

Abstract
Interferon (IFN) regulatory factors (IRFs) have crucial roles in immune regulation and oncogenesis. We have recently shown that IRF4 is activated through c-Src-mediated tyrosine phosphorylation in virus-transformed cells. However, the intracellular signaling pathway triggering Src activation of IRF4 remains unknown. In this study, we provide evidence that Epstein–Barr virus (EBV) latent membrane protein 1 (LMP1) promotes IRF4 phosphorylation and markedly stimulates IRF4 transcriptional activity, and that Src mediates LMP1 activation of IRF4. As to more precise mechanism, we show that LMP1 physically interacts with c-Src, and the phosphatidylinositol 3 kinase (PI3K) subunit P85 mediates their interaction. Depletion of P85 by P85-specific short hairpin RNAs disrupts their interaction and diminishes IRF4 phosphorylation in EBV-transformed cells. Furthermore, we show that Src is upstream of PI3K for activation of both IRF4 and Akt. In turn, inhibition of PI3K kinase activity by the PI3K-speicfic inhibitor LY294002 impairs Src activity. Our results show that LMP1 signaling is responsible for IRF4 activation, and further characterize the IRF4 regulatory network that is a promising therapeutic target for specific hematological malignancies.
INTRODUCTION
Epstein–Barr virus (EBV) latency programs are manifest as a large spectrum of lymphomas, and are associated with > 50% of AIDS-related lymphomas, including diffuse large B-cell lymphoma, post-transplant lymphoproliferative disease, Hodgkin’s lymphoma and non-Hodgkin’s lymphoma.1–4 EBV is also the etiological pathogen of Burkitt’s lymphoma, Hodgkin’s lymphoma, nasopharyngeal carcinoma and infectious mononucleosis in immunocompetent individuals.3 EBV latent infection induces expression of interferon (IFN) regulatory factor 2 (IRF2), −4 and −7, three members with oncogenic properties in the IRF family of transcription factors.5 However, their regulation and roles in EBV oncogenesis are largely unknown.6
The EBV principal oncoprotein latent membrane protein 1 (LMP1) is a pleiotropic factor that can cause cell transformation in vitro, as well as oncogenesis in transgenic mice.7 LMP1 is a member of the tumor necrosis factor receptor superfamily, and is similar to with CD40 and Toll-like receptors in signal transduction, including the activation of NFκB, mitogen-activated protein kinases and phosphatidylinositol 3 kinase (PI3K)/Akt. It has been well documented that all of these downstream pathways contribute to LMP1 oncogenesis.8–11 Along with others, we have shown compelling evidence that LMP1 also activates IRF7.12–14
The lymphocyte-specific factor IRF4 is overexpressed in EBV-infected cells,15–18 multiple myeloma,19,20 as well as in human T-cell leukemia virus-1-infected cell lines and associated adult T-cell leukemia.21–24 It is now understood that IRF4 is induced by LMP1,15,16,18 and stabilized by Epstein-Barr Virus Nuclear Antigen 3C,25 in EBV-infected cells. Overexpression of IRF4 is a hallmark of the Activated B-Cell like type of diffuse large B-cell lymphoma and multiple myeloma.26,27 Genetic alterations of IRF4 have been found in MM, peripheral T-cell lymphomas28 and chronic lymphocytic leukemia.19,29 Recently, IRF4 has been shown to be expressed in all LMP1-driven tumors in mice.30 In clinical practice, IRF4 is an important prognostic and diagnostic marker for different hematological malignancies.27,31–33 These lines of evidence strongly highlight the importance of IRF4 in hematological malignancies. However, the main body of these data has come from clinical observations. The lack of necessary molecular and mechanistic studies is a major barrier that prevents the development of therapeutic strategies to target the IRF4 network for treatment.
IRF4 is a quintessential ‘context-dependent’ factor that regulates unique targets in different contexts in cooperation with different co-regulators.32,34 We have identified B cell Integration Cluster (BIC), the gene encoding miR-155, as the first transcriptional target induced by IRF4 in virus-transformed cells.6 More recently, we have further identified IRF4-associated unique molecular signatures in a panel of hematological malignancies.35 Furthermore, we have recently demonstrated that IRF4 is activated through Src-mediated tyrosine phosphorylation in EBV-transformed cells.36 Correspondingly, Src is functional in these cells.36 These findings have prompted us to further identify the molecular mechanism underlying IRF4 activation in the context of EBV infection.
In this study, we aimed to identify the cellular signaling pathway responsible for IRF4 activation by c-Src in the context of EBV infection. We show that LMP1 interacts with c-Src via P85 and triggers c-Src-mediated IRF4 tyrosine phosphorylation and transcriptional activity, and a positive regulatory circuit between P83 and c-Src is required for both downstream Akt and IRF4 activation.
RESULTS
LMP1 stimulates IRF4 tyrosine phosphorylation and transcriptional activity
We initially tested the ability of LMP1 to promote IRF4 phosphorylation. 293 cells were transfected with the constructs shown in Figure 1a, and the cell lysates were used for immunoblotting with specific antibodies. c-Src was used as a positive control. As shown in Figure 1a, in the presence of LMP1, a specific band representing phosphorylated IRF4 was detected with the phospho-IRF4(Y121/124) antibody, indicating that LMP1 promotes IRF4 tyrosine phosphorylation.
Figure 1.
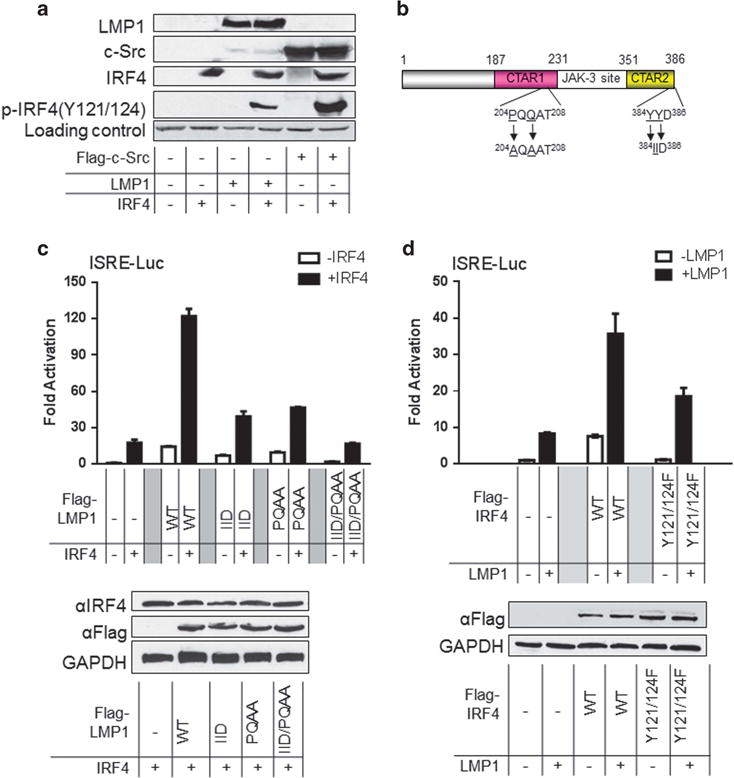
LMP1 promotes IRF4 transcriptional activity and phosphorylation. (a) LMP1 stimulates IRF4 phosphorylation. 293 cells in 60-mm dishes were transfected with 1 μg LMP1 (or Flag-c-Src) and 1 μg IRF4 expression plasmids. Cells were collected 48 h post-transfection and analyzed for IRF4 phosphorylation with p-IRF4(Y121/124). (b) A diagram showing the LMP1 point mutants. (c) and (d) LMP1 promotes IRF4 transcriptional activity. 293 cells in 24-well plates were transfected with 50 ng IRF4 or the mutant IRF4(Y121/124 F) and 50 ng LMP1 or its mutants expression plasmids, 40 ng pGL3/Interferon-Stimulated Responsive Element (ISRE)-Luc and 10 ng Renilla. Dual luciferase assay was performed. Results are the averages ± s.e. of duplicates. Representative results from at least three independent experiments are shown. The ability of the vector control to activate the promoter construct was set to 1.
