

Supplemental Digital Content is available in the text
Keywords: acute myocardial infarction, biomarkers, cytokines, inflammation, interleukin-16, pathophysiology
Abstract
Interleukin (IL)-16, a polypeptide cytokine, plays a crucial role in the inflammatory process, acting as a chemoattractant for peripheral immune cells and has been linked to various inflammatory diseases. However, its role in patients with acute myocardial infarction (AMI) is unclear.
We retrospectively analyzed serum levels of IL-16 in blood of patients with (STEMI, n = 45) and without ST-segment elevation myocardial infarction (NSTEMI, n = 42) compared with controls with excluded coronary artery disease (n = 55). Furthermore, correlation analysis with inflammatory cells, C-reactive protein (CRP) levels, dendritic cell precursors (DCPs), and other clinical and biochemical markers was performed.
Compared with controls, patients with STEMI and NSTEMI evidenced higher levels of IL-16 in pg/mL (STEMI: 759.38 ± 471.54, NSTEMI: 677.77 ± 438.8, control: 500.45 ± 432.21; P = .002). IL-16 correlated with CRP (r = 0.26, P = .001), leucocytes (r = 0.38, P < .001), NT-proBNP (r = 0.20, P = .02) and hsTnT (r = 0.25, P = .004). Circulating myeloid DCPs, plasmacytoid DCPs, and total DCPs showed a significant inverse correlation to IL-16 levels (r = −0.21, P = .01; r = −0.23, P = .005; r = −0.26, P = .002, respectively).
Interleukin-16 might play an important role in the inflammatory process of patients suffering from AMI and correlates with inflammatory cell activation and clinical and biochemical markers. The cytokine IL-16 might upregulate the proinflammatory response and recruitment of inflammatory cells into infarcted myocardium.
1. Introduction
Acute myocardial infarction (AMI) followed by cardiac remodeling and a decline in left ventricular contractility often leads to congestive heart failure. Local and systemic inflammation after AMI plays a paramount role in this remodeling process.[1,2] This response is mediated by inflammatory immune cells within the infarcted myocardium. They phagocytose the necrotic tissue and also secrete soluble factors such as cytokine-initiated homing of granulocytes, macrophages, and dendritic cells.[3–5] The inflammatory reactions observed after AMI might be necessary for tissue stabilization and formation of a robust scar.[6]
Interleukin-16 (IL-16) is considered a proinflammatory cytokine and is produced by not only T-cells, mast cells, eosinophils, fibroblasts, but also dendritic cells and epithelial cells.[7–11] IL-16 promotes migration of lymphocytes, induces the expression of proinflammatory factors, and modulates apoptosis.[11] As a potent chemoattractant for peripheral immune cells, IL-16 has been linked to inflammatory diseases such as asthma or Crohn disease.[12,13] Its role in the complex pathobiological pathways during and after AMI is unclear. In addition to established biomarkers, the incremental diagnostic and prognostic value of newer generation mediators is of high interest. Measurement of biomarker concentrations may help to assess infarct size, myocardial dysfunction, and clinical outcome, and may help to identify high-risk patients, establish individualized treatment and secondary prevention strategies.[14] The aim of the present study was to analyse (i) circulating IL-16 levels in patients with different types of AMI compared to controls and (ii) its correlation with systemic and local inflammatory processes, as well as (iii) with clinical and established biomarkers.
2. Materials and methods
2.1. Patients and controls
The study comprised 142 patients, who underwent a coronary angiography at the University Hospital in Jena. Based on standardized definitions,[15] patients were divided into 3 subgroups: patients with typical angina pectoris-like symptoms and elevated serum creatine kinase and cardiac troponin I were classified as AMI and were subdivided according to the presence or absence of ST-segment elevations (at least 0.2 mV in 2 or more contiguous precordial leads or at least 0.1 mV in the limbs lead) in the initial ECG into the STEMI (n = 61) or NSTEMI (n = 57) group. During the study enrolment period all patients with STEMI and all admitted patients with NSTEMI underwent emergent coronary angiography.[16] The control-group (n = 76) consisted of subjects who underwent a coronary angiography for angina pectoris-like symptoms, and coronary artery disease was excluded in the coronar angiogram. Patients with diseases that could interfere with analysis, including any kind of infections, malignancies, autoimmune diseases, hyperthyroidism, or medication with immunosuppressive agents, were excluded. The study protocol was performed in accordance with the Declaration of Helsinki and was approved by the local ethics committee. All patients provided written informed consent to participate in the study before enrollment. The present study is a substudy of previously published data that analyzed the role of myeloid dendritic cell precursors (mDCP) and plasmacytoid dendritic cell precursors (pDCP) in patients with AMI.[17] Data have now been complemented with a detailed analysis of IL-16 levels and its correlation with inflammatory cells and other clinical and serum parameters.
