

Abstract
Cathelicidin-BF, derived from the banded krait (Bungarus fasciatus), is a typically cationic, amphiphilic and α-helical antimicrobial peptide (AMP) with 30 amino acids that exerts powerful effects on multidrug-resistant (MDR) clinical isolates, including Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae, but whether it targets plasma membranes or intracellular targets to kill bacteria is still controversial. In the present study, we demonstrated that the disruption of bacterial membranes with high concentrations of cathelicidin-BF was the cause of bacterial death, as with conventional antibiotics at high concentrations. At lower concentrations, cathelicidin-BF did not cause bacterial plasma membrane disruption, but it was able to cross the membrane and aggregate at the nucleoid regions. Functional proteins of the transcription processes of P. aeruginosa and A. baumannii were affected by sublethal doses of cathelicidin-BF, as demonstrated by comparative proteomics using isobaric tags for relative and absolute quantification and subsequent gene ontology (GO) analysis. Analysis using the Kyoto Encyclopedia of Genes and Genomes showed that cathelicidin-BF mainly interferes with metabolic pathways related to amino acid synthesis, metabolism of cofactors and vitamins, metabolism of purine and energy supply, and other processes. Although specific targets of cathelicidin-BF must still be validated, our study offers strong evidence that cathelicidin-BF may act upon intracellular targets to kill superbugs, which may be helpful for further efforts to discover novel antibiotics to fight against them.
Keywords: antimicrobial peptide pressure, cathelicidin-BF, intracellular targets, multidrug-resistant, Pseudomonas aeruginosa, Acinetobacter baumannii, comparative proteomics
Introduction
The bacterial pathogens Pseudomonas aeruginosa and Acinetobacter baumannii are the top two causes of pneumonia acquired in intensive care units (ICUs) and ventilator-associated pneumonia (VAP), with mortality rates of 37.4 and 34.5%, respectively (Zhang et al., 2014). Emerging resistance to colistin and tigecycline, two of the few choices for last-resort treatment of P. aeruginosa and A. baumannii, may make this situation worse (Cai et al., 2012; Deng et al., 2014; Potron et al., 2015; Lee et al., 2016). Some antimicrobial peptides (AMPs) have shown excellent effects on these drug-resistant pathogens in vitro but are not able to be administered systemically due to their shortcomings, such as hemolytic activity or poor stability in vivo (Ramamoorthy et al., 2006; Zetterberg et al., 2011; Vila-Farres et al., 2012; Li et al., 2014; Liu et al., 2015). Though efforts have been made, mainly based on structural design, to overcome these disadvantages, no AMP is clinically available to date (Fjell et al., 2011; Andres, 2012). Instead of taking the original AMPs as structural templates, the understanding of their unique mechanisms of action, especially those whose targets may be different from those of the antibiotics available at present, may be more helpful, along with the development of advanced computer-aided drug discovery.
The well-known targets of AMPs are negatively charged prokaryotic cell membranes. Their “selective toxicity” induces transmembrane pores that cause the leakage of intracellular components and finally bacterial death while leaving the electrically neutral membranes of eukaryotic cells untouched (Matsuzaki, 1999). This hypothesis is challenged by the fact that some AMPs kill not only bacteria but also viruses, fungi, protozoa, parasites, and cancer cells, and some AMPs have hemolytic activities (Wang et al., 2016). In recent decades, more and more non-membrane targets of AMPs similar to those of conventional antibiotics have been reported. Examples include cell wall synthesis (mersacidin), DNA (tachyplesin, indolicidin), RNA (buforin II) and important proteins (microcin B17, microcin J25, pyrrhocoricin) (Yonezawa et al., 1992; Brotz et al., 1998; Park et al., 1998; Heddle et al., 2001; Kragol et al., 2001; Mukhopadhyay et al., 2004; Brogden, 2005; Hsu et al., 2005; Parks et al., 2007). Because some clinical isolates have gained resistance to nearly all of the antibiotics available yet some AMPs still work, in particular through non-membrane targets, elucidation of their unique modes of action is highly anticipated.
Cathelicidin-BF, derived from the banded krait (Bungarus fasciatus), is a typically cationic, amphiphilic and α-helical AMP with 30 amino acids that exerts powerful effects on multidrug-resistant (MDR) clinical isolates, including P. aeruginosa, A. baumannii and Klebsiella pneumoniae, but its mechanism of action is still controversial (Wang et al., 2008; Zhou et al., 2011; Hao et al., 2013; Liu et al., 2015; Azim et al., 2016). We have noticed that while some reports have tried to explain its mechanism using the membrane rupture thesis, the concentrations of AMP used to support such claims are always higher than their minimal inhibitory concentration (MIC), which makes the interpretation implausible (Zhou et al., 2011; Gao et al., 2015; Yu et al., 2015). Conversely, at concentrations that result in low toxicity to normal mammalian cells, including erythrocytes, cathelicidin-BF was reported to inhibit cancer cell proliferation, possibly via intracellular targets (Tian et al., 2013; Wang et al., 2013). These clues imply that cathelicidin-BF may act on intracellular targets to kill bacteria.
A systemic view of how bacteria react to antibiotics by comparative proteomics or proteome microarray may reflect pathways with which these antibiotics interfere and is helpful to elucidate undefined mechanisms of novel antibiotics, including AMPs (Kohanski et al., 2010; Hessling et al., 2013; Liu et al., 2014; Elnakady et al., 2016; Ho et al., 2016; Pulido et al., 2016). In the present study, we treated MDR P. aeruginosa and A. baumannii with sublethal doses of cathelicidin-BF, tested the membrane permeability using the DNA-binding fluorescent dye propidium iodide (PI), checked the localization of fluorescein isothiocyanate (FITC)-tagged AMP with confocal microscopy, and further analyzed the differentially expressed proteins by isobaric tags for relative and absolute quantification (iTRAQ) with standard bioinformatics analyses, such as gene ontology (GO) and use of the Kyoto Encyclopedia of Genes and Genomes (KEGG).
Materials and methods
Ethics statement
The animal experimental procedures were approved by the Ethics Committee of Animal Care and Welfare of the Institute of Medical Biology, Chinese Academy of Medical Sciences (CAMS) & Peking Union Medial College (PUMC) (Permit Number: SYXK (dian) 2010-0007), in accordance with the animal ethics guidelines of the Chinese National Health and Medical Research Council (NHMRC) and the Office of Laboratory Animal Management of Yunnan Province, China. All efforts were made to minimize animal suffering.
All participants submitted a signed informed consent form to participate in the study. The protocol complied with the Helsinki Declaration and was approved by the Institutional Review Board of the Institute of Medical Biology, CAMS & PUMC.