We then assayed the ability of LMP1 to potentiate IRF4-mediated transactivation. In addition to the wild-type LMP1, a panel of LMP1 point mutants were also used: LMP1-PQAA, in which C-terminal activation region 1 (CTAR1) is disabled, LMP1-IID, in which (CTAR2) is disabled, and LMP1-IID/PQAA, in which both CTAR1 and -2 are disabled (Figure 1b). As shown in Figure 1c, LMP1 or IRF4 alone only slightly activate the Interferon-Stimulated Responsive Element (ISRE) promoter, but together they markedly activate it, up to 120-fold, when compared with the vector control. Mutation in either CTAR1 or CTAR2 or both significantly reduces the ability of LMP1 to transactivate IRF4. These results indicate that LMP1 potentiates IRF4-mediated transactivation, and both CTAR1 and -2 contribute.
We also used the IRF4 mutant, IRF4(Y121/124F) that is deficient in response to Src,36 to test its ability to respond to LMP1. As shown in Figure 1d, activation of IRF4(Y121/124F) by LMP1 is significantly impaired, compared with wild-type IRF4. As IRF4 (Y121/124) is still activated by LMP1, other sites on IRF4 may contribute to its activation by LMP1.
Collectively, these results indicate that LMP1 signaling can promote IRF4 phosphorylation and activation.
LMP1 interacts with c-Src in vitro and in vivo
As a tyrosine protein kinase, Src is normally maintained in an inactive form and is activated by autophosphorylation of Y418 or dephosphorylation of Y527.37 We have previously provided strong evidence that c-Src activates IRF4, and Src(Y418) is phosphorylated and therefore Src is active, in EBV-transformed cells.36 We have also shown that the total endogenous Src protein levels and activity correlate with the LMP1 protein levels in EBV latency.36 These observations suggest that, in EBV latency, LMP1 can stimulate Src activation, which then leads to IRF4 phosphorylation.
To further explore the possibility that Src is involved in LMP1 activation of IRF4, we tested if LMP1 physically interacts with Src. We transfected 293T cells with HA-LMP1 and Flag-c-Src (or Flag-IRF4). Cell lysates were subjected to immunoprecipitation and then probed with HA antibody. As shown in Figure 2a, LMP1 clearly interacts with c-Src, but not with IRF4. We further demonstrated endogenous LMP1 and c-Src physically interact in EBV-transformed cell lines, including IB4, SavIII and JiJoye via IP (Figure 2b). We conclude that LMP1 physically interacts with Src.
Figure 2.
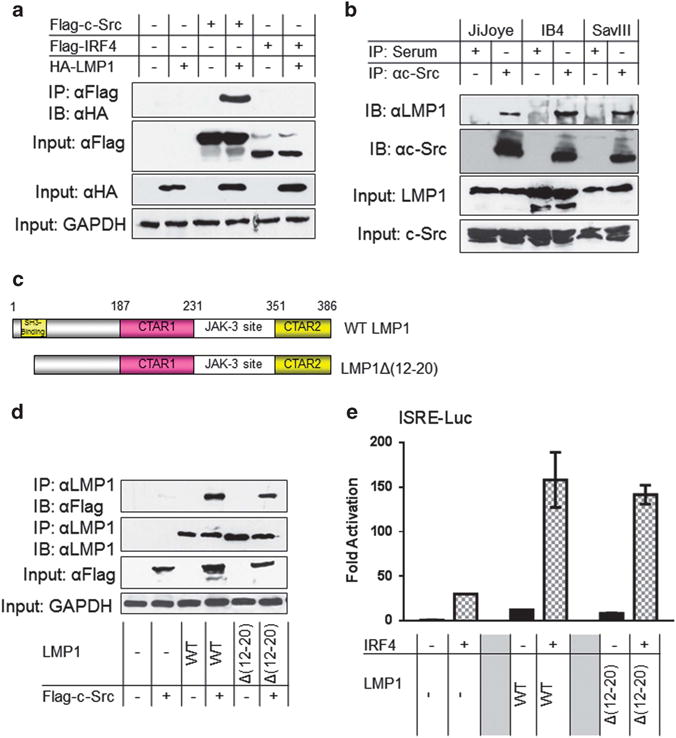
LMP1 interacts with Src. (a) LMP1 interacts with Src in vitro. 293T cells in 60-mm dishes were transfected with 1 μg HA-LMP1 (or Flag-c-Src) and 1 μg Flag-IRF4 expression plasmids. Cells were collected 48 h post-transfection and then subjected to immunoprecipitation with the Flag antibody clone M2. Beads were washed extensively and then subjected to immunoblotting with the HA antibody clone HA-7. Inputs (5%) were subjected to immunoblotting with the indicated antibodies. (b) Endogenous LMP1 and Src interact in EBV latency. Cell lysates (approximately 1 mg of total proteins in each sample) prepared from JiJoye, IB4 and SavIII cells were subjected to immunoprecipitation with the c-Src antibody clone N16 (or rabbit serum as control). Beads were washed extensively and probed with LMP1 and Src antibodies. Inputs (5%) were also probed with these antibodies. (c) A diagram showing the LMP1 deletion mutant LMP1Δ(12–20). (d) The LMP1 SH3-binding region is not required for Src interaction. 293T cells in 60-mm dishes were transfected with 1 μg LMP1 or LMP1Δ(12–20) and 1 μg Flag-c-Src expression plasmids. Cells were collected 48 h post-transfection and then subjected to immunoprecipitation with the LMP1 antibody clone CS1-4. Beads were washed extensively and then subjected to immunoblotting with the Flag antibody clone M2 and the LMP1 antibody. Inputs (5% of total lysates) were subjected to immunoblotting with indicated antibodies. (e) The LMP1 SH3-binding region is not required for IRF4 activation. The 293 cells in 24-well plates were transfected with 50 ng IRF4 and 50 ng LMP1 (or LMP1Δ(12–20)) expression plasmids, 40 ng pGL3/IFNβ-Luc and 10 ng Renilla. Dual luciferase assay was performed. Results are the averages ± s.e. of duplicates. Representative results from at least three independent experiments are shown. The ability of the vector control to activate the promoter construct was set to 1.
LMP1 has a SH3-binding region located in the N-terminal first 25 aa;38 whereas the Src kinase family contains an SH3 domain. We therefore then postulated that LMP1 interaction with Src is mediated by its SH3-binding region. To test this possibility, we performed IP with the use of LMP1Δ(12–20), an LMP1 deletion mutant in which the SH3-binding region is deleted (Figure 2c).38 Surprisingly, our results show that deletion of LMP1 SH3-binding region did not affect its ability to bind to c-Src (Figure 2d). Correspondingly, LMP1Δ(12–20) has a similar capability to the wild-type LMP1 in activating IRF4 transcription (Figure 2e). These results show that the LMP1 SH3-binding region is responsible for neither Src binding nor IRF4 activation.
Taken together, LMP1 interacts with c-Src, but the N-terminal SH3-binding motif is neither responsible for their interaction, nor for IRF4 activation.