2.2. Blood samples
Blood samples were collected from a cubital vein using a clean venipuncture under controlled venous stasis within the first 24 hours after hospital admission. Venous blood was obtained using blood collection tubes for heparin plasma. The tubes were centrifuged within 20 minutes after blood withdrawal and the obtained plasma samples were frozen at −80°C until further measurements were conducted. Routine blood analyses were performed immediately after blood withdrawal according to clinical standards at the Department of Clinical Chemistry at the University Hospital Jena. Analyses of high-sensitive troponin T (hsTnT) and N-terminal prohormone of brain natriuretic peptide (NT-proBNP) were measured on a COBAS 8000 device (Roche Diagnostics, Rotkreuz, Switzerland) using the Roche assays “Troponin T hs” and “proBNP II”, respectively.
2.3. Analysis of IL-16
Plasma levels of IL-16 were measured using a commercially available enzyme-linked immunosorbent assay (ELISA) kit (Duoset DY316; R&D Systems, Minneapolis, MN) according to the instructions supplied by the manufacturer. Plasma samples and standard protein were added to the wells and incubated for 2 hours. Then, plates were washed and a biotin-labeled secondary antibody was added to each well. Plates were incubated for another 2 hours at room temperature. Plates were washed once more and streptavidin–horseradish–peroxidase was added. A color reaction was achieved by adding tetramethylbenzidine (TMB; Sigma Aldrich, St. Louis, MO) to each well. Values of optical density were obtained by measurement at 450 nm on an ELISA reader (Bio-Rad Laboratories, Vienna, Austria).
2.4. Analysis of circulating dendritic cells precursors
Analysis of circulating myeloid, plasmacytoid and total dendritic cells precursors (DCPs) were performed using the Blood Dendritic Cell Enumeration kit (BDCA kit; Miltenyi Biotec, Bergisch Gladbach, Germany). Fluorescence-activated cell sorting (FACS) analysis was performed using FACSCalibur flow cytometer with CellQuest software (Becton Dickinson, Franklin Lakes, NJ). The detailed analysis technique is described in our previous study.[16]
2.5. Analysis of inflammatory cytokines
FACS was also utilized to screen the plasma of patients and controls for inflammatory cytokines (IL-2, -4, -5, -6, -10, -12, and TNFα). For this purpose, we used the BDTM Enhanced Sensitivity CBA (Cytometric Bead Array) Flex Set (BD Biosciences, Franklin Lakes, NJ). Analyses were performed according to the manufacturer's instructions. The manufacturer states a detection range of cytokine concentrations from 0.27 to 200 pg/mL. By using additional probes in the standard rows, it was possible to detect cytokine levels of even 0.01 pg/mL.
2.6. Immunohistochemical analysis
Myocardial specimens were collected from 12 patients with AMI (mean age 73 years, 75% male), who died during hospitalization, and from 10 accident victims serving as controls. The detailed immunohistochemical staining technique is described in our previous work.[16]
2.7. Statistical analysis
Statistical analysis was performed using SPSS (Version 22.0, SPSSS Inc., Armonk, NY) and MedCalc version 17.4.4 (MedCalc Software bvba, Ostend, Belgium). Data for nonparametric continuous variables were expressed as median ± interquartile range (IQR). Data for parametric continuous variables were expressed as mean ± standard error of the mean (SEM). A Kolmogorov–Smirnow test was performed for all variables to test whether the variables are parametric or nonparametric distributed. For nonparametric distributed variables the Kruskal–Wallis test was performed. Parametric distributed data were compared by one-way ANOVA test. Two-group comparison was performed with the Mann—Whitney U test or unpaired t test. Correlation analysis was performed using Spearman rank-order test. Receiver-operating characteristics (ROC) analysis was performed; area under the curve (AUC) and an optimal cutoff by means of the Youden Index were calculated. AUCs were compared according to Hanley and McNeil. A P value ≤.05 was considered to be statistically significant.