Serum stability of cathelicidin-BF
Female BALB/c mice (6–8 weeks old, 16–18 g) were purchased from Vital River Laboratory Animal Technology Co. Ltd., and raised and maintained in the Central Animal Care Services of our institute under specific pathogen-free (SPF) conditions. Mice were anesthetized, and blood samples were collected by cardiac puncture. After being kept at 37°C for 1 h and then at 4°C overnight, blood samples were centrifuged at 3,000 × g for 15 min, and sera were collected. A portion of the serum was inactivated by boiling in a water bath for 20 min. Escherichia coli strain DH5α was grown in Luria-Bertani (LB) medium at 37°C with constant shaking at 220 rpm overnight to reach the middle of the logarithmic growth phase and diluted with LB to 3 × 105 CFU/mL before use. For tests, cathelicidin-BF (purity ≥95%, synthesized by GL Biochem Ltd, Shanghai, China) was dissolved in sterile deionized water and mixed with serum, inactivated serum or sterile deionized water at a volume ratio of 1:4 to achieve a final concentration of 2 mg/mL. After incubation at 37°C, aliquots were taken at each time point, and the MIC for DH5α was taken as the lowest peptide concentration at which no microbial growth was observed visually after 18 h of incubation at 37°C.
Cytotoxicity assay
Cell viability was measured using cell proliferation kit II (XTT) (Roche). Cells, including A549 (adenocarcinomic human alveolar basal epithelial cells), 293FT (human embryonic kidney cells) and L929 (murine fibroblast cells), were cultured in Dulbecco's minimum essential medium (DMEM) supplemented with 10% fetal bovine serum, 100 U penicillin/mL, and 100 μg streptomycin/mL in a humidified 5% CO2 atmosphere at 37°C. After digestion with trypsin, the cells were diluted in serum-free DMEM without phenol red to a final concentration of 2 × 105 cell/mL, seeded in 96 well plates (100 μl/well) and cultured overnight until adhesion. Cathelicidin-BF dissolved in serum-free DMEM without phenol red was added to wells, and the plates were incubated for 24 h as previously described. Subsequent procedures were performed according to the kit. Briefly, XTT labeling reagent (sodium 3′-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis-4-methoxy-6-nitro) benzene sulfonic acid hydrate) in Roswell Park Memorial Institute (RPMI) 1640 medium without phenol red was mixed with electron-coupling reagent PMS (N-methyl dibenzopyrazine methyl sulfate) at a volume ratio of 50:1 to make the working solution. Fifty microliters of the XTT labeling mixture was added to each well and incubated at the same conditions for 6 h. Absorbance [A492 nm-A690 nm] stands for the quantification of viable cells.
For the hemolysis assay, blood samples from mice were mixed with Alsever's solution (8 g/L sodium citrate, 0.55 g/L citric acid, 20.5 g/L glucose, 4.2 g/L NaCl, pH 6.1) at a volume ratio of 1:5, centrifuged at 1,000 × g for 10 min, and washed three times with 0.9% saline. Erythrocytes were suspended in 0.9% saline at a volume ratio of 1:50. Cathelicidin-BF dissolved in 0.9% saline was added and incubation continued at 37°C for 30 min. Then, the samples were centrifuged at 1,000 × g for 15 min, the supernatants were diluted four times with 0.9% saline, and the absorbance at 540 nm was measured. Using 1% Triton X-100 (v/v) to determine 100% hemolysis and 0.9% saline as the negative control, the hemolysis rate of cathelicidin-BF is expressed as [(Absorbance sample-Absorbance control)/(Absorbance100%-Absorbance control)] × 100.
Membrane permeabilization assay
One MDR clinical isolate, P. aeruginosa 1409, was identified with a Vitek 32 system (bioMerieux, France) and further verified by sequencing of 16s rDNA with universal primers 27f and 1492R. For tests, the bacteria were incubated in LB at a concentration corresponding to an OD600 value of 0.5, and then cathelicidin-BF was added to 100 μL of culture to obtain final concentrations of 4 × MIC (32 μg/mL) or ¼ × MIC (2 μg/mL). Levofloxacin (Tokyo Chemicals Industry Co. Ltd.,) with final concentrations of 4 × MIC (64 μg/mL) or ¼ × MIC (4 μg/mL) was used as a control. After 1 h of incubation at 25°C, the culture was centrifuged at 3,000 × g for 5 min and resuspended in PBS (phosphate-buffered saline, 8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, 0.24 g/L KH2PO4, pH 7.4). Then, PI was added to a final concentration of 10 μg/mL. After 30 min of incubation at 25°C, the cells were washed 3 times with PBS and immediately imaged with a fluorescence microscope (Olympus, Japan) (Yu et al., 2015).
Localization of cathelicidin-BF in viable bacteria
One MDR clinical isolate, A. baumannii 1408, was identified with a Vitek 32 system (bioMerieux, France) and further verified by sequencing 16s rDNA with universal primers 27f and 1492R. For tests, bacterial strains A. baumannii 1408 and P. aeruginosa 1409 were incubated in LB at a concentration corresponding to an OD600 value of 0.5, and then cathelicidin-BF conjugated with FITC at its N terminus (purity ≥95%, synthesized by GL Biochem Ltd, Shanghai, China) was added to a final concentration of ¼×MIC (i.e., 4 μg/mL for A. baumannii 1408 and 2 μg/mL for P. aeruginosa 1409) and incubated at 25°C for 1 h. The culture was centrifuged at 3,000 × g for 5 min, resuspended in PBS, and incubated at room temperature for 20 min with Hoechst (Sigma) diluted with PBS to a final concentration of 20 μg/mL. Next, the culture was centrifuged at 3,000 × g for 5 min, incubated with SynaptoRed C2 (Tocris Bioscience), diluted with Hank's solution (8 g/L NaCl, 0.4 g/L KCl, 1 g/L glucose, 60 mg/L KH2PO4, 47.5 mg/L Na2HPO4, pH 7.2) to a final concentration of 20 μg/mL, and maintained on ice for 1 min. Microscopy was performed with excitation and emission wavelengths, respectively, of 488 nm and 530 nm for FITC, 352 nm and 461 nm for Hoechst, and 515 nm and 640 nm for SynaptoRed C2 (Olympus, Japan) (Wang et al., 2015).
Bacterial protein preparation
Bacterial strains A. baumannii 1408 and P. aeruginosa 1409 were grown overnight in LB medium at 37°C with constant shaking at 220 rpm to reach the middle of their logarithmic growth phase. Cathelicidin-BF was added to a final concentration of 1/2 MIC (i.e., 8 μg/mL for A. baumannii 1408 and 4 μg/mL for P. aeruginosa 1409), and the cultures were incubated at 37°C for 2 h. Samples were collected by centrifugation at 4,000 × g for 5 min at 4°C and washed 3 times with PBS. All samples were homogenized in lysis buffer (4% SDS, 1 mM DTT, 150 mM Tris-HCl, pH 8.0, protease inhibitor). After 5 min incubation in boiling water, the homogenate was sonicated on ice. The crude extract was then incubated in boiling water again and clarified by centrifugation at 16,000 × g at 25°C for 10 min before the supernatants were collected. The protein concentration in the supernatants was determined using the BCA protein assay (Beyotime, China).