P85 mediates LMP1 and Src interaction
It has been shown that LMP1 CTAR1 directly interacts with P85, the regulatory subunit of PI3K,39 and c-Src binds to P85 through their SH3 domains.40 Thus, we speculated that P85 mediates LMP1/Src interaction. To test this, we first confirmed the interaction between LMP1 and P85 in EBV-transformed cells (Figure 3a). Then, we determined whether LMP1 CTAR1, the region responsible for P85 interaction,39 is also responsible for c-Src interaction. To this end, a panel of LMP1 deletion mutants, including CTAR1, CTAR2 and double deletion (DD), were used for IP to assess their abilities to interact with c-Src. As expected, LMP1 CTAR1 is also responsible for c-Src interaction (Figure 3b).
Figure 3.
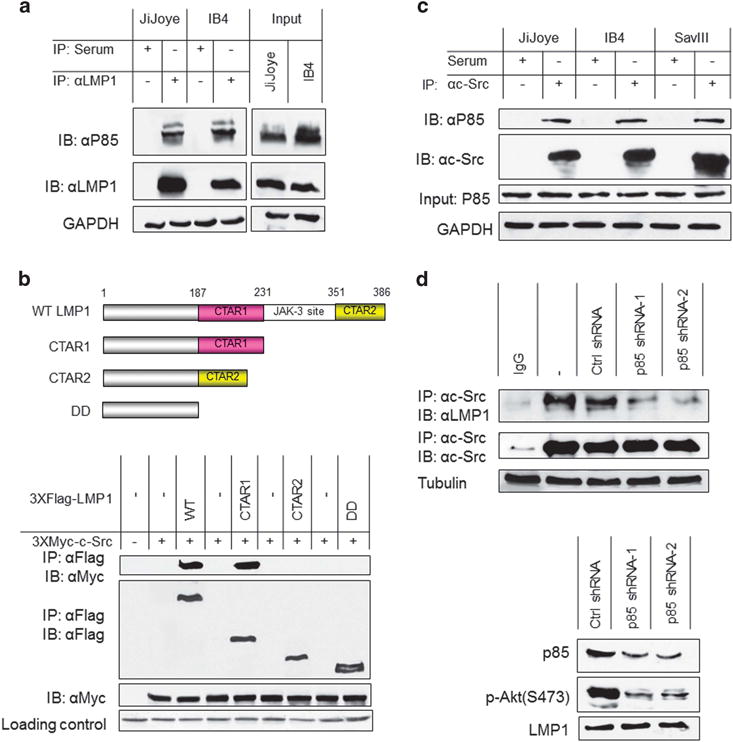
P85 mediates LMP1 and Src interaction. (a) Cell lysates (approximately 1 mg of total proteins in each sample) prepared from JiJoye and IB4 cells were subjected to immunoprecipitation with the LMP1 antibody clone CS1-4 (or mouse serum as control). Beads were washed extensively and probed with P85 and LMP1 antibodies. Inputs (5%) were also probed with these antibodies. (b) LMP1 CTAR1 is responsible for its interaction with Src. 293T cells in 60-mm dishes were transfected with 1 μg 3XMyc-Src and 1 μg Flag-LMP1 or its deletion mutants as indicated. Cells were collected 48 h post-transfection and then subjected to immunoprecipitation with 1.5 μg Myc antibody 9E10 (Roche). Beads were washed extensively and then subjected to immunoblotting with the Flag antibody clone M2 and Myc antibody 9E10. Loading control is Ponceau S staining. Upper panel shows a diagram showing the LMP1 protein structure. (c) Endogenous P85 and Src interact in EBV latency. Cell lysates (approximately 1 mg of total proteins in each sample) prepared from JiJoye, IB4 and SavIII cells were subjected to immunoprecipitation with the c-Src antibody clone N16 (or rabbit serum as control). Beads were washed extensively and probed with P85 and Src antibodies. (d) P85 depletion disrupts the interaction between LMP1 and Src. IB4 cells were infected with retrovirus expressing P85-specific shRNAs (or control). Stable transfectants were selected with 1 μg/ml puromycin for 2 weeks. shRNA expression was induced by 1 μg/ml doxycycline for 3 days. Knockdown efficiency is shown in the lower panel. Cells were then harvested and subjected to IP with the Src antibody clone N16, and then probed with the LMP1 antibody clone CS1-4. Representative results from at least three independent experiments are shown.
Furthermore, we show that endogenous Src and P85 interact in EBV-transformed cells (Figure 3c). More importantly, depletion of endogenous by short hairpin RNA (shRNA) in EBV-transformed cells disrupts the interaction between LMP1 and Src (Figure 3d). However, depletion of p110, the catalytic subunit of PI3K, had no detectable effect on LMP1/Src interaction (data not shown). These results indicate that P85 specifically mediates LMP1 interaction with and activation of Src.
Taken together, these results support our model that LMP1 CTAR1 recruits c-Src via P85.
PI3K and Src are required for LMP1 activation of IRF4
As LMP1 interacts with Src, we then assessed if Src activity is required for LMP1 activation of IRF4. To this end, we performed promoter–reporter assays with the use of the Src-specific inhibitor pyrazolo-[2,3-d]pyrimidine 2 (PP2), which we have shown to block Src(Y418) phosphorylation.36 Our results show that PP2 markedly decreases (up to 90%) IRF4 transcriptional activity stimulated by LMP1 (Figure 4a), indicating that Src activity is required for IRF4 transcriptional activity stimulated by LMP1.
Figure 4.
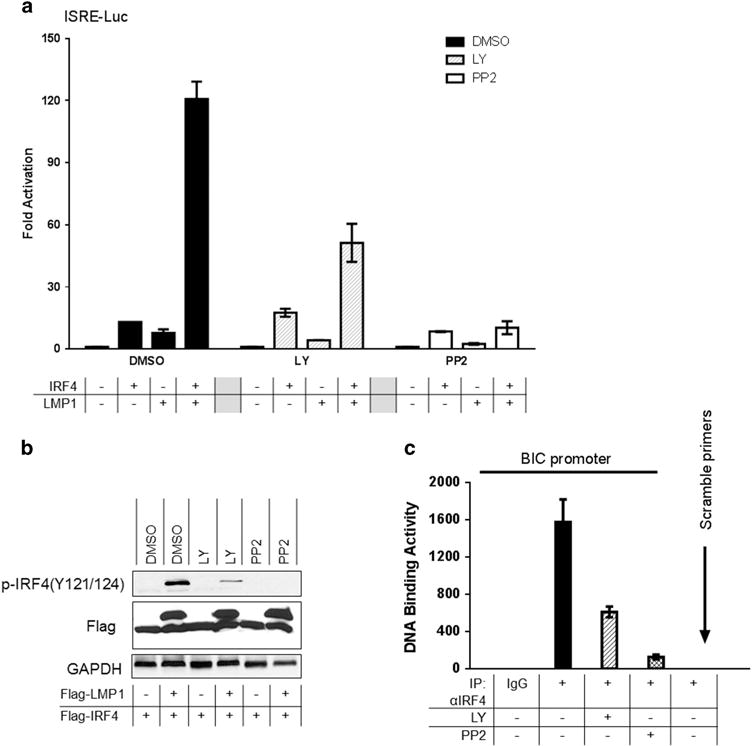
PI3K and Src are required for LMP1 activation of IRF4. (a) The PI3K-specific inhibitor LY294002, and the Src-specific inhibitor PP2, inhibit LMP1 activation of IRF4. 293 cells in 24-well plates were transfected with 50 ng IRF4 and 50 ng LMP1 expression plasmids, 40 ng pGL3/ISRE-Luc and 10 ng Renilla. Five hours later, cells were treated with the following inhibitors for 20 h: 20 μM LY or 40 μM PP2 (or DMSO as control). Dual luciferase assay was performed. Results are the averages ± s.e. of duplicates. Representative results from at least five independent experiments are shown. The ability of the vector control to activate the promoter construct in each treatment group was set to 1. (b) Inhibition of LMP1-promoted IRF4 phosphorylation by LY and PP2. 293T cells in six-well plates were transfected with 0.2 μg Flag-LMP1 and 0.2 μg Flag-IRF4 expression plasmids. Cells were then treated with 20 μM LY or 40 μM PP2 (or DMSO as control) for 20 h before harvest for immunoblotting with the indicated antibodies. (c) IB4 cells (2 × 106 for each treatment) were treated with 20 μM LY or 20 μM PP2 for 24 h. Cells were then subjected to ChIP assay. For each sample, DNA pellets were dissolved in 200 μl ddH2O, and 15 μl was used for quantitative PCR using the B cell Integration Cluster (BIC) and β-actin promoter primers. Representative results from at least three independent experiments are shown.
