

Abstract
AIM
To studied iron metabolism in liver, spleen, and serum after acute liver-damage, in relation to surrogate markers for liver-damage and repair.
METHODS
Rats received intraperitoneal injection of the hepatotoxin thioacetamide (TAA), and were sacrificed regularly between 1 and 96 h thereafter. Serum levels of transaminases and iron were measured using conventional laboratory assays. Liver tissue was used for conventional histology, immunohistology, and iron staining. The expression of acute-phase cytokines, ferritin light chain (FTL), and ferritin heavy chain (FTH) was investigated in the liver by qRT-PCR. Western blotting was used to investigate FTL and FTH in liver tissue and serum. Liver and spleen tissue was also used to determine iron concentrations.
RESULTS
After a short initial decrease, iron serum concentrations increased in parallel with serum transaminase (aspartate aminotransferase and alanine aminotransferase) levels, which reached a maximum at 48 h, and decreased thereafter. Similarly, after 48 h a significant increase in FTL, and after 72h in FTH was detected in serum. While earliest morphological signs of inflammation in liver were visible after 6 h, increased expression of the two acute-phase cytokines IFN-γ (1h) and IL-1β (3h) was detectable earlier, with maximum values after 12-24 h. Iron concentrations in liver tissue increased steadily between 1 h and 48 h, and remained high at 96 h. In contrast, spleen iron concentrations remained unchanged until 48 h, and increased mildly thereafter (96 h). Although tissue iron staining was negative, hepatic FTL and FTH protein levels were strongly elevated. Our results reveal effects on hepatic iron concentrations after direct liver injury by TAA. The increase of liver iron concentrations may be due to the uptake of a significant proportion of the metal by healthy hepatocytes, and only to a minor extent by macrophages, as spleen iron concentrations do not increase in parallel. The temporary increase of iron, FTH and transaminases in serum is obviously due to their release by damaged hepatocytes.
CONCLUSION
Increased liver iron levels may be the consequence of hepatocyte damage. Iron released into serum by damaged hepatocytes is obviously transported back and stored via ferritins.
Keywords: Iron metabolism, Ferritin, Liver, Cytokines, Acute liver damage
Core tip: In humans, an increase in hepatic iron concentration is caused by chronic hepatitis-C infection, alcohol abuse, and non-alcoholic fatty liver disease. The pathophysiology behind increased liver iron concentrations caused by acute liver damage has remained obscure. Using thioacetamide-injection in rats, we demonstrate that the increase in liver iron may be a consequence rather than the cause of hepatocyte damage. Thereby, iron is released into serum by damaged hepatocytes during acute liver damage, and reabsorbed by remaining hepatocytes (but not spleen) by means of ferritin L and ferritin H subunits. Our studies also show that ferritin H is a promising surrogate marker for damaged hepatocytes.
INTRODUCTION
Iron is an essential and the most abundant trace element for human physiology. It is required not only for erythropoiesis and oxygen transport, but also for enzymatic functions[1,2]. Human body contains 50 mg iron per kg body weight. The daily intestinal (duodenal) iron uptake is 1-2 mg, which compensates for the iron loss by sloughing off of intestinal and epidermal cells. In fact, 65% of the iron is bound to hemoglobin (2300 mg), and most of the non-hemoglobin iron is stored in the liver[1,3-5]. Hepatocytes are responsible for the uptake of ionic iron, which reaches the portal blood from the intestine, and is transported to the liver as transferrin-iron[1,3,6]. A small amount of iron is also taken up by Kupffer cells, which have the important function of clearing aged erythrocytes[2,5,7]. In normal plasma, transferrin iron-binding sites are saturated to about one third. The remaining iron-binding capacity serves as a buffer for conditions where iron concentrations increase acutely, which may induce tissue damage due to iron toxicity[8-10].
The hepatocytes capture about 80% of transferrin-bound iron through caging receptors, made up of the ferritin (FT) subunits ferritin heavy chain (FTH) and ferritin light chain (FTL)[6,8,11-13]. However, in contrast to other cells, hepatocytes not only synthesize FTL as a component of the iron cage, but also as a secreted serum protein[14]. As serum ferritin is reported to be exclusively FTL, and not FTH[14,15], its synthesis and secretion in the liver is differentially regulated[16], mainly at post-transcriptional levels[12].
In all tissues, iron is the main stimulus for FTL gene expression. An increase in synthesis and secretion of FTL has been demonstrated in the liver in a model of acute-phase response[14,17], and in hepatocytes in vitro by different acute-phase cytokines including interleukin-6 (IL-6)[14]. Cytokines are known to be responsible for dramatic changes of hepatic protein synthesis, including upregulation of hepcidin[17], lactoferrin[18], and lipocalin-2[19]. These low molecular weight proteins are involved in the control of iron metabolism, as a reaction to extrahepatic tissue damage.