Protein digestion and iTRAQ labeling
Protein digestion was performed based on a filter-aided sample preparation procedure (Wisniewski et al., 2009). The resulting peptide mixtures were labeled with the 4-plex iTRAQ reagent according to the manufacturer's instructions (Applied Biosystems). Briefly, 200 μg of proteins for each sample was incorporated into 30 μl STD buffer (4% SDS, 100 mM DTT, 150 mM Tris-HCl, pH 8.0). The detergent, DTT and other low-molecular-weight components were removed using UA buffer (8 M urea, 150 mM Tris-HCl, pH 8.0) by repeated ultrafiltration (Microcon units, 30 kD). Then, 100 μl of 0.05 M iodoacetamide in UA buffer was added to block reduced cysteine residues, and the samples were incubated in darkness for 20 min. The filters were washed with 100 μl UA buffer three times and then with 100 μl DS buffer (50 mM triethylammonium bicarbonate at pH 8.5) twice. Finally, the protein suspensions were digested with 2 μg trypsin (Promega) in 40 μl DS buffer overnight at 37°C, and the resulting peptides were collected as a filtrate. The peptide content was estimated by UV light spectral density at 280 nm. A standard pool comprising a mixture of an equal amount of protein derived from all samples served as an internal control (IS). For labeling, each iTRAQ reagent was dissolved in 70 μl of ethanol, added to the respective peptide mixture, and then multiplexed and vacuum dried.
Peptide fractionation with strong cation exchange (SCX) chromatography
iTRAQ-labeled peptides were fractionated by SCX chromatography using the AKTA Purifier system (GE Healthcare). The dried peptide mixture was dissolved in 2 mL buffer A (10 mM KH2PO4 in 25% acetonitrile, pH 3.0) and loaded onto a Polysulfoethyl column (4.6 × 100 mm, 5 μm, 200 Å, PolyLC Inc.). The peptides were eluted at a flow rate of 1 mL/min with a gradient of 0–10% buffer B (500 mM KCl, 10 mM KH2PO4 in 25% acetonitrile, pH 2.7) for 7 min, 10–20% buffer B for 10 min, 20–45% buffer B for 5 min, and 45–100% buffer B for 5 min. The eluates were monitored by absorbance at 214 nm and collected every 1 min. The collections were pooled in groups of 4 fractions and desalted separately on C18 Cartridges (Empore™ SPE Cartridges C18, standard density, bed I.D. 7 mm, volume 3 mL, Sigma). Each final fraction was dried in a vacuum concentrator and reconstituted in 40 μl of 0.1% (v/v) trifluoroacetic acid. All samples were stored at −80°C before the next analysis.
Liquid chromatography (LC)-electrospray ionization (ESI) tandem mass spectrometry (MS) analysis by Q exactive
MS experiments were performed on a Q Exactive mass spectrometer that was coupled to a nanoflow HPLC instrument (Easy nLC, Thermo Fisher Scientific). The peptide mixture (5 μg) was loaded onto a C18-reversed phase column (Thermo Scientific Easy Column, 10 cm long, 75 μm diameter, 3 μm resin) in buffer A (0.1% formic acid) and separated in a linear gradient of buffer B (80% acetonitrile and 0.1% formic acid) at a flow rate of 250 nL/min over 140 min, controlled by IntelliFlow technology. MS data were acquired using a data-dependent “top10” method, dynamically choosing the most abundant precursor ions from the survey scan (300–1800 m/z) for HCD fragmentation. Determination of the target value was based on predictive Automatic Gain Control (pAGC). The dynamic exclusion duration was 60 s. Survey scans were acquired at a resolution of 70,000 at m/z 200, and the resolution for the HCD spectra was set to 17,500 at m/z 200. The normalized collision energy was 30 eV, and the underfill ratio, which specifies the minimum percentage of the target value likely to be reached at maximum fill time, was defined as 0.1%. The instrument was run with peptide recognition mode enabled.
Sequence database search and data analysis
MS/MS spectra were searched using the MASCOT engine (Matrix Science, London, UK, version 2.2.) embedded into Proteome Discover 1.4 (Thermo Electron, San Jose, CA) against Uniprot_A. baumannii, Uniprot_P. aeruginosa and the corresponding decoy databases. Proteins were identified with the following parameters: Peptide mass tolerance = 20 ppm; MS/MS tolerance = 0.1 Da; Enzyme = trypsin; Missed cleavage = 2; Fixed modification: Carbamidomethyl (C), iTRAQ 4plex (K), iTRAQ 4plex (N-term); Variable modification: Oxidation (M). All reported data were based on 99% confidence intervals for protein identification as determined by a false discovery rate (FDR) ≤0.01 (Zheng et al., 2014).
The final ratios of proteins were normalized to the median average protein ratio of a mixture of equal volumes of differently labeled samples. Differentially expressed proteins were specified by a ratio of > ±1.2 and p < 0.05 (Cox and Mann, 2008). Screened proteins were loaded into Blast2GO (Version 2.7.0) for GO mapping and annotation (Ashburner et al., 2000; Gotz et al., 2008). These proteins were also mapped to KEGG pathways for further analysis (Kanehisa et al., 2012). Fisher's exact test was used to calculate p-values, and p < 0.05 indicated GO or KEGG pathways that were significantly enriched in differentially expressed proteins compared to the untreated group (Blüthgen et al., 2005).
Results
Cathelicidin-BF promptly lost its antibacterial activity in mouse serum
Although cathelicidin-BF was stable after incubation in sterile deionized water at 37°C for nearly 24 h, reflected by the fact that the MIC against DH5α increased slightly from 8 mg/L to 16 mg/L, it almost completely lost its antibacterial activity after 1 h incubation in mouse serum, reflected by the fact that the MIC against DH5α increased to more than 128 mg/L (Figure 1). This process was so rapid that cathelicidin-BF lost half of its antibacterial activity immediately after being mixed with mouse serum, reflected by the fact that the MIC against DH5α was 8 mg/L after being mixed with water, while the MIC against DH5α was 16 mg/L after being mixed with serum at incubation time 0. When the serum was heat inactivated, the antimicrobial activity of cathelicidin-BF was relatively stable, as indicated by the slight increase in the MIC against DH5α from 8 mg/L at time 0 to approximately 13.3 mg/L after 6 h of incubation. These results are consistent with previous reports that linear AMPs are unstable in vivo because of endogenous mammalian proteases and proteases secreted by pathogens (Pasupuleti et al., 2009; Li et al., 2012).
Figure 1.