We further determined if PI3K activity is required for IRF4 activation. To this end, we first show that inhibition of PI3K activity by the PI3K-specific inhibitor LY294002 (designated as LY) significantly decreases IRF4 transcriptional activity stimulated by LMP1 (Figure 4a). Further, LY decreases IRF4 phosphorylation stimulated by LMP1 (Figure 4b), indicating that the PI3K pathway is required for IRF4 phosphorylation and functional activation. Autophosphorylation of Y418 or dephosphorylation of Y527 is required to switch Src from inactive to active form.37 Interestingly, the kinase CSK, which inactivates Src by phosphorylating Src (Y527), did not affect LMP1 activation of IRF4 in promoter–reporter assays (data not shown), implicating that Src(Y527) phosphorylation status is not involved in Src activation by LMP1.
We further explored whether blockage of the LMP1/PI3K pathway affects IRF4 DNA-binding activity. To this end, IB4 cells were treated with LY or PP2, and cells were then subjected to chromosome immunoprecipitation (ChIP). IRF4-DNA complexes were pulled down with an IRF4 antibody, and the recovered DNA fragments were subjected to real-time PCR amplification for a fragment containing ISRE of the BIC promoter.6 Results indicate that inhibition of PI3K or Src significantly reduces IRF4 DNA-binding activity (Figure 4c).
Together, our results indicate that both PI3K and Src downstream of LMP1 are involved in IRF4 activation.
A positive regulatory circuit between Src and PI3K for LMP1 activation of Akt and IRF4
There are reports showing that Src triggers PI3K activation in distinct biological contexts.40–42 A previous report has also shown that the levels of LMP1 and Src activity positively correlate in EBV latency, and a chimeric NGFP-LMP1 was able to activate the Src family member Fyn.43 As we show here that LMP1 interacts with c-Src via P85 (Figures 2 and 3), we were interested in examining the possibility that c-Src mediates LMP1 activation of Akt. First, we show that transient expression of wild-type Flag-c-Src or the constitutively active mutant Flag-c-Src(Y527F), but not the kinase-dead mutant Flag-c-Src(Y418F), triggers Akt activation in DG75 B cells (Figure 5a). Further, we inhibited the Src activity in EBV-transformed cells using the Src-specific inhibitor PP2, and results show that Src inactivation markedly impairs Akt activity (Figure 5b). These results disclose that Src is upstream of P85 and promotes LMP1 activation of the PI3K/Akt pathway.
Figure 5.
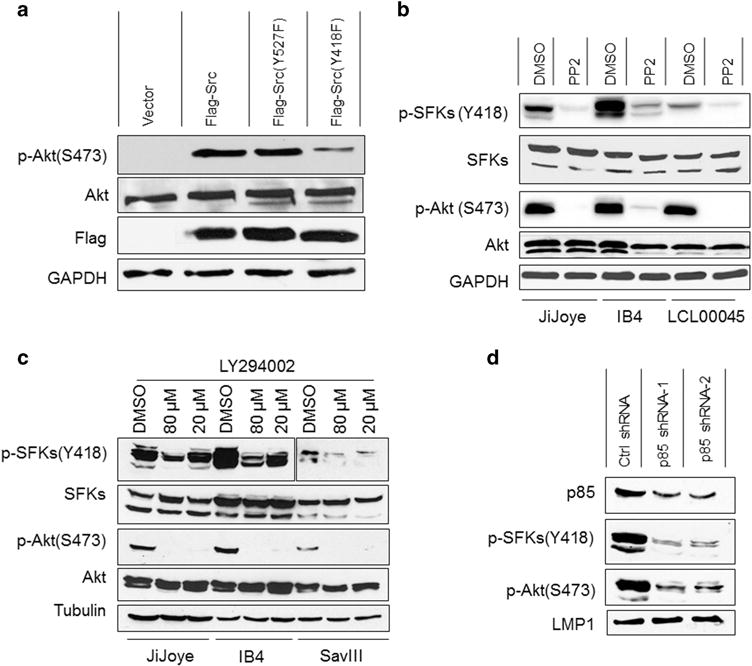
A positive circuit between Src and PI3K for LMP1 activation of Akt and IRF4. (a) Src triggers Akt phosphorylation and activation. DG75 cells (1 × 106) were transfected with Flag-Src or its mutants Flag-Src(Y527F) or Flag-Src(Y418F), or vector control. Cells were subjected to serum-free treatment for 24 h. Cells were then harvest for immunoblotting with the indicated antibodies. (b) Inhibition of Src activity abolishes Akt activity in EBV-transformed cells. JiJoye, IB4 and LCL00045 cells were treated with 20 μM Src-specific inhibitor PP2 for 24 h in serum-free medium before subjected to immunoblotting analysis. (c) LY inhibits endogenous Src Y418 phosphorylation. JiJoye, IB4 and SavIII were treated with 10 μM 20 or 80 μM LY (or DMSO control) for 48 h. Cell lysates were then prepared and probed with the indicated antibodies. (d) P85 depletion diminishes Src phosphorylation and activity. IB4 cells were infected with retrovirus expressing P85-specific shRNAs (or control). Stable transfectants were selected with 1 μg/ml puromycin for 2 weeks. shRNA expression was induced by 1 μg/ml doxycycline for 3 days. Cells were then harvested and subjected to immunoblotting with the indicated antibodies. Representative results from at least three independent experiments are shown.
We also evaluated whether, in turn, PI3K has any effect on Src activation. To this end, we first blocked the kinase activity of PI3K by LY in EBV-transformed cells, and the results show that Src Y418 phosphorylation is remarkably reduced at the LY concentration of 80 μM (Figure 5c). We further confirmed this finding using P85 shRNA, and results indicate that P85 has similar effect on Src activity (Figure 5d). Thus, PI3K and its kinase activity are involved in Src activation in the setting of EBV latent infection.
Together, these results (Figures 4 and 5) show that a positive regulatory circuit exists between Src and PI3K, and is required for the LMP1 signaling transduction to Akt and IRF4 activation.
The level of IRF4 phosphorylation correlates with LMP1 and is dampened by the Src-specific inhibitor PP2 in EBV+ cells
As the phospho-IRF4 (Y121/124) antibody we developed cannot detect endogenous IRF4 phosphorylation in EBV latency by immunoblotting, we attempted to use an alternative method, flow cytometry, to address this issue. To this end, two pairs of cell lines, SavI and SavIII, and P3HR1 and JiJoye, were subjected to flow analysis using p-IRF4(Y121/124) and LMP1 antibodies. SavI and P3HR1, which express neither LMP1 nor IRF4, were used to normalize the basal levels. As shown in Figure 6a, we detected a considerably higher population of SavIII cells with phosphorylated IRF4 (21.7% compared with 0.049% in SavI), which positively correlate with their LMP1 expression levels. Similar results were obtained from P3HR1 and JiJoye cells (Figure 6a).
Figure 6.