3. Results
3.1. Study population
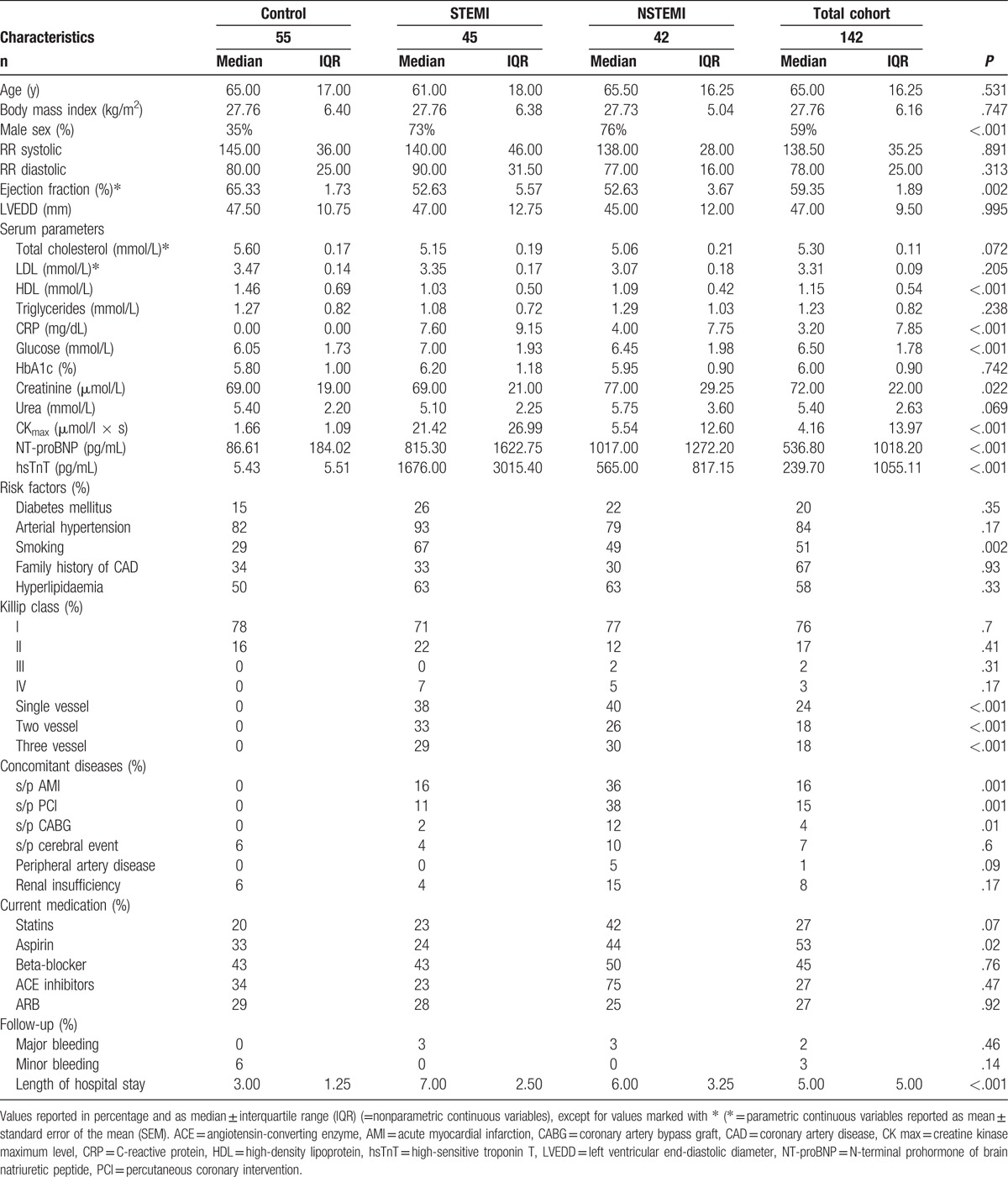
Baseline characteristics for all 142 participants in this study are summarized in Table 1. P values for comparison between the 3 subgroups are shown in supplemental Table 1. The control group consisted of 55 patients with exclusion of coronary artery disease by coronary angiography compared with 45 patients with STEMI 42 patients diagnosed with NSTEMI and. There were no statistically significant differences concerning age, atherogenic risk factors, and medications, except for aspirin. Incidence of previous AMI or percutaneous coronary intervention (PCI) or coronary artery bypass graft (CABG) was significantly different among groups. Compared with controls, male sex was dominant in the AMI population (STEMI: 73%, NSTEMI: 76%, control: 40%; P < .001, respectively). Ejection fraction, calculated with the Simpson formula by echocardiography, was lower in patients with STEMI (52.63 ± 5.57%) and NSTEMI (52.63 ± 3.67%) compared with the control group (65.33 ± 1.73%; P < .002). Values of peak creatine kinase (CK) in μmol/L × s (STEMI: 21.42 ± 26.99, NSTEMI: 5.54 ± 12.60 vs control: 1.66 ± 1.09; P < .001) group and peak hsTnT in ng/mL (STEMI: 1676.00 ± 3015.40, NSTEMI: 565.00 ± 817.15 vs control: 5.43 ± 5.51; P < .001) were significantly more increased in the AMI population compared with the control. The same encountered for NT-proBNP (STEMI: 815.30 ± 1622.75, NSTEMI: 1017.00 ± 1272.20 vs control: 86.61 ± 184.02; P = .001). Markers for inflammation, such as C-reactive protein (CRP) in mg/dL (STEMI: 7.60 ± 9.15, NSTEMI: 4.00 ± 7.75 vs control: 0.00 ± 0.00; P < .001) and such as leucocytes in s ×103/μL (STEMI: 9.50 ± 3.40, NSTEMI: 8.30 ± 3.4 vs control: 7.10 ± 1.80; P < .001) were significantly higher in the AMI groups compared with controls. The same encountered for granulocytes, monocytes, basophils, and thrombocytes.
Table 1.
Patient characteristics.