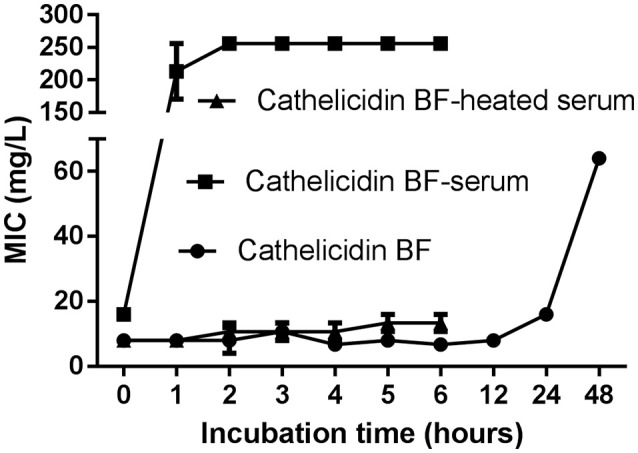
Serum stability of cathelicidin-BF. Cathelicidin-BF dissolved in sterile deionized water was mixed with mouse serum, heat-inactivated serum, or sterile deionized water at a volume ratio of 1:4 to reach a final concentration of 2 mg/mL. After incubation at 37°C, aliquots were taken at each time point, and the MIC against DH5α was defined as the lowest peptide concentration at which no microbial growth was observed visually after 18 h of incubation at 37°C. The stability of the MIC is an indicator of the stability of cathelicidin-BF.
Cathelicidin-BF caused slight cytotoxicity to specific eukaryotic cells
Less than 1% hemolysis was observed when cathelicidin-BF was at a concentration of 800 mg/L (Figure 2A). Regarding other cells, cathelicidin-BF inhibited the proliferation of human embryonic kidney 293FT cells at concentrations >50 mg/L (Figure 2B) while leaving the murine fibroblast cell line L929 and adenocarcinomic human alveolar basal epithelial cell line A549 intact at 400 mg/L (Figures 2C, D). Our results are consistent with previous reports that a cathelicidin-BF mutant is toxic to Madin-Daby canine kidney (MDCK) cells at 20 μM (~72 mg/L) and may cause renal injury when applied systemically (Tian et al., 2013).
Figure 2.

Cytotoxicity of cathelicidin-BF. Hemolysis and cell viability assays were conducted to test the cytotoxicity of cathelicidin-BF to mammalian cells. (A) For the hemolysis assay, cathelicidin-BF dissolved in 0.9% saline was added to mouse erythrocytes diluted in 0.9% saline and incubated at 37°C for 30 min. Supernatants were collected by centrifugation at 1,000 × g for 15 min and further diluted four times with 0.9% saline to test the absorbance at 540 nm. Using 1% Triton X-100 (v/v) to determine 100% hemolysis and 0.9% saline as the negative control, the hemolysis rate of cathelicidin-BF is expressed as [(Absorbance sample-Absorbance control)/(Absorbance100%-Absorbance control)]×100. The cell proliferation kit II (XTT) (Roche) was used to test the effects of cathelicidin-BF on the viability of (B) 293FT (human embryonic kidney cells), (C) L929 (mice fibroblast cell line) and (D) A549 (adenocarcinomic human alveolar basal epithelial cells) cells. Absorbance [A492 nm-A690 nm] was used to quantify viable cells.
Cathelicidin-BF could cross the bacterial membrane at low concentrations without detectable membrane disruption
PI is a DNA-binding fluorescent dye that can penetrate broken membranes but not intact membranes. As shown in Figure 3, neither levofloxacin nor cathelicidin-BF at low concentrations (¼ × MIC) affects the integrity of bacterial membranes, as no red dyes were detected under these conditions. When the concentrations increased to 4 × MIC (a lethal concentration), both levofloxacin- and cathelicidin-BF-treated bacteria showed red fluorescence after incubation with PI, which indicated that the membranes of these bacteria were broken and that PI entered these cells and formed complexes with the DNA inside. Cathelicidin-treated membranes were noticeably more thoroughly disrupted than levofloxacin-treated ones. We are not sure whether these differences are due to the greater efficiency of cathelicidin-BF on bacteria or its potential actions on membranes or DNA compared with that of levofloxacin.
Figure 3.

Bacterial plasma membrane permeabilization assay of cathelicidin-BF. Cathelicidin-BF and levofloxacin were incubated with MDR P. aeruginosa in Luria-Bertani medium at final concentrations of 4 × MIC (minimal inhibitory concentration) or ¼ × MIC. After incubation at 25°C for 1 h, bacteria were collected by centrifugation at 3,000 × g for 5 min and resuspended with PBS. The DNA-binding fluorescent dye PI was added to a final concentration of 10 μg/mL. After 30 min incubation at 25°C, the cells were washed 3 times with PBS and immediately imaged using a fluorescence microscope.
Indeed, cathelicidin-BF passed through bacterial membranes without disruption at low concentrations (Figure 4). Rather than localizing at membranes (stained with SynaptoRed C2 and shown in red in Figure 4, S) as we had previously expected according to the rupture thesis, FITC-tagged cathelicidin-BF (green in Figure 4, F) localized at nuclear regions, which is indicated by its co-localization with DNA (stained with Hoechst and shown in blue in Figure 4, H). Notably, FITC-tagged cathelicidin-BF seems to be more concentrated compared with the distribution of DNA at the nuclear region, suggesting that cathelicidin-BF may act on specific regions of the nucleoid.
Figure 4.

Localization of cathelicidin-BF in viable bacteria. N-terminus FITC (fluorescein isothiocyanate)-tagged cathelicidin-BF was added to final concentrations of ¼ × MIC for P. aeruginosa and A. baumannii. After incubation at 25°C for 1 h, bacteria were collected by centrifugation at 3,000 × g for 5 min, resuspended with PBS and then incubated with Hoechst at a final concentration of 20 μg/mL at room temperature for 20 min. After collection by centrifugation as described above, the bacteria were incubated with SynaptoRed in Hank's solution at a final concentration of 20 μg/mL on ice for 1 min. Microscopy was performed with excitation and emission wavelengths as follows: 488 nm and 530 nm for FITC (F, green color), 352 nm and 461 nm for Hoechst (H, blue color), 515 nm and 640 nm for SynaptoRed C2 (S, red color), respectively.
Go suggested potential intracellular targets of cathelicidin-BF
The functional interpretation of differentially expressed proteins (Tables S1–S3) was enriched via GO analysis. Interestingly, cathelicidin-BF- and levofloxacin-treated P. aeruginosa shared many GO categories concerning the transcription process (Table 1). These similarities also existed in cathelicidin-BF-treated A. baumannii, with shared processes, including core RNA polymerase binding (Table 2). Notably, both cathelicidin-BF- and levofloxacin-treated P. aeruginosa have their own specific GO categories. For example, GO categories concerning nucleic acids from cathelicidin-BF-treated P. aeruginosa mainly involve RNA (i.e., tRNA 3′-terminal CCA addition, RNA repair, etc.,), while GO categories concerning nucleic acids from levofloxacin-treated P. aeruginosa primarily involved DNA (i.e., primosome complex, replisome, double-strand break repair, etc.,) (Table 3). These results are consistent with the mechanisms of levofloxacin-mediated killing of bacteria (inhibition of two type II topoisomerases, namely, DNA gyrase and topoisomerase IV, which are involved in DNA separation and supercoiling, respectively) and implied that although both molecules target the nucleoid, cathelicidin-BF may have different mechanisms from levofloxacin (Drlica and Zhao, 1997; Ferrandiz and de la Campa, 2014).