Experimentally, the generation of a systemic acute-phase reaction, by means of turpentine oil-induced sterile intramuscular abscesses, was shown to be accompanied by elevated hepatic iron concentrations. This was attributed to increased iron-uptake into hepatocytes[20], which could also be observed in vitro[15]. Similarly, increased hepatic iron storage, in addition to elevated serum cytokine levels, has been reported in human liver diseases caused by viruses, alcohol consumption or overweight[21,22]. These alterations were shown to be reversible after elimination of the noxae, as is the case after successful treatment of hepatitis C[23,24]. An increased absorption of dietary-iron in the intestine has been postulated to cause NAFLD/NASH in humans[25], however, the mechanisms underlying the increase of the iron deposition in the liver during acute-phase reaction are still poorly understood.
Here, we have studied changes in the expression of FTH and FTL, various acute-phase cytokines, as well as iron concentrations in liver and spleen after intraperitoneal administration of thioacetamide (TAA), in parallel with serological markers for liver damage and repair. We provide evidence for the hypothesis that the increase of liver iron concentrations may be rather the consequence than the cause of hepatocyte damage.
MATERIALS AND METHODS
Materials and chemicals were bought from Merck (Darmstadt, Germany), Applichem (Darmstadt, Germany) and Sigma (Steinheim, Germany). Details for each chemical and material are noted in the appropriate sections.
Rat model of hepatic acute-phase reaction
Male Sprague-Dawley rats weighing 200-220 g were purchased from Charles River (Sulzfeld, Germany) and Harlan Winkelmann (Borchen, Germany). The animals were kept according to our institutional guidelines and the rules of the German Law for the Protection of Animals. The experiments were approved by the committee on animal protection of the University of Goettingen and the Lower Saxony State Office for Consumer Protection and Food Safety (Study no. 33.9-42502-04-13/1086).
The rats were divided into a control group (n = 12) and an experiment group (n = 32). Five animals were kept per cage following a 12:12-hour (h) light-dark cycle. They had unrestricted access to food and water. An intraperitoneal (ip) injection of thioacetamide (TAA) (prepared freshly, dissolved in sterile NaCl) with a dose of 500 mg/kg body weight was given to each rat as described before[26]. Control rats were injected with the same volume of NaCl. Rats were sacrificed after 1, 3, 6, 12, 24, 48, 72, and 96 h under pentobarbital anesthesia. Serum, liver and spleen were snap frozen in liquid nitrogen and stored at -80 °C.
Measurement of liver enzymes in serum
Blood samples from the inferior vena cava were collected from control and experimental rats after 1, 3, 6, 12, 24, 48, 72, and 96 h. Serum was used for the determination of alkaline phosphatase with a standard p-nitrophenolphosphate photometric assay (Roche, Mannheim, Germany). Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activities were measured by using analysis kits (DiaSys, Karben, Germany) according to the manufacturer’s instructions.
Histopathology
Histopathology was performed on 5 μm acetone-fixed cryostat sections (Reichert Jung, Wetzlar, Germany) as described previously[20,27]. Briefly, the cryostat sections were washed with phosphate-buffered saline (PBS), and stained with hematoxylin for 5 minutes. Thereafter, tissues were stained with 1% eosin followed by short washing with 100% alcohol. Tissues were then transferred into xylol and mounted.
Measurement of serum and tissue iron levels
Serum and tissue iron levels were measured with a colorimetric ferrozine-based assay[28] following the company’s protocol, through facility service provided by the University Medical School, Goettingen. The hepatic tissue iron concentrations were measured from liver homogenates, which were prepared as described earlier[20].
Prussian blue iron staining of liver and spleen
5 μm thick air-dried cryostat sections (Reichert Jung, Wetzlar, Germany) were stained for iron using the Prussian blue method. Fixation was performed by incubating the slides in ice-cold methanol (-20 °C) for 10 min, and ice-cold acetone (-20 °C) for 10 s. The sections were stained for iron by using iron-stain kit from Sigma-Aldrich (Munich, Germany), according to the manufacturer’s instructions.
RNA isolation from liver tissue and real-time PCR
RNA was isolated from all control and TAA-treated rat livers as described previously[17]. Briefly, mRNA was converted into cDNA with Superscript kit (Invitrogen, CA). ABI Prism Sequence Detection System 7000 (Applied Biosystems, Foster City, CA) was used for real-time PCR. The primers used in this study are presented in Table 1. They were synthesized by Invitrogen. Ubiquitin C (UBC) served as housekeeping gene, and was used to normalize the values of the genes of interest. The normalized values for TAA-treated rats were compared with saline-treated controls, and the relative expression was plotted against the observation time. The results represent at least three experiments performed as duplicates.