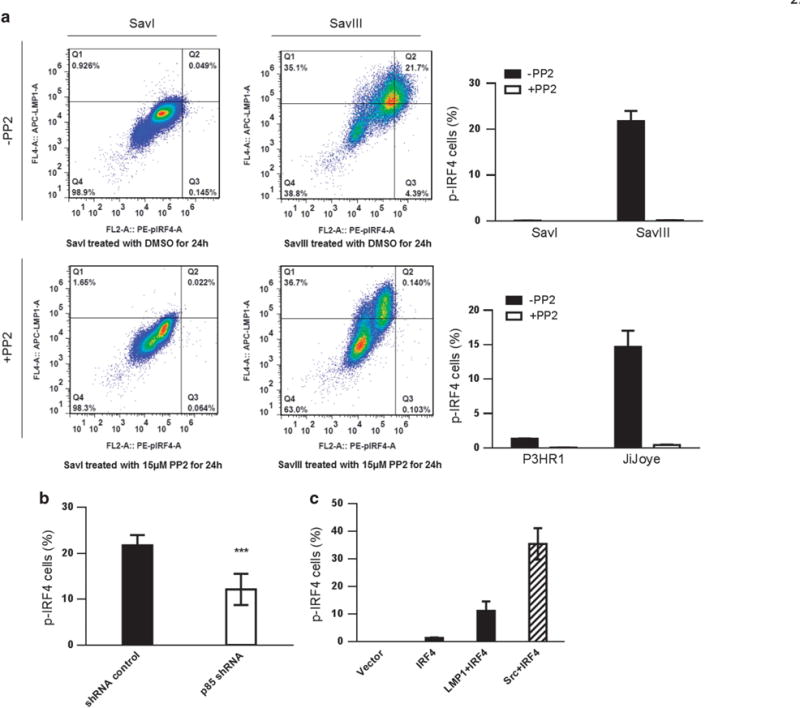
The level of IRF4 phosphorylation correlates with LMP1 and is dampened by the Src-specific inhibitor PP2 in EBV+ cells. (a) Cells were collected, washed and fixed after treatment with 15 μM PP2 or DMSO (control) for 24 h. Cells were then permeabilized, and incubated with mouse anti-LMP1 antibody (Dako) and rabbit anti-p-IRF4 antibody (21st Century Biochemicals). After wash, cells were incubated with anti-mouse IgG APC (eBioscience) and anti-rabbit PE (eBioscience) before subjected to flow cytometry analysis. Q4 represents the portion of LMP1+/p-IRF4+ cells. (b) SavIII cells were transfected with p85 shRNA (#1) (or control). shRNA expression was induced by 1 μg/ml doxycycline for 3 days. p-IRF4 was then evaluated by flow. P = 0.0007 (unpaired t-test). (c) BJAB cells were transfected with IRF4, IRF4 plus LMP1 (or c-Src as a positive control). Cells were subjected to flow analysis after 48 h. Statistical analysis was performed for three independent flow analyses. Results are the averages ± s.d. Representative results from at least three independent experiments are shown.
We then treated these cells with the Src-specific inhibitor PP2, and assessed endogenous IRF4 phosphorylation by flow cytometry. As shown in Figure 6a, inhibition of Src with 15 μM PP2 for 24 h results in marked decreases of endogenous IRF4 phosphorylation in these cells, with a 99% reduction observed in SavIII, and a 97% reduction in JiJoye cells. These results confirm that Src activity mediates LMP1 activation of IRF4.
We further evaluated IRF4 phosphorylation in SavIII cells after P85 depletion by shRNA. Our results show that p85 depletion results in significant reduction in p-IRF4-positive cell portion (Figure 6b). On the other hand, either LMP1 or Src promotes IRF4 phosphorylation when transiently expressed in EBV-negative BJAB cells (Figure 6c).
These results indicate that the levels of LMP1 and IRF4 phosphorylation positively correlate in EBV latency.
DISCUSSION
We have recently shown that IRF4 is activated through c-Src-mediated tyrosine phosphorylation, and is constitutively activated in EBV-transformed cells.36 In this study, we aimed to disclose the intracellular signaling pathway responsible for IRF4 activation in EBV-transformed cells. Our results definitively show that LMP1 promotes IRF4 tyrosine phosphorylation and markedly stimulates its transcriptional activity through recruiting Src via P85. Furthermore, we show a positive regulatory circuit between PI3K and P85 that is required for activation of both IRF4 and Akt downstream of LMP1 signaling. Our results support a model that LMP1 recruits Src via P85, resulting in Src autophosphorylation and activation, and then leads to IRF4 phosphorylation and activation (Figure 7).
Figure 7.
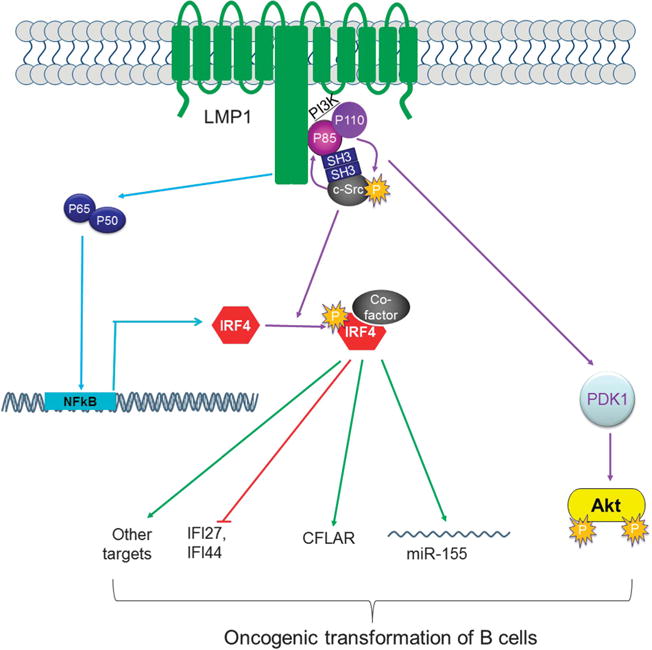
A diagram showing LMP1 signaling transduction to IRF4 activation. LMP1 is known to induce IRF4 expression via NFκB. In our current study, we show that LMP1 recruits Src through P85 that is a subunit of the PI3K kinase complex, and that these two kinases constitutes a positive regulatory circuits, leading to Akt and IRF4 activation. Activated IRF4, in cooperation with a co-factor, regulates expression of its targets, such as IFI27, IFI44, CFLAR35 and BIC/miR-155.6
It is notable that mutation in the TRAF-binding motif in LMP1 CTAR1 reduces the ability of LMP1 to activate IRF4 (Figure 1). This motif is well known to interact with TRAF1, -2, -3, -5 and -6. Previous reports have shown that TRAF2 and -6, together with P85, are in the signalosome for Akt activation by CD40 and OX40,44–46 which share signaling transduction mechanisms with LMP1. However, our preliminary results suggest that knockdown of either TRAF2 or -6 did not significantly impair the interaction between CTAR1 and P85; rather, these two ubiquitin E3 ligases may interact with IRF4 directly and enhance its transcriptional activity by promoting its K63-linked ubiquitination (data not shown). As both CTAR1 and -2 are known to interact with both TRAF2 and -6,47–49 the possible roles of TRAF2 and -6 in IRF4 ubiquitination and activation also account for our promoter–reporter results that both CTARs contribute to IRF4 transcriptional activity (Figure 1c). Further investigation is needed to make a definite conclusion whether any of these TRAFs mediates the interaction between LMP1 CTAR1 and P85 leading to downstream IRF4 and Akt activation, and to disclose K63-linked ubiquitination as a novel mechanism that cooperates with phosphorylation to activate IRF4.