3.2. Analysis of circulating cytokines
Plasma concentrations of interleukins, DCPs, and blood cell counts in the 3 different patient groups are presented in Table 2 and supplemental Table 2. Median IL-16 serum level in pg/mL was significantly increased in STEMI (759.38 ± 471.54) and NSTEMI patients (677.77 ± 438.82) compared with the control group (500.45 ± 432.21, P < .002) (Fig. 1), as were plasma concentrations of IL-2 (P = .036), IL-6 (P <.001), IL-10 (P <.001), and IL-12p70 (P <.001). No significant difference between study groups was found for IL-4, IL-5, and tumor necrosis factor alpha. The IL-16 levels in patients with AMI were not affected by sex (P = .46). In both AMI groups compared with the control group a significant decrease in circulating DCPs in %/cells per μL was found (P = <.001).
Table 2.
Levels of interleukins, dentritic cell precursors, and blood cells for all three subgroups and total cohort.
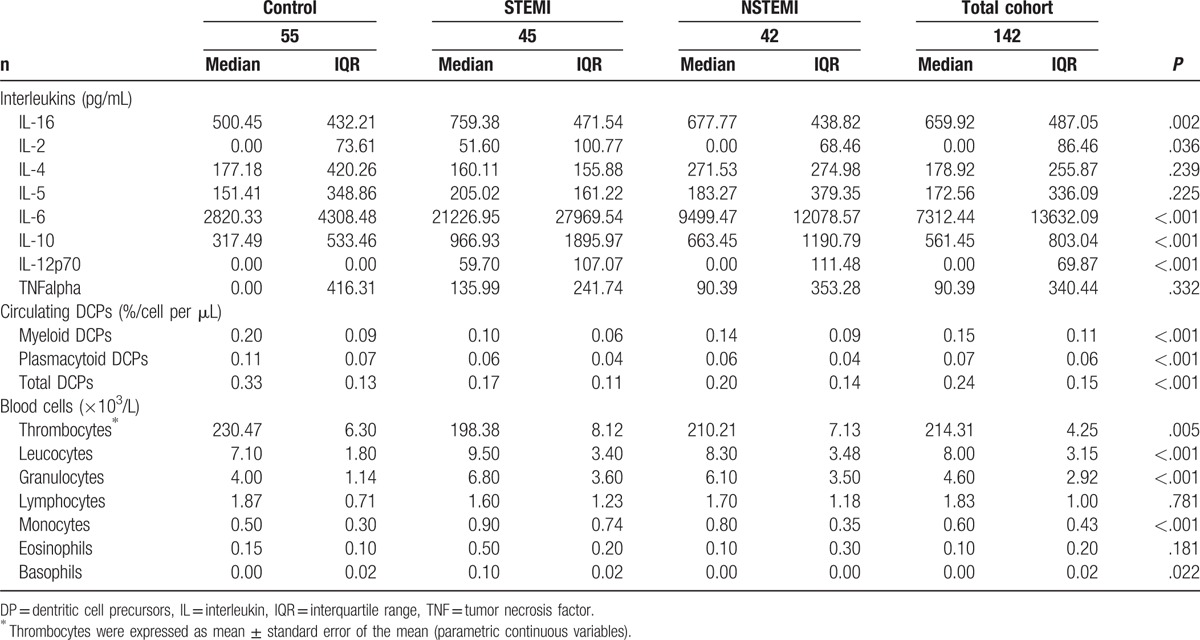
Figure 1.
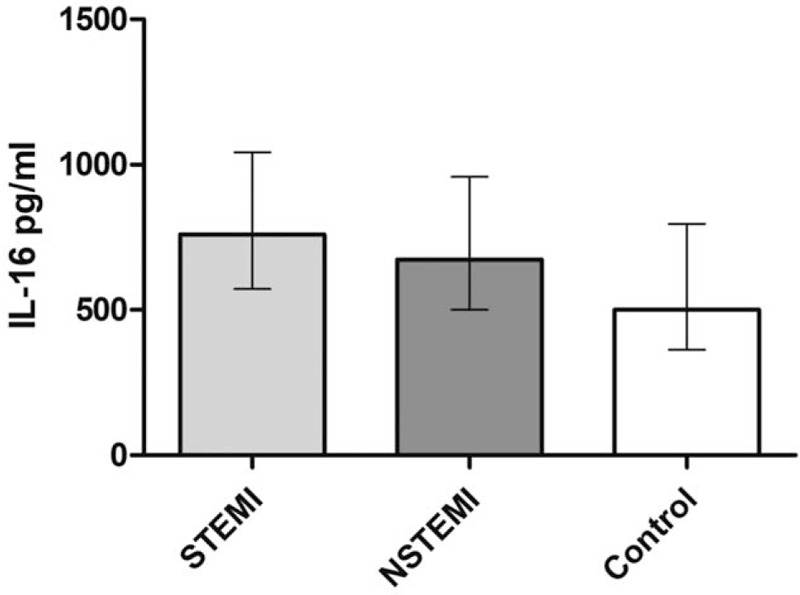
Serum concentrations of IL-16 in patients with (STEMI) and without ST-segment elevation myocardial infarction (NSTEMI) compared with a control group. Median IL-16 serum levels in pg/mL (±IQR) were significantly higher in the AMI population compared with the control group (P = .002).