Table 1.
Primers used for polymerase chain reaction
| Primer | 5’ → 3’ Forward | 5’ → 3’ Reverse |
| UBC | CAC CAA GAA GGT CAA ACA GGA A | AAG ACA CCT CCC CAT CAA ACC |
| Ferritin H | GCCCTGAAGAACTTTGCCAAAT | TGCAGGAAGATTCGTCCACCT |
| Ferritin L | AACCACCTGACCAACCTCCGTA | TCAGAGTGAGGCGCTCAAAGAG |
| IL-6 | GTCAACTCCATCTGCCCTTCAG | GGCAGTGGCTGTCAACAACAT |
| IL-1β | TACCTATGTCTTGCCCGTGGAG | ATCATCCCACGAGTCACAGAGG |
| TNF-α | ACAAGGCTGCCCCGACTAT | CTCCTGGTATGAAGTGGCAAATC |
| IFN-γ | GAACTGGCAAAAGGACGGTA | CTGATGGCCTGGTTGTCTTT |
Western blot analysis of liver tissue and serum proteins
Serum and total liver protein from all control and TAA-treated rats (time points: 1, 3, 6, 12, 24, 48, 72, 96 h) was used for Western blot analysis as described before[20]. Shortly, for total liver protein isolation, hepatic tissue probes were lysed in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mmol/L NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS). The Bradford method was used to measure protein concentrations. 50 μg of protein either from tissue or serum samples were used in sodium dodecyl sulfate polyacrylamide gel electrophoresis (NuPAGE, Novex 4%-12%, Bis-Tris gel, Invitrogen), under reducing conditions. Then, proteins were transferred to nitrocellulose membranes, and Western blot analysis was performed with antibodies listed in Table 2. Immunodetection was performed according to the ECL Western blotting protocol (Amersham, GE Healthcare, United States).
Table 2.
Antibodies used in this study
| Antibodies | Company | Reference | Dilution |
| FTH | Santa Cruz | SC-25617 | 1:100 |
| FTH | LS Bio | LS-C23537 | 1:250 |
| FTL | Abcam | AB-69090 | 1:1000 |
| FTL | Santa Cruz | SC-14422 | 1:100 |
| FTL peptide | Santa Cruz | SC-14422 | 1:50 |
| β-actin | Sigma | A-2228 | 1:4000 |
Immunohistochemistry of liver tissue
5 μm hepatic cryostat sections (Reichert Jung, Wetzlar, Germany) from control and TAA-injected animals were used for immunodetection of FTL and FTH as described before[27]. Fixation was performed with ice-cold methanol (-20 °C) for 10 min, and ice-cold acetone (-20 °C) for 10 s. The sections were incubated with PBS containing glucose/glucose oxidase/sodium azide to inhibit endogenous peroxidase reactions. Nonspecific staining was minimized by incubation with fetal calf serum (FCS) for 30 min. Commercially available antibodies for FTL (Abcam, UK and Santa Cruz, United States) and FTH (LS Bio and Santa Cruz, United States) were used for immunostaining. The sections were incubated with primary antibodies overnight at 4 °C. (For details see Table 2). Negative controls were performed with isotype-specific IgGs. Next day, sections were washed with PBS, and incubated with peroxidase-conjugated anti-rabbit/anti-mouse IgG (Dako, Hamburg, Germany), pre-absorbed with normal rat serum to minimize cross-reactivities. Peroxidase reaction was performed with 3,3’-diaminobenzidine (0.5 mg/mL) and H2O2 (0.01%) for 10 min. Meyer’s hemalaun solution was used for counter-staining.
Statistical analyses
Student’s t test and One-way ANOVA with Dennett’s post-test were performed using Graph Pad Prism version 4.00 for Windows (Graph Pad Software, San Diego, CA, www.graphpad.com). Experimental errors are shown as SEM. Statistical significance was accepted at: aP ≤ 0.05; bP ≤ 0.01; cP ≤ 0.001.
RESULTS
Measurement of liver damage in TAA-treated animals
Liver damage was analyzed by measuring the amount of circulating liver enzymes at various times after TAA injection. The serum levels of aspartate aminotransferase (AST) reached to their peak (8301 ± 1007 U/L) after 48 h of TAA injection with a statistical significant of P = 0.001. Similarly, compared to controls (54 ± 1.4 U/L), serum activity of alanine aminotransferase (ALT) was significantly elevated with a maximum at 48 h (1389 ± 121 U/L; P < 0.001) (Figure 1 A and B). Correspondingly, in liver tissue sections of TAA-injected animals we have previously observed immigration of inflammatory cells over time, staring in portal areas and extending into pericentral areas, reaching a maximal infiltration at 48 h, and declining thereafter until complete disappearance at 96 h[26].
