzang NJ, Small EJ, Harzstark AL, Gordon MS, et al.. Cabozantinib in patients with advanced prostate cancer: Results of a phase II randomized discontinuation trial. J Clin Oncol. 2013;31:412–9. doi: 10.1200/JCO.2012.45.0494. PMID:23169517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basilico C, Pennacchietti S, Vigna E, Chiriaco C, Arena S, Bardelli A, Valdembri D, Serini G, Michieli P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin Cancer Res. 2013;19:2381–92. doi: 10.1158/1078-0432.CCR-12-3459. PMID:23532890 [DOI] [PubMed] [Google Scholar]
- 14.Michieli P, Basilico C, Pennacchietti S. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET–response. Clin Cancer Res. 2013;19:4291. doi: 10.1158/1078-0432.CCR-13-1534. PMID:23766360 [DOI] [PubMed] [Google Scholar]
- 15.Jayaram A, Zafeiriou Z, de Bono JS. Cabozantinib-getting under the skin of cutaneous toxicity. JAMA Oncol. 2015;1:535–6. doi: 10.1001/jamaoncol.2015.0702. PMID:26181263 [DOI] [PubMed] [Google Scholar]
- 16.Belum VR, Serna-Tamayo C, Wu S, Lacouture ME. Incidence and risk of hand-foot skin reaction with cabozantinib, a novel multikinase inhibitor: A meta-analysis. Clin Exp Dermatol. 2016;41:8–15. doi: 10.1111/ced.12694. PMID:26009777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenall SA, Adams TE, Johns TG. Incomplete target neutralization by the anti-cancer antibody rilotumumab. mAbs. 2016;8:246–52. doi: 10.1080/19420862.2015.1122149. PMID:26750997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon MS, Sweeney CS, Mendelson DS, Eckhardt SG, Anderson A, Beaupre DM, Branstetter D, Burgess TL, Coxon A, Deng H, et al.. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res. 2010;16:699–710. doi: 10.1158/1078-0432.CCR-09-1365. PMID:20068101 [DOI] [PubMed] [Google Scholar]
- 19.Ryan CJ, Rosenthal M, Ng S, Alumkal J, Picus J, Gravis G, Fizazi K, Forget F, Machiels JP, Srinivas S, et al.. Targeted MET inhibition in castration-resistant prostate cancer: A randomized phase II study and biomarker analysis with rilotumumab plus mitoxantrone and prednisone. Clin Cancer Res. 2013;19:215–24. doi: 10.1158/1078-0432.CCR-12-2605. PMID:23136195 [DOI] [PubMed] [Google Scholar]
- 20.Cunningham D, Tebbutt NC, Davidenko I, Murad AM, Al-Batran S-E, Ilson DH, Tjulandin S, Gotovkin E, Karaszewska B, Bondarenko I.. Phase III, randomized, double-blind, multicenter, placebo (P)-controlled trial of rilotumumab (R) plus epirubicin, cisplatin and capecitabine (ECX) as first-line therapy in patients (pts) with advanced MET-positive (pos) gastric or gastroesophageal junction (G/GEJ) cancer: RILOMET-1 study. J Clin Oncol. 2015;33:4000–4000. abstract 4000. [Google Scholar]
- 21.Merchant M, Ma X, Maun HR, Zheng Z, Peng J, Romero M, Huang A, Yang NY, Nishimura M, Greve J, et al.. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci U S A. 2013;110:E2987–96. doi: 10.1073/pnas.1302725110. PMID:23882082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finisguerra V, Prenen H, Mazzone M. Preclinical and clinical evaluation of MET functions in cancer cells and in the tumor stroma. Oncogene. 2016;35(42):5457–5467. doi: 10.1038/onc.2016.36. PMID:26996670 [DOI] [PubMed] [Google Scholar]
- 23.Spigel DR, Edelman MJ, O'Byrne K, Paz-Ares L, Mocci S, Phan S, Shames DS, Smith D, Yu W, Paton VE, et al.. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: METLung. J Clin Oncol. 2017;35:412–20. doi: 10.1200/JCO.2016.69.2160. PMID:27937096 [DOI] [PubMed] [Google Scholar]
- 24.Rolfo C, Van Der Steen N, Pauwels P, Cappuzzo F. Onartuzumab in lung cancer: The fall of Icarus? Expert Rev Anticancer Ther. 2015;15:487–9. doi: 10.1586/14737140.2015.1031219. PMID:25818471 [DOI] [PubMed] [Google Scholar]
- 25.Fiedler U, Ekawardhani S, Cornelius A, Gilboy P, Bakker TR, Dolado I, Stumpp MT, Dawson KM.. MP0250, a VEGF and HGF neutralizing DARPin® molecule shows high anti-tumor efficacy in mouse xenograft and patient-derived tumor models. OncoTarget [in press]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol. 2003;332:489–503. PMID:12948497 [DOI] [PubMed] [Google Scholar]
- 27.Stahl A, Stumpp MT, Schlegel A, Ekawardhani S, Lehrling C, Martin G, Gulotti-Georgieva M, Villemagne D, Forrer P, Agostini HT, et al.. Highly potent VEGF-A-antagonistic DARPins as anti-angiogenic agents for topical and intravitreal applications. Angiogenesis. 2013;16:101–11. doi: 10.1007/s10456-012-9302-0. PMID:22983424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zahnd C, Kawe M, Stumpp MT, de Pasquale C, Tamaskovic R, Nagy-Davidescu G, Dreier B, Schibli R, Binz HK, Waibel R, et al.. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: Effects of affinity and molecular size. Cancer Res. 2010;70:1595–605. doi: 10.1158/0008-5472.CAN-09-2724. PMID:20124480 [DOI] [PubMed] [Google Scholar]
- 29.Ståhl S, Nygren PA. The use of gene fusions to protein A and protein G in immunology and biotechnology. Pathol Biol. 1997;45:66–76. PMID:9097850 [PubMed] [Google Scholar]
- 30.Steiner D, Merz FW, Sonderegger I, Gulotti-Georgieva M, Villemagne D, Phillips DJ, Forrer P, Stumpp MT, Zitt C, Binz HK.. Half-life extension using serum albumin-binding DARPin® domains. Prot Eng Des Selection. [in press]. [DOI] [PubMed] [Google Scholar]
- 31.Ings RM. Interspecies scaling and comparisons in drug development and toxicokinetics. Xenobiotica. 1990;20:1201–31. doi: 10.3109/00498259009046839. PMID:2275215 [DOI] [PubMed] [Google Scholar]
- 32.Ling J, Zhou H, Jiao Q, Davis HM. Interspecies scaling of therapeutic monoclonal antibodies: Initial look. J Clin Pharmacol. 2009;49:1382–402. doi: 10.1177/0091270009337134. PMID:19837907 [DOI] [PubMed] [Google Scholar]
- 33.Lin YS, Nguyen C, Mendoza JL, Escandon E, Fei D, Meng YG, Modi NB. Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of a humanized monoclonal antibody against vascular endothelial growth factor. J Pharmacol Exp Ther. 1999;288:371–8. PMID:9862791 [PubMed] [Google Scholar]
- 34.Kawe M, Horn U, Plückthun A. Facile promoter deletion in Escherichia coli in response to leaky expression of very robust and benign proteins from common expression vectors. Microb Cell Fact. 2009;8:8. doi: 10.1186/1475-2859-8-8. PMID:19171063 [DOI] [PMC free article] [PubMed] [Google Scholar]