3.3. Correlation of IL-16 with serum and clinical parameters
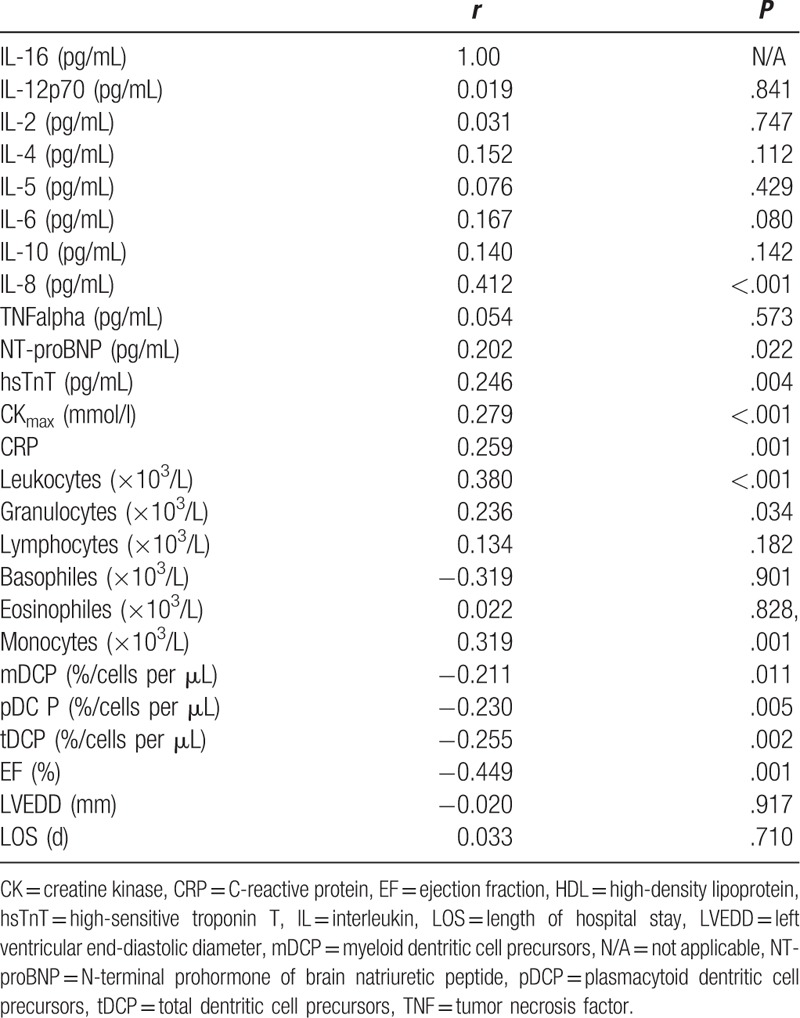
To further explore the association of IL-16 and cell concentrations and serum parameters, a detailed correlation analysis was performed (Table 3). A significant inverse correlation between IL-16 plasma levels and mDCP (r = –0.21, P = .01), pDCP (r = –0.23, P = .005), and tDCP (r = –0.26, P = .002) in % was observed. Notably, indicators for inflammatory status such as CRP (r = 0.26, P = .001) concentrations as well as leukocytes (r = 0.38, P < .001), granulocytes (r = 0.24, P = .034), and monocytes (r = 0.32, P = .001) were found to be significantly correlated with IL-16. Peak CK levels in mmol/L (r = 0.28, P < .001), hsTropT (r = 0.25, P = .004), pro-BNP (0.20, P = .02) were significantly correlated with IL-16 serum concentrations. Notably, a significant inverse correlation between ejection fraction in % and IL-16 was found (r = –45, P = .001). Besides IL-8 (r = 0.41, P < .001), no other interleukins showed a significant correlation with IL-16.
Table 3.
Correlation analysis of interleukin-16 with biochemical and clinical markers.
3.4. Diagnostic value for AMI of IL-16
AUC was 0.68 (95% confidence interval 0.59–0.75) and an optimal cutoff for detection of AMI for our study cohort was calculated by means of the Youden Index for IL-16 with 565.65 ng/mL and showed a 72% sensitivity and 62% specificity.
4. Discussion
In the present study, we demonstrated that IL-16 protein is elevated in patients with AMI compared with controls; a significant correlation between IL-16 and peripheral immune cells—producing IL-16, but also chemoattracted by IL-16—occurs in patients with STEMI/NSTEMI; an inverse correlation of IL-16 and DCPs in the plasma and significantly higher numbers of DCPs in the infarcted myocardium indicates a possible role of IL-16 in chemoattracting DCPs to infarcted myocardium; a significant correlation of IL-16 exists with inflammatory markers (leukocytes, CRP) and established biomarkers (hsTropT, CKmax, NT-proBNP) and clinical parameters (EF).