Figure 1.
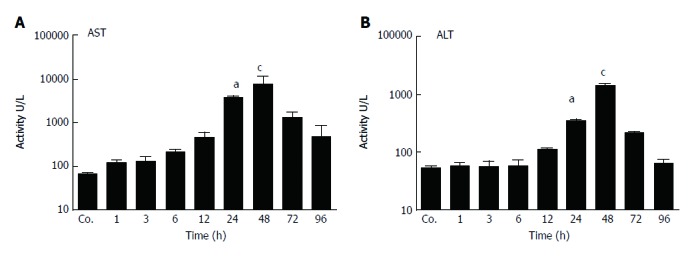
Serum enzyme levels in control and thioacetamide-injected rats. Serum aspartate aminotransferase (AST) (A) and alanine aminotransferase (ALT) (B) levels showed a statically significant (aP ≤ 0.05; cP ≤ 0.001) increase at 24 and 48 h followed by a decrease thereafter. Results represent mean values ± SEM, measured in duplicate in four animals for each time point, compared to controls (Co.).
Changes in hepatic gene expression of major acute-phase cytokines
The changes in mRNA levels of major acute-phase cytokines (APC) were measured by qPCR in TAA-treated rats at different time points compared to controls. An early up-regulation of the expression of several APCs was observed (Figure 2). IFN-γ started to increase first. A significant induction was observed after 1h, and a maximum (13.7 ± 2.64 - fold) after 12 h of TAA-administration. The levels of IFN-γ decreased thereafter, and turned to normal after 24 h. We also measured a significant induction of the expression of IL-1β with a maximum (41 ± 2.2 - fold) after 12 h. Thereafter, levels decreased but remained significantly elevated throughout the course of the study (until 96 h). Upregulation of both IL-6 and TNF-α started later (6 h) and reached a maximum after 24 h with an increase of 55 ± 2.1 and 69 ± 5 - fold, respectively. Expression decreased thereafter (Figure 2).
Figure 2.
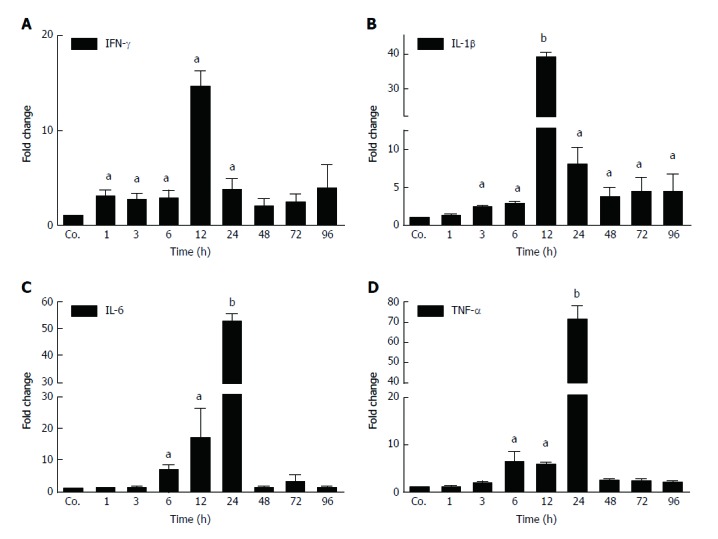
Early up-regulation of the expression of several acute-phase cytokines was observed. A-D: Significant increase of mRNA expression of acute-phase cytokines (IFN-γ, IL-1β, IL-6, TNF-α) in liver of thioacetamide -injected rats after various time points (aP ≤ 0.05; bP ≤ 0.01). Real-time PCR was normalized to GAPDH. Results represent mean values ± SEM, measured in duplicate in four animals for each time point, compared to controls (Co.).
Measurement of serum, hepatic and splenic iron levels
Compared to control serum iron levels (1.7 ± 0.03 mg/L), a slight but non-significant decrease (1.4 ± 0.1 mg/L) was observed shortly (1 h) after TAA injection. Values remained below controls until 6 h. Thereafter, serum iron level started to increase (2.4 ± 0.04 mg/L at 12h after TAA injection), reached a maximum after 24 h (3.97 ± 0.2 mg/L; P = 0.0010), and remained significantly elevated until 48 h. Then, serum iron decreased and returned to control levels at 72 - 96 h (Figure 3A). Compared to control animals (15.2 ± 0.9 mg/g), hepatic iron concentrations started to increase immediately (1 h) after TAA-administration, and progressively increased throughout the course of the study. The most prominent elevation (38 ± 3.1 mg/g; P = 0.030) was measured during 48 - 96 h after TAA injection (Figure 3A).