In our earlier studies, we have provided solid evidence that LMP1 signaling activates another IRF family member, IRF7, and the underlying mechanism involves TRAF6- and RIP-mediated, K63-linked, regulatory ubiquitination.12–14 Although the Ser/Thr kinase responsible for LMP1 activation of IRF7 has not been identified, the PI3K axis is likely not involved. Thus, LMP1 may use different but overlapping mechanisms to control activation of IRF7 and IRF4.
In addition to LMP1 signaling, other unidentified lymphocyte-specific pathways may also impact the outcomes of IRF4 transcriptional activation, considering the following facts: (i) IRF4 is also phosphorylated in other contexts, such as human T-cell leukemia virus-1-infected cells, Hodgkin’s lymphoma cells50 and multiple myeloma cells,51 where LMP1 is absent; (ii) we have previously shown that, in addition to Src, other kinases including Alk and TNK also stimulate IRF4 transcriptional activity.36 Thus, IRF4 may be activated through different lymphocyte-specific signaling pathways that involve distinct kinases in different contexts; (iii) in the context of EBV latent infection, in addition to LMP1, LMP2A also activates the PI3K/Akt pathway in response to BCR signaling.52–55 It will be interesting to investigate if LMP2A stimulates IRF4 transcriptional activity through the Src/PI3K pathway axis; and (iv) the inhibitors LY294002 and PP2 are known to have cross-reactivity.
We have published a series of original findings related to this important project.32,35,36 The long-term efforts will characterize the IRF4-specific regulatory network that includes unique kinases involved in its activation in different contexts. From a clinical perspective, results from these studies will identify novel targets for potential therapeutic applications. For example, the Src inhibitors, Dasatinib, Sacracatinib and Bosutinib, which are used clinically, may have the potential to treat EBV-associated lymphomas by targeting the LMP1/IRF4 pathway.
MATERIALS AND METHODS
Cell lines
SavI, SavIII, JiJoye, P3HR1, IB4 and LCL00045 are EBV-transformed B-cell lines. DG75 and BJAB are EBV-negative human B-cell lines. All B-cell lines are cultured in RPMI1640 medium plus 10% fetal bovine serum and antibiotics. 293 and 293T cells are cultured with Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum and antibiotics.
Plasmids, reagents and antibodies
IRF4 and LMP1 expression plasmids were described in our recent publication.36 Flag-tagged LMP1 point mutants IID (384-YYD-386 mutated to 384-IID-386), PQAA (204-PQQAT-208 mutated to 204-AQAAT-208) and IID/PQAA were generated by site-directed mutagenesis. 3XFlag-tagged LMP1 deletion mutants (LMP1 WT, LMP1(1–231), LMP1Δ(187–351) and LMP1(1–187)) were generated by mutagenesis and cloned in pCMV7.1/3XFlag. pSV2–LMP1 and its deletion mutant pSV2–LMP1Δ(12–20) were gifts from Dr Bill Sugden.38 P85-specific shRNA expression plasmids cloned in the vector TRIPz and control were purchased from Open Biosystems (Lafayette, CO, USA). pGL3/ISRE-Luc (IFN-stimulated response element-Luc) and Renilla (pRL-TK) were purchased from Stratagene (San Diego, CA, USA) and Promega (Fitchburg, WI, USA), respectively. Phospho-IRF4(Y121/124) was developed in rabbit and characterized in our recent publication.36 Mouse anti-Flag M2 (Sigma, St Louis, MO, USA), mouse anti-HA-7 (Roche, Indianapolis, IN, USA), mouse anti-LMP1 CS1-4 (Dako, Carpinteria, CA, USA), anti-IRF4 H140 (rabbit) and M17 (goat) (Santa Cruz, Dallas, TX, USA), anti-c-Src B12 (mouse) and N16 (rabbit) (Santa Cruz), rabbit anti-phospho-Src family (Y418) (Cell Signaling), Akt and p-Akt(S473) (Cell Signaling), and mouse anti-GAPDH (Santa Cruz) were used for immunoblotting and immunoprecipitation. Anti-mouse IgG APC and anti-rabbit IgG PE were purchased from eBioscience (San Diego, CA, USA). All second antibodies for immunoblotting were purchased from Cell Signaling. LY294002 and PP2 were purchased from Sigma and EMD Millipore (Billerica, MA, USA), respectively.
Promoter–reporter assay
293 cells in 24-well plates were transfected with 0.2 μg expression plasmids as indicated together with 40 ng ISRE-Luc and 10 ng Renilla as internal transfection control. Empty vector was used to equalize the total amounts of DNA in all transfections. Cells were collected 24 h after transfection. Luciferase activity was measured with equal amounts (10% of total for each sample) of protein lysates with the use of a Dual Luciferase Assay kit (Promega), on a Turner BioSystems Modulus II Microplate Multimode Reader (Promega).
Transfection
293 and 293T cells were transfected with Effectene reagent (Qiagen, Valencia, CA, USA), and B cells were transfected with Lonza (Walkersville, MD, USA) Human B cell Nucleofector Kits following the manufacturer’s instructions. P85 shRNA in TRIPz were introduced into cells by retrovirus-mediated transfection. Retrovirus packing, production and infection were detailed in our previous publication.6 Stable transfectants were selected with 1 μg/ml puromycin for 2 weeks. Expression of TRIPz/shRNA is induced by 1 μg/ml doxycycline (Sigma).
Flow cytometry
Cell pellets were washed with phosphate-buffered saline and fixed in fixation buffer (Biolegend, San Diego, CA, USA), permeabilized with permeabilization solution (eBioscience), and then incubated with mouse LMP1 CS1-4 and rabbit p-IRF4(Y121/124) antibodies for 40 min. After washing with flow cytometry buffer, cells were incubated further with anti-mouse IgG APC and anti-rabbit IgG PE for 25 min, and then followed by flow cytometry analysis with a Flow Cytometer (Accuri, BD Biosciences, Franklin Lakes, NJ, USA) and BD Accuri C6 Software (BD Biosciences, San Jose, CA, USA). Data were further analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA). The fluorescence minus one strategy was used to determine background level and to adjust multicolor compensation for cell gating.
In vitro immunoprecipitation
293T cells in 60-mm dishes were transfected with 1 μg each indicated expression plasmids. Cells were harvested 48 h after transfection, and lysed in 1 ml NP-40 lysis buffer (10 mM Tris, pH7.5, 0.5% NP-40, 0.5% TritonX-100, 2.5 mM KCl, 150 mM NaCl, 30 mM β-glycerophosphate, 50 mM NaF, 1 mM NaOV4, and cocktail protease inhibitors (Sigma)). Supernatants were incubated with indicated antibodies. Protein A/G beads (Santa Cruz) were then added and incubated for 1 h and then subjected to extensive washes with NP-40 lysis buffer. Immunoblotting was performed with antibodies indicated.
In vivo immunoprecipitation
EBV-transformed cells (5 × 106 for each) were lysed in NP-40 lysis buffer. Supernatants were collected and pre-cleared with normal serum (2 μl for 1 ml lysates containing about 1 mg total proteins). The lysates were then incubated overnight with 2 μg rabbit Src antibody N16 or LMP1 antibody CS1-4 or normal serum (21st Century Biochemicals Inc., Marlborough, MA, USA). In all, 50 μl protein A/G beads (Santa Cruz) were then added and incubated for 30 more minutes. Beads were extensively washed with NP-40 lysis buffer and subjected to immunoblotting analysis.