Table 1.
GO categories shared between cathelicidin-BF- and levofloxacin-treated P. aeruginosa (p < 0.05).
| GO ID | GO category | p-value |
|---|---|---|
| GO:0016989 | Sigma factor antagonist activity | 0.003 |
| 0.005 | ||
| GO:0000989 | Transcription factor activity, transcription factor binding | 0.003 |
| 0.005 | ||
| GO:0006355 | Regulation of transcription, DNA-templated | 0.005 |
| 0.014 | ||
| GO:2001141 | Regulation of RNA biosynthetic process | 0.012 |
| 0.032 | ||
| GO:1903506 | Regulation of nucleic acid-templated transcription | 0.012 |
| 0.032 | ||
| GO:0010468 | Regulation of gene expression | 0.015 |
| 0.018 | ||
| GO:2000112 | Regulation of cellular macromolecule biosynthetic process | 0.016 |
| 0.019 | ||
| GO:0010556 | Regulation of macromolecule biosynthetic process | 0.016 |
| 0.019 | ||
| GO:0031326 | Regulation of cellular biosynthetic process | 0.017 |
| 0.020 | ||
| GO:0006351 | Transcription, DNA-templated | 0.017 |
| 0.041 | ||
| GO:0097659 | nucleic acid-templated transcription | 0.017 |
| 0.042 | ||
| GO:0009889 | Regulation of biosynthetic process | 0.019 |
| 0.023 | ||
| GO:0051252 | Regulation of RNA metabolic process | 0.020 |
| 0.047 | ||
| GO:0032774 | RNA biosynthetic process | 0.020 |
| 0.020 | ||
| GO:0019219 | regulation of nucleobase-containing compound metabolic process | 0.023 |
| 0.023 | ||
| GO:0000988 | Transcription factor activity, protein binding | 0.025 |
| 0.036 | ||
| GO:0016070 | RNA metabolic process | 0.025 |
| 0.009 | ||
| GO:0051171 | Regulation of nitrogen compound metabolic process | 0.027 |
| 0.032 | ||
| GO:0060255 | Regulation of macromolecule metabolic process | 0.028 |
| 0.033 | ||
| GO:0080090 | Regulation of primary metabolic process | 0.029 |
| 0.034 | ||
| GO:0031323 | Regulation of cellular metabolic process | 0.031 |
| 0.037 | ||
| GO:0072509 | Divalent inorganic cation transmembrane transporter activity | 0.033 |
| 0.041 | ||
| GO:0015197 | Peptide transporter activity | 0.033 |
| 0.041 | ||
| GO:0015095 | Magnesium ion transmembrane transporter activity | 0.033 |
| 0.041 | ||
| GO:0015693 | Magnesium ion transport | 0.033 |
| 0.041 | ||
| GO:0019222 | Regulation of metabolic process | 0.036 |
| 0.043 | ||
| GO:0090304 | Nucleic acid metabolic process | 0.039 |
| 0.019 |
The p-values of levofloxacin-treated P. aeruginosa are shaded, whereas those of cathelicidin-BF-treated cells are not.
Table 2.
GO categories for cathelicidin-BF-treated A. baumannii.
| GO ID | GO category | p-value |
|---|---|---|
| GO:0016987 | Sigma factor activity | 0.008 |
| GO:0006352 | DNA-templated transcription, initiation | 0.008 |
| GO:0000996 | Core DNA-dependent RNA polymerase binding promoter specificity activity | 0.008 |
| GO:0000990 | Transcription factor activity, core RNA polymerase binding | 0.008 |
| GO:0000988 | Transcription factor activity, protein binding | 0.008 |
Only GO categories with p < 0.01 are shown.
Table 3.
Differences in GO categories related to nucleic acids between cathelicidin-BF- and levofloxacin-treated P. aeruginosa (p < 0.05).
| GO ID | GO category | p-value |
|---|---|---|
| GO:0010629 | Negative regulation of gene expression | 0.031077 |
| GO:0001680 | tRNA 3′-terminal CCA addition | 0.033263 |
| GO:0017148 | Negative regulation of translation | 0.033263 |
| GO:0042245 | RNA repair | 0.033263 |
| GO:0001071 | Nucleic acid binding transcription factor activity | 0.044257 |
| GO:0003700 | transcription factor activity, sequence-specific DNA binding | 0.044257 |
| GO:0005667 | Transcription factor complex | 0.044257 |
| GO:2000104 | Negative regulation of DNA-dependent DNA replication | 0.041047 |
| GO:1990077 | Primosome complex | 0.041047 |
| GO:0006302 | double-strand break repair | 0.041047 |
| GO:0090329 | Regulation of DNA-dependent DNA replication | 0.041047 |
| GO:0006269 | DNA replication, synthesis of RNA primer | 0.041047 |
| GO:0030894 | Replisome | 0.041047 |
| GO:0030174 | Regulation of DNA-dependent DNA replication initiation | 0.041047 |
| GO:0032297 | Negative regulation of DNA-dependent DNA replication initiation | 0.041047 |
The GO categories enriched in levofloxacin-treated P. aeruginosa are shaded, whereas those enriched in cathelicidin-BF-treated cells are not.
KEGG analysis confirmed intracellular targets
KEGG pathway maps represent experimental knowledge on metabolism and various other functions of the cell and organism. Localization of differentially expressed proteins in these pathways may offer hints to how drugs work by interfering with key metabolic activities. As seen in Table 4, differentially expressed proteins from P. aeruginosa after cathelicidin-BF or levofloxacin treatment shared many pathways, including amino acid synthesis and pyrimidine and purine metabolism. Some of these pathways were further confirmed by their existence in cathelicidin-BF treated A. baumannii (KEGG categories with MapIDs are underlined in Table 4). Notably, while some of these pathways were not “enriched” (p > 0.05), they share some differently expressed proteins with other enriched pathways. For example, proteins Q9HVA1 and Q9I3S7 are shared in six pathways, although they are considered enriched in only 4 of them. Q9HVA1 is the acetolactate synthase isozyme III small subunit with functions in branched-chain amino acid biosynthesis and metabolism of cofactors and vitamins. It is overexpressed in both cathelicidin-BF- and levofloxacin-treated P. aeruginosa. Q9I3S7 is a likely decarboxylase that may interact selectively and non-covalently with thiamine pyrophosphate (the diphosphoric ester of thiamine, TPP). It is overexpressed in levofloxacin-treated P. aeruginosa but downregulated in cathelicidin-BF-treated P. aeruginosa. A similar phenomenon occurred with Q9HUU8, the urease subunit gamma that is involved in arginine biosynthesis and purine metabolism.