Figure 3.
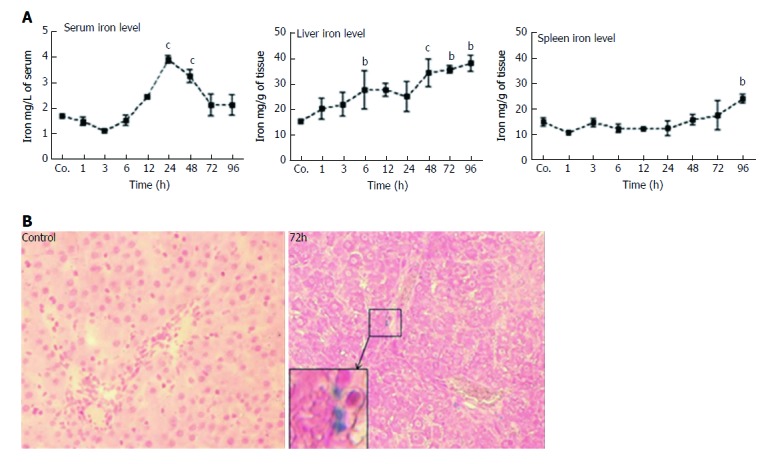
Measurement of serum, hepatic and splenic iron levels. A: Serum, liver and spleen iron levels at various time points after TAA injection in rats, as compared to controls (Co.). A mild decrease in serum iron levels was followed by a significant (cP ≤ 0.001) increase at 24 h and 48 h after TAA injection. Liver iron levels showed a significant (bP ≤ 0.01; cP ≤ 0.001) increase almost throughout the whole course of the study (a clear tendency is seen already after 1 h and 3 h). However, splenic iron content increased significantly (bP ≤ 0.01) only after 96 h. Results show mean ± SEM of each four animals; B: Prussian blue iron staining in liver of control and TAA-injected rats. Inset shows a higher magnification of the boxed area. A few positive cells are visible in rats after 72 h. Original magnification × 200.
In contrast, splenic iron showed a tendency to decrease 1 h after TAA administration, and compared to control levels of 15 ± 1 mg/g, a mild decrease was observed during most of the investigated time points (3 - 24 h). Splenic iron concentrations reached normal levels after 48 - 72 h, which was followed by a mild but significant increase (23 ± 1 mg/g) at 96 h after TAA injection (Figure 3A).
Prussian blue staining of hepatic and splenic tissue
After having measured a significant increase in iron concentration in liver homogenates, we sought to detect iron in liver cells by histological staining methods. However, the applied method did not consistently detect iron in the specimens, obviously due to insufficient sensitivity. We could only detect iron in a few nucleated cells 72 h after TAA-administration in liver (Figure 3B), but never in spleen.
Changes in hepatic and serum levels of FTL and FTH
Our qRT-PCR analysis of total liver RNA showed a mild increase of mRNA expression of the iron storage protein FTH after 3 h - 48 h of TAA-injection, and a significant increase after 72 h (4.9 ± 2.6 - fold; P ≤ 0.01) (Figure 4A). Additionally, a significant increase of the expression of FTL was noted after 48 h (7.8 ± 3.18 - fold; P ≤ 0.001) (Figure 4A).
Figure 4.
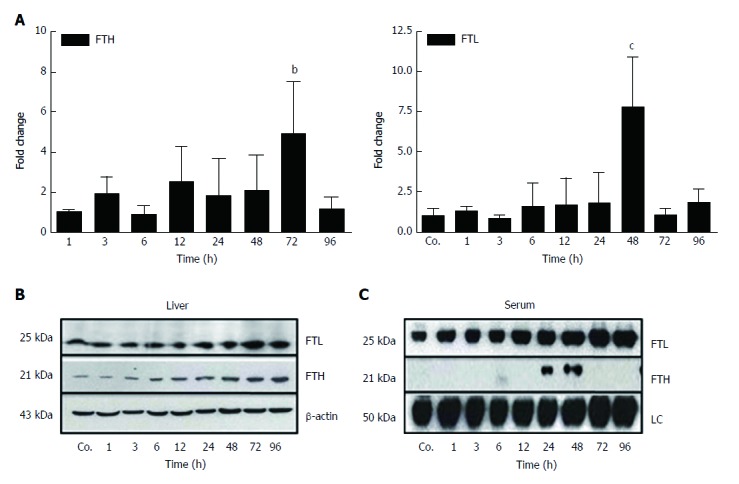
Changes in hepatic and serum levels of ferritin heavy chain and ferritin light chain after thioacetamide-induced liver damage. A: Significant (bP ≤ 0.01; cP ≤ 0.001) fold-change of hepatic mRNA expression of ferritin heavy chain (FTH) and ferritin light chain (FTL) in TAA-injected animals vs controls (Co.) after 72 h and 48 h, respectively, analyzed by qPCR and normalized against the house-keeping gene UBC. Results represent mean values ± SEM of each four animals per group. Controls were set as 1; B, C: Western blot analysis of FTL and FTH in total liver and in serum of control (Co.) and TAA-injected animals at indicated time points. β-actin (about 43 kDa) was used as loading control for liver tissue, while in serum the loading control (LC) represents an internal control (about 50 kDa).