In the setting of AMI, a complex cascade of inflammatory processes plays a major role for reparation, scar formation, and remodeling of the infarcted tissue, and is involved in the development of heart failure.[4,6] In the interaction of cell apoptosis, recruitment or migration of inflammatory cells and cell repair, cytokines function as essential proinflammatory triggers. Cytokines are key mediators of the inflammatory response facilitating cell activation and migration and constitute ischemic injury mechanisms.[5,18] The present study aimed to expand knowledge about cytokine regulation with the main focus on the IL-16 axis in the setting of AMI. IL-16 has been linked to inflammatory diseases such as airway hyperresponsiveness,[12] Crohn disease,[13] or rheumatoid arthritis.[19] In one of our previous studies, we were able to demonstrate elevated IL-16 levels besides IL-12p40 levels in overweight adolescents, indicative of a state of permanent low-grade inflammation in such individuals.[20] Schwab et al demonstrated in 20 brains with focal cerebral infarctions a significant infiltration of IL-16 expressing immune cells such as granulocytes, CD8 +lymphocytes, and microglia and macrophages in the necrotic perilesional brain area.[21] We now extend these observations demonstrating significantly higher IL-16 levels in patients with NSTEMI and STEMI compared with controls, indicating a role in the inflammatory processes in AMI as well.
Previous studies have demonstrated significant roles of IL-6, IL-10, IL-12, IL-18 in patients with AMI.[22–25] In the present study, we also analyzed plasma concentrations of other cytokines and found in addition to IL-16 significantly differences between the AMI groups and control group for IL-2, IL-6, IL-10, IL12p70, but not for TNF-alpha. It has been shown that IL-16 is a growth factor for resting CD4+ cells, stimulating the expression of IL-6, IL-1β, and TNF-alpha.[21] One could speculate that the IL-16 axis might be a predominant proinflammatory signaling pathway in myocardial ischemia and that at the relatively early time point (within 24 hours after PCI) the immunologic response upon IL-16 stimulation is still ongoing and incomplete.
Underlying the inflammatory theory, in accordance with previous papers[26,27] in the presented study, CRP levels in the AMI cohort were significantly higher compared with controls and a significant correlation between CRP and IL-16 levels was shown. Furthermore, leucocytes significantly correlated with IL-16.
IL-16 is produced by not only T cells, mast cells, eosinophils, and fibroblasts, but also dendritic cells and epithelial cells. It functions as a potent chemoattractant for all peripheral immune cells including CD4+ monocytes, eosinophils, and dendritic cells, and exerts functional roles via binding to cell surface CD4 glycoprotein or CD4 independently for induction of all its bioactivities.[7,24,28,29] In the setting of AMI necrotic cardiomyocytes and damaged extracellular matrix release trigger the complement system and activate IL-1 signaling. Degranulated mast cells and activated endothelial cells and fibroblasts induce cytokines, such as IL-16.[4,18] Moreover, it was shown that apoptotic immune cells are also a direct source of IL-16. In our previous studies, we found that IL-16 secretion can be augmented via various stimuli of apoptosis such as irradiation and activation-induced cell death.[30,31] In the context of AMI, it has been shown that mononuclear cells evidence higher rates of apoptosis[32] which allows speculation about different IL-16 sources. We therefore propose the following hypothetical causal mechanism: apoptosis and necrosis of cardiomyocytes within the ischemic myocardiums attracts inflammatory cells and triggers mechanisms of local inflammation. During the hours after AMI, fractions of these inflammatory cells become apoptotic and release IL-16. In this early inflammatory process, IL-16 could function as a major gear by further triggering the synthesis of a whole spectrum of further inflammatory factors that might maintain a state of local and also systemic inflammation.
In addition, in the inflammatory pathways in AMI, DCPs were shown to play a crucial role. We were able to demonstrate in a previous work that in AMI circulating DCPs were significantly reduced and hypothesized an enhanced recruitment in the infarcted myocardium.[17] IL-16 has been found to play an important role in the cross-talk between DCPs and T cells. Kaser et al identified IL-16 as a substantial chemoattractant for DCPs and noted that DCPs themselves constitutively synthesize IL-16 in cell cultures.[8] Strikingly, in accordance to this, in our AMI cohort circulating mDCPs, pDCPs, and tDCPs were significantly reduced compared with the control group and showed a significant inverse correlation to IL-16 levels. The phenomenon was accompanied by the fact that DCPs showed an enhanced appearance in the infarcted myocardium stains compared with unaffected myocardial tissue. Furthermore, as also previously described, significantly higher numbers of macrophages and T cells were detected in the infarcted tissue corresponding to the inflammatory hypothesis of myocardial damage.[17]
Cardiac troponins, especially new generation of hsTropT, CK, and pro-BNP markers are widely used for AMI diagnosis and post-MI risk stratification.[14,33] All were increased in patients with AMI and correlated significantly with IL-16. IL-16 showed no superiority to these established markers, but might, possibly, evidence an incremental prognostic value in circumstances when hsTnT is less specific, such as in patients with renal failure, cardiac decompensation, or tachycardia. It may help identify patients at high risk for death or developing heart failure enabling more individualized treatment strategies for these patients to improve outcomes.