Table 4.
KEGG categories shared between cathelicidin-BF- and levofloxacin-treated P. aeruginosa.
| Map ID | Map name | Genes shared | p-value |
|---|---|---|---|
| ko02010 | ABC transporters | Q9HVR6 Q9I33L L9 Q9I5T5 | 0.025 |
| 0.046 | |||
| ko00920 | Sulfur metabolism | Q9I33L L9 | |
| ko00660 | C5-branched dibasic acid metabolism | Q9HVA1 Q9I3S7 | 0.025 |
| 0.003 | |||
| ko01230 | Biosynthesis of amino acids | Q9HVA1 Q9I3S7 | |
| ko00770 | Pantothenate and CoA biosynthesis | Q9HVA1 Q9I3S7 | |
| 0.013 | |||
| ko00220 | Arginine biosynthesis | Q9HUU8 | |
| ko00650 | Butanoate metabolism | Q9HVA1 Q9I3S7 | |
| ko02020 | Two-component system | ||
| ko00791 | Atrazine degradation | Q9HUU8 | |
| ko01210 | 2-oxocarboxylic acid metabolism | Q9HVA1 Q9I3S7 | |
| 0.016 | |||
| ko00290 | Valine, leucine and isoleucine biosynthesis | Q9HVA1 Q9I3S7 | |
| 0.001 | |||
| ko00240 | Pyrimidine metabolism | ||
| ko00230 | Purine metabolism | Q9HUU8 | |
The p-values of levofloxacin-treated P. aeruginosa are shaded, whereas those of cathelicidin-BF-treated cells are not. Underlined map IDs are also shared with cathelicidin-BF-treated A. baumannii.
Unique pathways were identified for each treatment. Q9I5V3, the multifunctional CCA protein, is downregulated in cathelicidin-BF-treated P. aeruginosa and enriched in the RNA transport pathway. Ubiquinone and other terpenoid-quinone biosynthesis pathways were enriched in levofloxacin-treated P. aeruginosa. Q9I298, a putative 3-methylglutaconyl-ConA hydratase, and Q9HTV3, a 3-octaprenyl-4-hydroxybenzoate carboxy-lyase, were downregulated in this pathway. Notably, Q9I5V3 was downregulated in levofloxacin-treated P. aeruginosa, and both Q9I298 and Q9HTV3 were downregulated in cathelicidin-BF-treated P. aeruginosa but they are not categorized as “differentially expressed proteins” according to the criteria set in the method section.
Interestingly, we found one protein, Q9I523, a nucleoside-triphosphate diphosphatase that is involved in both purine and pyrimidine metabolism, that was downregulated in cathelicidin-BF-treated P. aeruginosa. This protein was reported to interact with Era, an essential GTPase identified in various bacteria and some eukaryotes (Zhang and Inouye, 2002). Recently, a study reported that AMPs can kill bacteria by inhibiting E. coli ATP synthase (Azim et al., 2016). Considering that Q9I523 could hydrolyze all eight of the canonical ribo- and deoxynucleoside triphosphates to their respective monophosphates and PP (i), downregulation of this “energy switch” may play an important role in regulating the energy supply of a bacterial or tumor cell.
Discussion
AMPs are effective for some of the clinical superbugs, including P. aeruginosa and A. baumannii, but they have not been able to be administered systemically, mainly because of their poor stability in vivo and possible toxic effects, such as hemolysis or immunoregulation. Although AMPs were discovered several decades ago, the mechanism by which they kill bacteria is still controversial. In recent years, the membrane rupture thesis has been challenged by the fact that some AMPs can kill not only bacteria but also viruses, fungi, protozoa, parasites and cancer cells. In addition, more and more AMPs have been reported to have intracellular targets (Shah et al., 2016).
Cathelicidin-BF is a typical cationic, amphiphilic and α-helical AMP that has powerful effects on MDR clinical superbugs but cannot be applied systemically due to its sensitivity to proteases in vivo. In this report, we demonstrated that the disruption of the bacterial membrane at high concentrations of cathelicidin-BF was the result of bacteria death, as is the case for conventional antibiotics at high concentrations (Figure 3). In fact, lower concentrations of cathelicidin-BF did not cause bacterial membrane damage but could cross the membrane and aggregate at nucleoid regions (Figure 4). Therefore, we used levofloxacin for comparison in further analysis because it kills bacteria by interfering with DNA separation and supercoiling, which also occurs at nucleoid regions. Comparative proteomics showed that cathelicidin-BF may affect transcription in a similar way as levofloxacin (Tables 1, 2), which is consistent with its localization in the nucleoid regions after incubation. However, cathelicidin-BF tends to affect RNA synthesis, while levofloxacin tends to affect DNA replication (Table 3), which implies that these molecules may have different mechanisms of killing bacteria. These similarities and differences could also be seen from the KEGG analysis. While the mechanisms shared genes involved in the biosynthesis of amino acids and purine metabolism, cathelicidin-BF specifically affects pathways involving RNA transport, and levofloxacin has unique effects on quinone biosynthesis (Table 4). Moreover, we found one downregulated nucleoside-triphosphate diphosphatase in cathelicidin-BF-treated P. aeruginosa, which offered hints that AMPs may kill bacteria by controlling the energy supply.
Although our study provided evidence that cathelicidin-BF may act on intracellular targets instead of membranes to kill bacteria, several questions still need to be answered: (1) What are the specific targets of cathelicidin-BF when it kills the bacteria? We are not sure whether it kills the bacteria by affecting key enzymes involved in translation, as levofloxacin does, or considering the absence of nuclear membranes in bacteria, whether it simply binds to the negatively charged nucleic acids after crossing plasma membranes because of its cationic characteristic and subsequently interrupts the functions of these nucleic acids. (2) If cathelicidin-BF has protein targets, does it target specific multifunctional enzymes or multiple key enzymes to kill bacteria? We have noticed that cathelicidin-BF interferes with multiple metabolic processes of bacteria, including amino acid synthesis, metabolism of cofactors and vitamins, and metabolism of purines and energy supply. Further steps are still needed to confirm the contributions of the bactericidal effects of these differentially expressed proteins. (3) What is the contribution of the plasma membrane-crossing activity of cathelicidin-BF in killing bacteria? Considering that cathelicidin-BF may have similar targets as levofloxacin but that A. baumannii 1408 and P. aeruginosa 1409 have gained resistance to levofloxacin (possibly by efflux pumps), understanding how cathelicidin-BF crosses the bacteria plasma membranes (e.g., receptors, biophysical characteristics, etc.,) may also contribute to further discoveries of novel antibiotics targeting superbugs.