Western blot analysis of total protein from hepatic tissue lysates demonstrated an almost constant increase in the content of FTL and FTH beginning early (3 h - 6 h after TAA-injection), reaching a maximum at 72 h and 96 h, respectively (Figure 4B). Thereby, hepatic expression of FTL was more abundant compared to FTH (Figure 4B). Such quantitative difference became even more clearly visible in sera from control and TAA-treated animals (Figure 4C). Western blots revealed expression of FTL, but not FTH, in control animals. Thereby, FTL increased progressively after TAA injection with a maximum level at 96 h, whereas FTH became positive only at 24 h and 48 h after TAA injection (Figure 4C). These data clearly show differential regulation and release of FTH and FTL in damaged liver.
Immunolocalization of FTL and FTH in liver of control and TAA-injected animals
Immunohistochemical analysis of normal liver revealed weak granular FTL positivity mainly in the cytoplasm of hepatic cells. Immunoreactivity strongly increased in non-damaged liver areas after 48 h in TAA-treated rats. An intense signal was still visible at 72 h (Figure 5), although at this time point liver recovery has started to take place (Figure 1).
Figure 5.
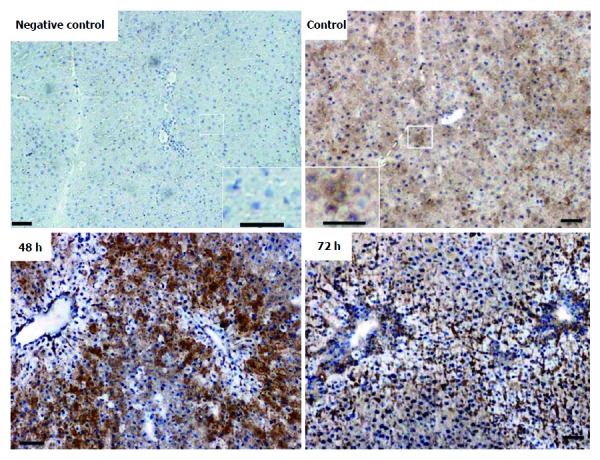
Immunoperoxidase staining of ferritin light chain in cryostat sections of rat liver from control and thioacetamide-injected animals. Insets show higher magnification of corresponding areas. Negative control represents the omission of primary antibodies. Note massive increase in signal intensity after 48 h and 72 h. Original magnification × 100; bar = 50 μm.
Immunostaining for FTH in liver tissue revealed a weak and almost homogenous staining pattern in control rats, and positivity in specific areas was found to increase most strongly at 48 h after TAA injection (Figure 6). Thereby, the regions of strongest staining obviously correlate to the pattern of inflammatory cells that invade the liver after TAA administration.
Figure 6.
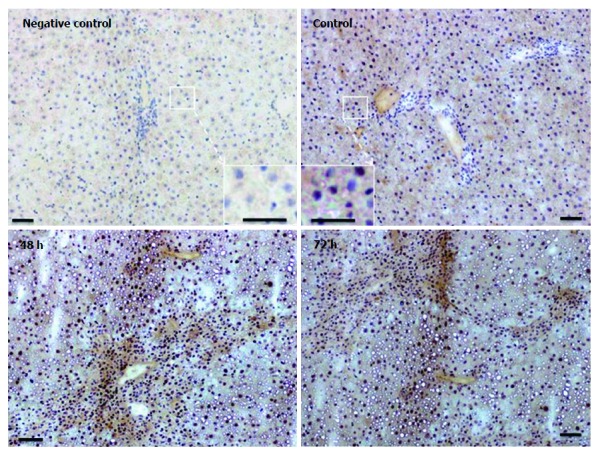
Immunodetection of ferritin heavy chain in cryostat sections of rat liver from control and thioacetamide-injected animals utilizing peroxidase-conjugated secondary antibodies. Insets show higher magnification of corresponding areas. Negative control represents the omission of primary antibodies. Note increase in signal intensity after 48 h and 72 h. Original magnification × 100; bar = 50 μm.