Immunoblotting
Proteins were separated by 10% Acr:bis gel, and then transferred to nitrocellulose membranes followed by immunoblotting with corresponding antibodies. Signals were detected with an enhanced chemiluminescence kit following the manufacturer’s protocol (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
Chromosome immunoprecipitation
ChIP was performed with the use of ChIP-IT Express Enzymatic kit (Active Motif, Carlsbad, CA, USA). Briefly, IB4 cells were treated with LY or PP2, and harvested after 24 h. Cells were subjected to crosslinking by adding formaldehyde to a final concentration of 1% for 30 min at room temperature with slow rotation. Crosslinking was stopped by adding glycine to a final concentration of 125 mM for 5 min. Shearing and enzymatic digestion of chromatin, IP with IRF4 antibody M17, and DNA recovery were performed following the manufacturers’ instructions. Quantitative PCR was performed with the human BIC promoter ISRE primers: 5′-CCCCTCCAGCCGACTG-3′ (forward) and 5′-AACACACGCCGT GTAC-3′ (reverse), and β-actin promoter primers (control): 5′-CCAAC AAAGCACTGTGG-3′ (forward) and 5′-GGGCGAAGGCAACGC-3′ (reverse).6
Acknowledgments
This work was supported by an NIH NIDDK grant to ZQY/JPM (R01DK093526), an NIH NIAID grant to ZQY/JPM (R01AI114748), the American Society of Hematology Scholar Award to SN, and in part by the NIH grant C06RR0306551. We thank Dr Bill Sugden for providing pSV2-LMP1 and its deletion mutant pSV2-LMP1Δ(12-20). This publication is the result of work supported with resources and the use of facilities at the James H Quillen Veterans Affairs Medical Center. The contents in this publication do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Boshoff C, Weiss R. Aids-related malignancies. Nat Rev Cancer. 2002;2:373–382. doi: 10.1038/nrc797. [DOI] [PubMed] [Google Scholar]
- 2.Carbone A, Gloghini A, Dotti G. EBV-associated lymphoproliferative disorders: classification and treatment. Oncologist. 2008;13:577–585. doi: 10.1634/theoncologist.2008-0036. [DOI] [PubMed] [Google Scholar]
- 3.Pagano JS, Blaser M, Buendia MA, Damania B, Khalili K, Raab-Traub N, et al. Infectious agents and cancer: criteria for a causal relation. Semin Cancer Biol. 2004;14:453–471. doi: 10.1016/j.semcancer.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Saddawi-Konefka R, Crawford JR. Chronic viral infection and primary central nervous system malignancy. J Neuroimmun Pharmacol. 2010;5:387–403. doi: 10.1007/s11481-010-9204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ning S, Pagano J, Barber G. IRF7: activation, regulation, modification, and function. Genes Immun. 2011;12:399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L, Toomey NL, Diaz LA, Walker G, Ramos JC, Barber GN, et al. Oncogenic IRFs provide a survival advantage for EBV- or HTLV1-transformed cells through induction of BIC expression. J Virol. 2011;85:8328–8337. doi: 10.1128/JVI.00570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thornburg NJ, Kulwichit W, Edwards RH, Shair KHY, Bendt KM, Raab-Traub N. LMP1 signaling and activation of NF-kappaB in LMP1 transgenic mice. Oncogene. 2006;25:288–297. doi: 10.1038/sj.onc.1209023. [DOI] [PubMed] [Google Scholar]
- 8.Kieser A. Signal transduction by the Epstein-Barr virus oncogene latent membrane protein 1 (LMP1) Signal Transduction. 2007;7:20–33. [Google Scholar]
- 9.Li HP, Chang YS. Epstein-Barr virus latent membrane protein 1: structure and functions. J Biomed Sci. 2003;10:490–504. doi: 10.1007/BF02256110. [DOI] [PubMed] [Google Scholar]
- 10.Middeldorp JM, Pegtel DM. Multiple roles of LMP1 in Epstein-Barr virus induced immune escape. Semin Cancer Biol. 2008;18:388–396. doi: 10.1016/j.semcancer.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Soni V, Cahir-McFarland E, Kieff E. LMP1 TRAFficking activates growth and survival pathways. In: Wu H, editor. Advances in Experimental Medicine and Biology vol. 597. Springer; New York, NY, USA: 2007. pp. 173–187. [DOI] [PubMed] [Google Scholar]
- 12.Huye LE, Ning S, Kelliher M, Pagano JS. IRF7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol Cell Biol. 2007;27:2910–2918. doi: 10.1128/MCB.02256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ning S, Campos AD, Darnay B, Bentz G, Pagano JS. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol Cell Biol. 2008;28:6536–6546. doi: 10.1128/MCB.00785-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Zhang J, Lambert Q, Der CJ, Del Valle L, Miklossy J, et al. Interferon regulatory factor 7 is associated with Epstein-Barr virus-transformed central nervous system lymphoma and has oncogenic properties. J Virol. 2004;78:12987–12995. doi: 10.1128/JVI.78.23.12987-12995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cahir-McFarland ED, Carter K, Rosenwald A, Giltnane JM, Henrickson SE, Staudt LM, et al. Role of NF- k B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr Virus latency III-infected cells. J Virol. 2004;78:4108–4119. doi: 10.1128/JVI.78.8.4108-4119.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin HJ, Lee JM, Walls D, Hayward SD. Manipulation of the Toll-like receptor 7 signaling pathway by Epstein-Barr virus. J Virol. 2007;81:9748–9758. doi: 10.1128/JVI.01122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spender LC, Lucchesi W, Bodelon G, Bilancio A, Karstegl CE, Asano T, et al. Cell target genes of Epstein-Barr virus transcription factor EBNA-2: induction of the p55á regulatory subunit of PI3-kinase and its role in survival of EREB2.5 cells. J Gen Virol. 2006;87:2859–2867. doi: 10.1099/vir.0.82128-0. [DOI] [PubMed] [Google Scholar]
- 18.Xu D, Zhao L, Del Valle L, Miklossy J, Zhang L. Interferon regulatory factors 4 is involved in Epstein-Barr virus-mediated transformation of human B lymphocytes. J Virol. 2008;82:6251–6258. doi: 10.1128/JVI.00163-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iida S, Rao PH, Butler M, Corradini P, Boccadoro M, Klein B, et al. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat Genet. 1997;17:226–230. doi: 10.1038/ng1097-226. [DOI] [PubMed] [Google Scholar]
- 20.Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454:226–231. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grandvaux N, Servant MJ, tenOever BR, Sen GC, Balachandran S, Barber GN, et al. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol. 2002;76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mamane Y, Sharma S, Grandvaux N, Hernandez E, Hiscott J. IRF-4 activities in HTLV-I-induced T cell leukemogenesis. J Interferon Cytokine Res. 2002;22:135–143. doi: 10.1089/107999002753452746. [DOI] [PubMed] [Google Scholar]
- 23.Ramos JC, Ruiz P, Jr, Ratner L, Reis IM, Brites C, Pedroso C, et al. IRF4 and c-Rel expression in antiviral-resistant adult T-cell leukemia/lymphoma. Blood. 2007;109:3060–3068. doi: 10.1182/blood-2006-07-036368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma S, Grandvaux N, Mamane Y, Genin P, Azimi N, Waldmann T, et al. Regulation of IFN regulatory factor 4 expression in human T cell leukemia virus-I-transformed T cells. J Immunol. 2002;169:3120–3130. doi: 10.4049/jimmunol.169.6.3120. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee S, Lu J, Cai Q, Saha A, Jha HC, Dzeng RK, et al. The EBV latent antigen 3C inhibits apoptosis through targeted regulation of interferon regulatory factors 4 and 8. PLoS Pathog. 2013;9:e1003314. doi: 10.1371/journal.ppat.1003314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rui L, Schmitz R, Ceribelli M, Staudt LM. Malignant pirates of the immune system. Nat Immunol. 2011;12:933–940. doi: 10.1038/ni.2094. [DOI] [PubMed] [Google Scholar]