Furthermore, our findings offer a better understanding of the complex pathobiological cascade in AMI and the results are in accordance with the theory of a major inflammatory component in the phathophysiologic changes after AMI. To date, clinically applicable therapeutic strategies to mitigate the inflammatory processes in the setting of AMI are lacking. IL-16, as a proinflammatory and chemokine-like chemoattractant, might constitute an interesting target for future therapeutic approaches in patients with AMI. It has been shown that its expression can be downregulated by dexamethasone.[7,34] However, the detection of other anti-inflammatory and more selective agents, without the adverse cardiovascular safety profile of corticosteroids, is essential.[35] Determination of IL-16 may furthermore be useful when monitoring future anti-inflammatory therapies.
5. Limitations
Based on our results we cannot definitively distinguish the main origin of IL-16, as we only analyzed plasma samples and not cell or tissue samples of study subjects. Further studies are needed to decode mechanisms leading to the expression of IL-16 and other inflammatory factors in the context of AMI. It would be of great interest to identify other cell types responsible for the proinflammatory response in AMI patients, to attain a better understanding of pathophysiological mechanisms induced by inflammation that might aid the development of new treatment strategies to counteract disease progression. This was a single-center retrospective investigation, and the relatively small sample size might lead to selection bias. We want to underline the principal problem of multiple testing: the main emphasis of our study was to compare IL-16 levels of patients with and without AMI. Still, as we compared several other parameters, the general problem of type I error cannot be excluded. U and T test P values are reported in supplemental Tables 1 and 2. Follow-up data regarding adverse clinical events are lacking precluding an investigation of prognostic value for IL-16.
6. Conclusions
IL-16 might play a crucial role in the understanding of inflammatory processes occurring in patients after AMI. Infalmmatory cells—chemoattracted by IL-16, but also synthesizing IL-16 and inflammatory markers (CRP, leukocytes)—showed a significant correlation with IL-16 levels in infarct patients underlining its role as chemokine in AMI.
Acknowledgments
We thank Kristen Kopp for her help in language editing of the manuscript.
Supplementary Material
Footnotes
Abbreviations: AMI = acute myocardial infarction, AUC = area under the curve, CABG = coronary artery bypass graft, CK = creatine kinase, CRP = C-reactive protein, DCP = dentritic cell precursors, EF = ejection fraction, FACS = fluorescence-activated cell sorting, hsTnT = high-sensitive troponin T, IL = interleukin, mDCP = myeloid dentritic cell precursors, NSTEMI = non-ST-segment elevation myocardial infarction, NT-proBNP = N-terminal prohormone of brain natriuretic peptide, PCI = percutaneous coronary intervention, pDCP = plasmacytoid dentritic cell precursors, SD = standard deviation, STEMI = ST-segment elevation myocardial infarction, tDCP = total dentritic cell precursors, TNF = tumor necrosis factor.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Nian M, Lee P, Khaper N, et al. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res 2004;94:1543–53. [DOI] [PubMed] [Google Scholar]
- [2].Anversa P, Nadal-Ginard B. Myocyte renewal and ventricular remodelling. Nature 2002;415:240–3. [DOI] [PubMed] [Google Scholar]
- [3].Sun M, Opavsky MA, Stewart DJ, et al. Temporal response and localization of integrins beta1 and beta3 in the heart after myocardial infarction: regulation by cytokines. Circulation 2003;107:1046–52. [DOI] [PubMed] [Google Scholar]
- [4].Frangogiannis NG. Inflammation in cardiac injury, repair and regeneration. Curr Opin Cardiol 2015;30:240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res 2015;116:354–67. [DOI] [PubMed] [Google Scholar]
- [6].Laskarin G, Zaputovic L, Persic V, et al. Harmful immune reactions during acute myocardial infarction. Med Hypotheses 2012;78:703–6. [DOI] [PubMed] [Google Scholar]
- [7].Cruikshank WW, Kornfeld H, Center DM. Interleukin-16. J Leukoc Biol 2000;67:757–66. [DOI] [PubMed] [Google Scholar]
- [8].Kaser A, Dunzendorfer S, Offner FA, et al. A role for IL-16 in the cross-talk between dendritic cells and T cells. J Immunol 1999;163:3232–8. [PubMed] [Google Scholar]
- [9].Liu Y, Cruikshank WW, O’Loughlin T, et al. Identification of a CD4 domain required for interleukin-16 binding and lymphocyte activation. J Biol Chem 1999;274:23387–95. [DOI] [PubMed] [Google Scholar]
- [10].Liu C, Mills J, Dixon K, et al. IL-16 signaling specifically induces STAT6 activation through CD4. Cytokine 2007;38:145–50. [DOI] [PubMed] [Google Scholar]
- [11].Mathy NL, Scheuer W, Lanzendörfer M, et al. Interleukin-16 stimulates the expression and production of pro-inflammatory cytokines by human monocytes. Immunology 2000;100:63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Teran LM, Ramirez-Jimenez F, Soid-Raggi G, et al. Interleukin 16 andCCL17/thymus and activation-regulated chemokine in patients with aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol 2017;118:191–6. [DOI] [PubMed] [Google Scholar]