DISCUSSION
In animals, biochemical and histological effects of TAA-induced liver damage have been widely studied, and are highly similar to human acute liver damage[29]. However, the iron metabolism in the liver during acute-phase reaction is still poorly understood. In the present study, we administered TAA to rats using single intraperitoneal injection to induce reversible liver damage. The success of the method was confirmed by measurements of circulating hepatic enzymes (AST, ALT), and the typical pattern of inflammatory cells invading the liver from portal to central fields. Serum levels of AST and ALT were found to be highest at 24 h and 48h after TAA injection, and decreased to baseline levels at 72 h and 96 h, indicating liver damage followed by recovery. Previous studies have demonstrated similar results when TAA was administered in rats intravenously[30] or intraperitoneally[26]. Here we have also shown upregulation of hepatic gene expression of acute-phase cytokines (APCs) IFN-γ, IL-1β, IL6, and TNF-α; starting with IFN-γ after 1 h and IL-1β after 3 h of TAA injection. IL-6 and TNF-α expression increased after 6 h, when hepatic damage becomes overt in histological sections.
Increased serum iron levels may be attributed to damage of liver cells
In parallel with the early increase of IFN-γ and IL-1β expression, a mild decrease of the serum iron concentration, together with an increase of liver iron concentrations was detected. However, serum iron levels increased after 24 h and 48 h of TAA administration, corresponding to the peak levels of serum transaminases. Interestingly, in contrast to FTL, which increased almost constantly, FTH protein was only detectable in serum at these time points. We assume that the extensive damage of hepatocytes, demonstrated by the massive release of intracellular enzymes and of FTH into the serum may also be responsible for the increased serum iron levels after 24 h and 48 h of TAA injection.
The extremely high expression of APCs after 12 h - 24 h may additionally have induced the increase of tissue iron concentrations, which is also supported by results obtained with intramuscular administration of turpentine oil[17,31]. Hepatic iron accumulates in parallel to the increase of the two ferritin subtypes FTH and FTL. Both FTH and FTL have been reported to be intracellular proteins, and in fact, until recently the mature form of serum ferritin was unidentified, as it was its source. We recently identified FTL as the sole ferritin species in serum, and also demonstrated that it is a secreted acute-phase protein[14]. In contrast, FTH remains intracellularly located, despite its similar reactivity under acute-phase conditions. In fact, gene expression of both of the FT forms is modulated not only by iron itself, but also by acute-phase mediators[14,15]. The increase in ferritin expression and production may be attributed to the increased uptake of iron, released from damaged liver cells, by healthy hepatocytes. The role of FTL appears to be more important in this case, because it does not only store increasing amounts of iron in the ‘stressed’ hepatocytes together with FTH, but is also secreted to transport iron in the serum. This phenomenon seems to be specific for liver and not for other organs.
Indeed, ferritin was discovered as a cytosolic iron storage protein[32-34]. However, the localization of ferritins within the cell is discussed controversially. Previously, we reported the nuclear localization of iron transport proteins including FTH in rat liver under normal and acute-phase conditions[20,16]. In the same line of evidence, presence of nuclear FTH has been described in human astrocytoma cell-lines[35], in corneal epithelial cells[36], and in murine hepatocytes in iron overload settings[37]. Differential localization of FTH and FTL as well as quantitative differences are obviously related to their distinct roles in the liver. Thereby, nuclear FTH suggests an important role of iron for the activation of nuclear enzymes, which are involved in DNA synthesis and repair, and also in the modulation and initiation of transcription[38].
Spleen does not accumulate iron in acute liver damage
We also sought to study the functions of the spleen in iron metabolism after TAA-induced hepatocellular damage. The spleen represents a major organ of the reticuloendothelial system, and is responsible for the uptake and clearing of corpuscular matter from the blood circulation. In contrast to liver, we could not detect increased iron levels in the spleen, neither at early time points nor during the period of most severe liver damage. Only after 96 h, a mild but significant increase was noted. Although iron is supposed to be sequestered in the cells of the reticuloendothelial system we assume that under acute-phase conditions most of the serum iron is taken up by ‘stressed’ hepatocytes[15,20]. Although our direct iron staining in liver tissue was hardly successful, two significant findings support our assumption: I) the increase of ferritins in hepatocytes, and II) the lack of increase of splenic iron concentrations when liver iron is increased.