- 27.Shaffer AL, Emre NC, Romesser PB, Staudt LM. IRF4: immunity. malignancy! therapy? Clin Cancer Res. 2009;15:2954–2961. doi: 10.1158/1078-0432.CCR-08-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feldman AL, Law M, Remstein ED, Macon WR, Erickson LA, Grogg KL, et al. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia. 2009;23:574–580. doi: 10.1038/leu.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Havelange V, Pekarsky Y, Nakamura T, Palamarchuk A, Alder H, Rassenti L, et al. IRF4 mutations in chronic lymphocytic leukemia. Blood. 2011;118:2827–2829. doi: 10.1182/blood-2011-04-350579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B, Kracker S, Yasuda T, Casola S, Vanneman M, Homig-Holzel C, et al. Immune surveillance and therapy of lymphomas driven by Epstein-Barr Virus protein LMP1 in a mouse model. Cell. 2012;148:739–751. doi: 10.1016/j.cell.2011.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang CC, Lorek J, Sabath DE, Li Y, Chitambar CR, Logan B, et al. Expression of MUM1/IRF4 correlates with clinical outcome in patients with B-cell chronic lymphocytic leukemia. Blood. 2002;100:4671–4675. doi: 10.1182/blood-2002-01-0104. [DOI] [PubMed] [Google Scholar]
- 32.Ning S. IRF4 as an oncogenic biomarker for hematological malignancies. J Oncobiomarkers. 2013;1:6. [Google Scholar]
- 33.Sundram U, Harvell JD, Rouse RV, Natkunam Y. Expression of the B-cell proliferation marker MUM1 by melanocytic lesions and comparison with S100, gp100 (HMB45), and MelanA. Mod Pathol. 2003;16:802–810. doi: 10.1097/01.MP.0000081726.49886.CF. [DOI] [PubMed] [Google Scholar]
- 34.Biswas PS, Bhagat G, Pernis AB. IRF4 and its regulators: evolving insights into the pathogenesis of inflammatory arthritis? Immunol Rev. 2010;233:79–96. doi: 10.1111/j.0105-2896.2009.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Yao ZQ, Moorman JP, Xu Y, Ning S. Gene expression profiling identifies IRF4-associated molecular signatures in hematological malignancies. PLoS One. 2014;9:e106788. doi: 10.1371/journal.pone.0106788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Ning S. IRF4 is activated through c-Src-mediated tyrosine phosphorylation in virus-transformed cells. J Virol. 2013;87:9672–9679. doi: 10.1128/JVI.01435-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alvarez RH, Kantarjian HM, Cortes JE. The role of Src in solid and hematologic malignancies. Cancer. 2006;107:1918–1929. doi: 10.1002/cncr.22215. [DOI] [PubMed] [Google Scholar]
- 38.Bloss T, Kaykas A, Sugden B. Dissociation of patching by latent membrane protein-1 of Epstein-Barr virus from its stimulation of NF-kappaB activity. J Gen Virol. 1999;80:3227–3232. doi: 10.1099/0022-1317-80-12-3227. [DOI] [PubMed] [Google Scholar]
- 39.Dawson CW, Tramountanis G, Eliopolous AG, Young LS. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J Biol Chem. 2003;278:3694–3704. doi: 10.1074/jbc.M209840200. [DOI] [PubMed] [Google Scholar]
- 40.Pleiman C, Hertz W, Cambier J. Activation of phosphatidylinositol-3′ kinase by Src-family kinase SH3 binding to the p85 subunit. Science. 1994;263:1609–1612. doi: 10.1126/science.8128248. [DOI] [PubMed] [Google Scholar]
- 41.Arron JR, Vologodskaia M, Wong BR, Naramura M, Kim N, Gu H, et al. A positive regulatory role for Cbl family proteins in tumor necrosis factor-related activation-induced cytokine (TRANCE) and CD40L-mediated Akt activation. J Biol Chem. 2001;276:30011–30017. doi: 10.1074/jbc.M100414200. [DOI] [PubMed] [Google Scholar]
- 42.Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H, et al. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell. 1999;4:1041–1049. doi: 10.1016/s1097-2765(00)80232-4. [DOI] [PubMed] [Google Scholar]
- 43.Hatton O, Lambert SL, Krams SM, Martinez OM. Src kinase and Syk activation initiate PI3K signaling by a chimeric latent membrane protein 1 in Epstein-Barr virus (EBV)+ B cell lymphomas. PLoS One. 2012;7:e42610. doi: 10.1371/journal.pone.0042610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davies CC, Mak TW, Young LS, Eliopoulos AG. TRAF6 is required for TRAF2-dependent CD40 signal transduction in nonhemopoietic cells. Mol Cell Biol. 2005;25:9806–9819. doi: 10.1128/MCB.25.22.9806-9819.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deregibus MC, Buttiglieri S, Russo S, Bussolati B, Camussi G. CD40-dependent activation of phosphatidylinositol 3-kinase/Akt pathway mediates endothelial cell survival and in vitro angiogenesis. J Biol Chem. 2003;278:18008–18014. doi: 10.1074/jbc.M300711200. [DOI] [PubMed] [Google Scholar]
- 46.So T, Choi H, Croft M. OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. J Immunol. 2011;186:3547–3555. doi: 10.4049/jimmunol.1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawson CW, Port RJ, Young LS. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC) Semin Cancer Biol. 2012;22:144–153. doi: 10.1016/j.semcancer.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 48.Kieser A, Sterz KR. The latent membrane protein 1 (LMP1) In: Münz C, editor. Epstein Barr Virus Volume 2: One Herpes Virus: Many Diseases. Springer International Publishing; Cham, Switzerland: 2015. pp. 119–149. [Google Scholar]
- 49.Stunz LL, Bishop GA. Latent membrane protein 1 and the B lymphocyte-A complex relationship. Crit Rev Immunol. 2014;34:177–198. doi: 10.1615/critrevimmunol.2014010041. [DOI] [PubMed] [Google Scholar]
- 50.Gu TL, Cherry J, Tucker M, Wu J, Reeves C, Polakiewicz RD. Identification of activated Tnk1 kinase in Hodgkin’s lymphoma. Leukemia. 2010;24:861–865. doi: 10.1038/leu.2009.293. [DOI] [PubMed] [Google Scholar]
- 51.St-Germain JR, Taylor P, Tong J, Jin LL, Nikolic A, Stewart II, et al. Multiple myeloma phosphotyrosine proteomic profile associated with FGFR3 expression, ligand activation, and drug inhibition. Proc Natl Acad Sci USA. 2009;106:20127–20132. doi: 10.1073/pnas.0910957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukuda M, Longnecker R. Latent membrane protein 2 A inhibits transforming growth factor-á1-induced apoptosis through the phosphatidylinositol 3-kinase/ Akt pathway. J Virol. 2004;78:1697–1705. doi: 10.1128/JVI.78.4.1697-1705.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Portis T, Longnecker R. Epstein-Barr virus (EBV) LMP2 A mediates B-lymphocyte survival through constitutive activation of the Ras//PI3K//Akt pathway. Oncogene. 2004;23:8619–8628. doi: 10.1038/sj.onc.1207905. [DOI] [PubMed] [Google Scholar]
- 54.Scholle F, Bendt KM, Raab-Traub N. Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J Virol. 2000;74:10681–10689. doi: 10.1128/jvi.74.22.10681-10689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Swart R, Ruf IK, Sample J, Longnecker R. Latent membrane protein 2A-mediated effects on the phosphatidylinositol 3-kinase/Akt pathway. J Virol. 2000;74:10838–10845. doi: 10.1128/jvi.74.22.10838-10845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]