- [13].Glass WG, Sarisky RT, Vecchio AM. Not-so-sweet sixteen: the role of IL-16 in infectious and immune-mediated inflammatory diseases. J Interferon Cytokine Res 2006;26:511–20. Review. [DOI] [PubMed] [Google Scholar]
- [14].Feistritzer HJ, Klug G, Reinstadler SJ, et al. Novel biomarkers predicting cardiac function after acute myocardial infarction. Br Med Bull 2016;119:63–74. [DOI] [PubMed] [Google Scholar]
- [15].Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J 2012;33:2551–67. [DOI] [PubMed] [Google Scholar]
- [16].Roffi M, Patrono C, Collet JP, et al. Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J 2016;37:267–315. [DOI] [PubMed] [Google Scholar]
- [17].Kretzschmar D, Betge S, Windisch A, et al. Recruitment of circulating dendritic cell precursors into the infracted myocardium and pro-inflammatory response in acute myocardial infarction. Clin Sci (Lond) 2012;123:387–98. [DOI] [PubMed] [Google Scholar]
- [18].Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest 2013;43:986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Murota A, Suzuki K, Kassai Y, et al. Serum proteomic analysis identifies interleukin 16 as a biomarker for clinical response during early treatment of rheumatoid arthritis. Cytokine 2016;78:87–93. [DOI] [PubMed] [Google Scholar]
- [20].Lichtenauer M, Franz M, Fritzenwanger M, et al. Elevated plasma levels of interleukin-12p40 and interleukin-16 in overweight adolescents. Biomed Res Int 2015;940910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schwab JM, Nguyen TD, Meyermann R, et al. Human focal cerebral infarctions induce differential lesional interleukin-16 (IL-16) expression confined to infiltrating granulocytes, CD8+ T-lymphocytes and activated microglia/macrophages. J Neuroimmunol 2001;114:232–41. [DOI] [PubMed] [Google Scholar]
- [22].Biswas S, Ghoshal PK, Mandal SC, et al. Relation of anti- to pro-inflammatory cytokine ratios with acute myocardial infarction. Korean J Intern Med 2010;25:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zykov MV, Barbarash OL, Kashtalap VV, et al. Interleukin-12 serum level has prognostic value in patients with ST-segment elevation myocardial infarction. Heart Lung 2016;45:336–40. [DOI] [PubMed] [Google Scholar]
- [24].Mathy NL, Bannert N, Norley SG, et al. Cutting edge: CD4 is not required for the functional activity of IL-16. J Immunol 2000;164:4429–32. [DOI] [PubMed] [Google Scholar]
- [25].Opstad TB, Arnesen H, Pettersen AÅ, et al. Combined elevated levels of the proinflammatory cytokines IL-18 and IL-12 are associated with clinical events in patients with coronary artery disease: an observational study. Metab Syndr Relat Disord 2016;14:242–8. [DOI] [PubMed] [Google Scholar]
- [26].Ridker PM, Hennekens CH, Buring JE, et al. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000;342:836–43. [DOI] [PubMed] [Google Scholar]
- [27].Kaptoge S, Seshasai SR, Gao P, et al. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Eur Heart J 2014;35:578–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rider P, Voronov E, Dinarello CA, et al. Alarmins: feel the stress. J Immunol 2017;198:1395–402. [DOI] [PubMed] [Google Scholar]
- [29].Elssner A, Doseff AI, Duncan M, et al. IL-16 is constitutively present in peripheral blood monocytes and spontaneously released during apoptosis. J Immunol 2004;172:7721–5. [DOI] [PubMed] [Google Scholar]
- [30].Lichtenauer M, Mildner M, Hoetzenecker K, et al. Secretome of apoptotic peripheral blood cells (APOSEC) confers cytoprotection to cardiomyocytes and inhibits tissue remodelling after acute myocardial infarction: a preclinical study. Basic Res Cardiol 2011;106:1283–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lichtenauer M, Mildner M, Werba G, et al. Anti-thymocyte globulin induceets neoangiogenesis and preserves cardiac function after experimental myocardial infarction. PLoS One 2012;7:e52101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Konstantinova EV, Khomyakova NF, Konstantinova NA, et al. Relationship between apoptosis and expression of heat shock proteins in peripheral blood lymphocytes of patients with myocardial infarction. Bull Exp Biol Med 2011;150:682–4. [DOI] [PubMed] [Google Scholar]
- [33].Mueller C, Giannitsis E, Christ M, et al. TRAPID-AMI Investigators. Multicenter evaluation of a 0-hour/1-hour algorithm in the diagnosis of myocardial infarction with high-sensitivity cardiac troponin T. Ann Emerg Med 2016;68:76–87.e4. [DOI] [PubMed] [Google Scholar]
- [34].Zhang Z, Fauser U, Schluesener HJ. Early attenuation of lesional interleukin-16 up-regulation by dexamethasone and FTY720 in experimental traumatic brain injury. Neuropathol Appl Neurobiol 2008;34:330–9. [DOI] [PubMed] [Google Scholar]
- [35].Ridker PM. From C-reactive protein to interleukin-6 to interleukin-1: moving upstream to identify novel targets for atheroprotection. Circ Res 2016;118:145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.