Author contributions
Conceived and designed the experiments: CL; performed the experiments: CL, BS, and JQ; analyzed the data: CL and BS; contributed reagents/materials/analysis tools: BS; contributed to the writing of the manuscript: CL and YM.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was financially supported by the National Natural Science Foundation of China (grant number 81503117, 81460322), the CAMS Initiative for Innovative Medicine (grant number 2017-I2M-3-022), and the Fundamental Research Funds for the Central Universities of China (grant number 2016ZX350073 and 3332013084).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2017.00466/full#supplementary-material
Peptide identification.
Protein identification.
Quantivative and differential analysis of proteins.
References
- Andres E. (2012). Cationic antimicrobial peptides in clinical development, with special focus on thanatin and heliomicin. Eur. J. Clin. Microbiol. Infect. Dis. 31, 881–888. 10.1007/s10096-011-1430-8 [DOI] [PubMed] [Google Scholar]
- Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., et al. (2000). Gene ontology: tool for the unification of biology. Gene Ontol. Consort. Nat. Genet. 25, 25–29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azim S., McDowell D., Cartagena A., Rodriguez R., Laughlin T. F., Ahmad Z. (2016). Venom peptides cathelicidin and lycotoxin cause strong inhibition of Escherichia coli ATP synthase. Int. J. Biol. Macromol. 87, 246–251. 10.1016/j.ijbiomac.2016.02.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blüthgen N., Brand K., Cajavec B., Swat M., Herzel H., Beule D. (2005). Biological profiling of gene groups utilizing Gene Ontology. Genome Inform. 16, 106–115. 10.11234/gi1990.16.106 [DOI] [PubMed] [Google Scholar]
- Brogden K. A. (2005). Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250. 10.1038/nrmicro1098 [DOI] [PubMed] [Google Scholar]
- Brotz H., Bierbaum G., Leopold K., Reynolds P. E., Sahl H. G. (1998). The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 42, 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Chai D., Wang R., Liang B., Bai N. (2012). Colistin resistance of Acinetobacter baumannii: clinical reports, mechanisms and antimicrobial strategies. J. Antimicrob. Chemother. 67, 1607–1615. 10.1093/jac/dks084 [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- Deng M., Zhu M. H., Li J. J., Bi S., Sheng Z. K., Hu F. S., et al. (2014). Molecular epidemiology and mechanisms of tigecycline resistance in clinical isolates of Acinetobacter baumannii from a Chinese university hospital. Antimicrob. Agents Chemother. 58, 297–303. 10.1128/AAC.01727-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica K., Zhao X. (1997). DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61, 377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnakady Y. A., Chatterjee I., Bischoff M., Rohde M., Josten M., Sahl H. G., et al. (2016). Investigations to the antibacterial mechanism of action of kendomycin. PLoS ONE 11:e0146165. 10.1371/journal.pone.0146165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandiz M. J., de la Campa A. G. (2014). The fluoroquinolone levofloxacin triggers the transcriptional activation of iron transport genes that contribute to cell death in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 58, 247–257. 10.1128/AAC.01706-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell C. D., Hiss J. A., Hancock R. E., Schneider G. (2011). Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 11, 37–51. 10.1038/nrd3591 [DOI] [PubMed] [Google Scholar]
- Gao W., Xing L., Qu P., Tan T., Yang N., Li D., et al. (2015). Identification of a novel cathelicidin antimicrobial peptide from ducks and determination of its functional activity and antibacterial mechanism. Sci. Rep. 5:17260. 10.1038/srep17260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz S., Garcia-Gomez J. M., Terol J., Williams T. D., Nagaraj S. H., Nueda M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. 10.1093/nar/gkn176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Q., Wang H., Wang J., Dou J., Zhang M., Zhou W., et al. (2013). Effective antimicrobial activity of Cbf-K16 and Cbf-A7 A13 against NDM-1-carrying Escherichia coli by DNA binding after penetrating the cytoplasmic membrane in vitro. J. Pept. Sci. 19, 173–180. 10.1002/psc.2488 [DOI] [PubMed] [Google Scholar]
- Heddle J. G., Blance S. J., Zamble D. B., Hollfelder F., Miller D. A., Wentzell L. M., et al. (2001). The antibiotic microcin B17 is a DNA gyrase poison: characterisation of the mode of inhibition. J. Mol. Biol. 307, 1223–1234. 10.1006/jmbi.2001.4562 [DOI] [PubMed] [Google Scholar]
- Hessling B., Bonn F., Otto A., Herbst F. A., Rappen G. M., Bernhardt J., et al. (2013). Global proteome analysis of vancomycin stress in Staphylococcus aureus. Int. J. Med. Microbiol. 303, 624–634. 10.1016/j.ijmm.2013.08.014 [DOI] [PubMed] [Google Scholar]
- Ho Y. H., Shah P., Chen Y. W., Chen C. S. (2016). Systematic analysis of intracellular-targeting antimicrobial peptides, bactenecin 7, hybrid of pleurocidin and dermaseptin, proline-arginine-rich peptide, and lactoferricin b, by using Escherichia coli proteome microarrays. Mol. Cell. Proteomics 15, 1837–1847. 10.1074/mcp.M115.054999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C. H., Chen C., Jou M. L., Lee A. Y., Lin Y. C., Yu Y. P., et al. (2005). Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 33, 4053–4064. 10.1093/nar/gki725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. 10.1093/nar/gkr988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski M. A., Dwyer D. J., Collins J. J. (2010). How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 8, 423–435. 10.1038/nrmicro2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragol G., Lovas S., Varadi G., Condie B. A., Hoffmann R., Otvos L., Jr. (2001). The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 40, 3016–3026. 10.1021/bi002656a [DOI] [PubMed] [Google Scholar]
- Lee J. Y., Park Y. K., Chung E. S., Na I. Y., Ko K. S. (2016). Evolved resistance to colistin and its loss due to genetic reversion in Pseudomonas aeruginosa. Sci. Rep. 6:25543 10.1038/srep25543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Wang Q., Li H., Yuan M., Yuan M. (2014). Preparation, characterization, in vitro release and degradation of cathelicidin-BF-30-PLGA microspheres. PLoS ONE 9:e100809. 10.1371/journal.pone.0100809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. A., Lee W. H., Zhang Y. (2012). Efficacy of OH-CATH30 and its analogs against drug-resistant bacteria in vitro and in mouse models. Antimicrob. Agents Chemother. 56, 3309–3317. 10.1128/AAC.06304-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. B., Shan B., Bai H. M., Tang J., Yan L. Z., Ma Y. B. (2015). Hydrophilic/hydrophobic characters of antimicrobial peptides derived from animals and their effects on multidrug resistant clinical isolates. Zool. Res. 36, 41–47. 10.13918/j.issn.2095-8137.2015.1.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Hu Y., Pai P. J., Chen D., Lam H. (2014). Label-free quantitative proteomics analysis of antibiotic response in Staphylococcus aureus to oxacillin. J. Proteome Res. 13, 1223–1233. 10.1021/pr400669d [DOI] [PubMed] [Google Scholar]
- Matsuzaki K. (1999). Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1462, 1–10. 10.1016/S0005-2736(99)00197-2 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay J., Sineva E., Knight J., Levy R. M., Ebright R. H. (2004). Antibacterial peptide microcin J25 inhibits transcription by binding within and obstructing the RNA polymerase secondary channel. Mol. Cell 14, 739–751. 10.1016/j.molcel.2004.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C. B., Kim H. S., Kim S. C. (1998). Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 244, 253–257. 10.1006/bbrc.1998.8159 [DOI] [PubMed] [Google Scholar]
- Parks W. M., Bottrill A. R., Pierrat O. A., Durrant M. C., Maxwell A. (2007). The action of the bacterial toxin, microcin B17, on DNA gyrase. Biochimie 89, 500–507. 10.1016/j.biochi.2006.12.005 [DOI] [PubMed] [Google Scholar]
- Pasupuleti M., Schmidtchen A., Chalupka A., Ringstad L., Malmsten M. (2009). End-tagging of ultra-short antimicrobial peptides by W/F stretches to facilitate bacterial killing. PLoS ONE 4:e5285. 10.1371/journal.pone.0005285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potron A., Poirel L., Nordmann P. (2015). Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int. J. Antimicrob. Agents 45, 568–585. 10.1016/j.ijantimicag.2015.03.001 [DOI] [PubMed] [Google Scholar]
- Pulido M. R., Garcia-Quintanilla M., Gil-Marques M. L., McConnell M. J. (2016). Identifying targets for antibiotic development using omics technologies. Drug Discov. Today 21, 465–472. 10.1016/j.drudis.2015.11.014 [DOI] [PubMed] [Google Scholar]
- Ramamoorthy A., Thennarasu S., Tan A., Gottipati K., Sreekumar S., Heyl D. L., et al. (2006). Deletion of all cysteines in tachyplesin I abolishes hemolytic activity and retains antimicrobial activity and lipopolysaccharide selective binding. Biochemistry 45, 6529–6540. 10.1021/bi052629q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P., Hsiao F. S., Ho Y. H., Chen C. S. (2016). The proteome targets of intracellular targeting antimicrobial peptides. Proteomics 16, 1225–1237. 10.1002/pmic.201500380 [DOI] [PubMed] [Google Scholar]
- Tian Y., Wang H., Li B., Ke M., Wang J., Dou J., et al. (2013). The cathelicidin-BF Lys16 mutant Cbf-K16 selectively inhibits non-small cell lung cancer proliferation in vitro. Oncol. Rep. 30, 2502–2510. 10.3892/or.2013.2693 [DOI] [PubMed] [Google Scholar]
- Vila-Farres X., Garcia de la Maria C., Lopez-Rojas R., Pachon J., Giralt E., Vila J. (2012). In vitro activity of several antimicrobial peptides against colistin-susceptible and colistin-resistant Acinetobacter baumannii. Clin. Microbiol. Infect. 18, 383–387. 10.1111/j.1469-0691.2011.03581.x [DOI] [PubMed] [Google Scholar]
- Wang G., Li X., Wang Z. (2016). APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44, D1087–D1093. 10.1093/nar/gkv1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Ke M., Tian Y., Wang J., Li B., Wang Y., et al. (2013). BF-30 selectively inhibits melanoma cell proliferation via cytoplasmic membrane permeabilization and DNA-binding in vitro and in B16F10-bearing mice. Eur. J. Pharmacol. 707, 1–10. 10.1016/j.ejphar.2013.03.028 [DOI] [PubMed] [Google Scholar]
- Wang J., Han Y., Yang R., Zhao X. (2015). Optimization of labeling and localizing bacterial membrane and nucleus with FM4-64 and Hoechst dyes. Wei Sheng Wu Xue Bao 55, 1068–1073. 10.13343/j.cnki.wsxb.20140603 [DOI] [PubMed] [Google Scholar]
- Wang Y., Hong J., Liu X., Yang H., Liu R., Wu J., et al. (2008). Snake cathelicidin from Bungarus fasciatus is a potent peptide antibiotics. PLoS ONE 3:e3217. 10.1371/journal.pone.0003217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. 10.1038/nmeth.1322 [DOI] [PubMed] [Google Scholar]
- Yonezawa A., Kuwahara J., Fujii N., Sugiura Y. (1992). Binding of tachyplesin I to DNA revealed by footprinting analysis: significant contribution of secondary structure to DNA binding and implication for biological action. Biochemistry 31, 2998–3004. 10.1021/bi00126a022 [DOI] [PubMed] [Google Scholar]
- Yu H., Lu Y., Qiao X., Wei L., Fu T., Cai S., et al. (2015). Novel cathelicidins from pigeon highlights evolutionary convergence in avain cathelicidins and functions in modulation of innate immunity. Sci. Rep. 5:11082. 10.1038/srep11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg M. M., Reijmar K., Pranting M., Engstrom A., Andersson D. I., Edwards K. (2011). PEG-stabilized lipid disks as carriers for amphiphilic antimicrobial peptides. J. Control. Release. 156, 323–328. 10.1016/j.jconrel.2011.08.029 [DOI] [PubMed] [Google Scholar]
- Zhang J., Inouye M. (2002). MazG, a nucleoside triphosphate pyrophosphohydrolase, interacts with Era, an essential GTPase in Escherichia coli. J. Bacteriol. 184, 5323–5329. 10.1128/JB.184.19.5323-5329.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Yao Z., Zhan S., Yang Z., Wei D., Zhang J., et al. (2014). Disease burden of intensive care unit-acquired pneumonia in China: a systematic review and meta-analysis. Int. J. Infect. Dis. 29, 84–90. 10.1016/j.ijid.2014.05.030 [DOI] [PubMed] [Google Scholar]
- Zheng B. B., Fang Y. N., Pan Z. Y., Sun L., Deng X. X., Grosser J. W., et al. (2014). iTRAQ-based quantitative proteomics analysis revealed alterations of carbohydrate metabolism pathways and mitochondrial proteins in a male sterile cybrid pummelo. J. Proteome Res. 13, 2998–3015. 10.1021/pr500126g [DOI] [PubMed] [Google Scholar]
- Zhou H., Dou J., Wang J., Chen L., Wang H., Zhou W., et al. (2011). The antibacterial activity of BF-30 in vitro and in infected burned rats is through interference with cytoplasmic membrane integrity. Peptides 32, 1131–1138. 10.1016/j.peptides.2011.04.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Peptide identification.
Protein identification.
Quantivative and differential analysis of proteins.