Acute-phase reaction can be subdivided into early and late stages
TAA administration induces an acute-phase reaction in the liver, which starts very early[39,40]. Thereby, liver macrophages obviously act as a first-line defense barrier. Inhibition of macrophages has been reported to aggravate liver damage in the rat model[41]. We and others have shown that liver macrophages are the first source of acute-phase mediators, e.g., when toxins like CCl4[42], acetaminophen[43], corpuscular matter, gadolinium or zymosan[44], and the bacterial polysaccharide (LPS)[18] are used to induce an acute-phase response in liver. At the same time, a massive production of chemoattractants is induced by the toxins in cells of the portal vessels and the portal fields, which may account for the recruitment of inflammatory cells and for hepatocellular damage[26]. We assume that hepatocytes “spared” from this damage take over the functions of damaged cells and maintain the acute-phase response. This may explain why we observed changes in the synthesis of ferritins after liver damage, which are similar to those in the liver when the tissue damage takes place in extrahepatic sites[17]. The hypothesis is further supported by the detection of FTH and increased levels of FTL in serum, and in non-damaged areas of the liver. Thereby, although histological examinations of the liver indicate complete recovery 96 h after TAA administration, the increased iron and ferritin concentrations suggest that this is not yet the case.
In conclusion, our results show that damaging noxae like TAA induce a hepatic acute-phase reaction before and during the appearance of acute hepatocellular damage. Iron is taken up by the surviving (spared) liver cells during the phases of damage and recovery. Iron released into serum by damaged cells may be transported to and stored in liver cells via FTL and FTH. Moreover, FTL is not only a marker of iron deposition but also a potential clinical marker for both acute and “chronic” liver damage, as it is not only a component of the iron “cage” within the tissue, but also a secretory protein in response to locally produced acute-phase cytokines.
COMMENTS
Background
The liver has a pivotal role in the homeostasis of iron under physiological conditions, including its storage and distribution to various organs according to their actual needs. The liver also has a central role in iron metabolism during the acute-phase reaction following tissue damage in extra-hepatic organs. Under these conditions, liver does not only increase its iron uptake, but also synthesizes the major iron-transport proteins ferritin (FT) and transferrin, in addition to lipocalin-2, lactoferrin and hepcidin, which are acute-phase proteins involved in systemic and local iron metabolism. Changes of iron metabolism induced by directly liver-damaging noxae like viruses, alcohol, and xenobiotics still need to be investigated.
Research frontiers
The authors provide evidence that the increase in liver iron concentrations after acute liver damage may be rather the consequence than the cause of hepatocyte loss; and that ferritin heavy chain (FTH) is a surrogate marker for hepatocyte damage in serum.
Innovations and breakthroughs
They results show that liver damage by thioacetamide induces a hepatic acute-phase reaction, as seen by early upregulation of cytokines followed by morphologically visible acute hepatocellular damage. Iron is taken up by the surviving (spared) hepatocytes during the phases of damage and recovery. Iron released into serum by dying cells is obviously transported back and stored in liver cells via FTL and FTH.
Applications
The current study increases our understanding of hyperferritinemia and hypersideremia associated with hepatic iron-overload found in non-alcoholic fatty liver disease, non-alcoholic steato-hepatitis, alcoholic liver disease, and chronic hepatitis-C. Moreover, we show that FTL is not only a marker for iron deposition, but also a potential clinical marker for both acute and chronic liver damage, as it is not only a component of the iron ‘cage’ within the tissue, but also a secreted protein in response to locally produced acute-phase cytokines.
Terminology
Ferritin is an intracellular protein, mainly functioning as an iron storage protein. It is made up of two subunits known as FTH and FTL. The two subunits are highly conserved, but independently and differently regulated both at transcriptional and posttranscriptional levels.
Peer-review
Article “Reabsorption of iron into acutely damaged rat liver: a role for ferritins” by Malik et al according to my opinion, is acceptable for publication without additional revision. This article is very interesting for persons involved in the field of hepatology This basic study try to explain what's happened with the iron during acute liver injury.
ACKNOWLEDGMENTS
We’d like to acknowledge the expert technical assistance of Mrs. D. Gerke.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Germany
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair):
Grade E (Poor): 0
Supported by the German Research Foundation, and the Open Access Publication Funds of the Göttingen University.
Institutional review board statement: The studies were performed according to the guidelines of good scientific practice.
Institutional animal care and use committee statement: The animal studies were reviewed and approved by the committee of the Central Institute for Animal Experiments of the University of Goettingen, and the Lower Saxony State Office for Consumer Protection and Food Safety (Study No. 33.9-42502-04-13/1086).
Conflict-of-interest statement: The authors declare that no actual or potential conflict-of-interest in relation to this article exists.
Data sharing statement: The data were obtained, analyzed and used by the authors only.
Peer-review started: July 21, 2017
First decision: August 10, 2017
Article in press: September 13, 2017
P- Reviewer: Markic D, Paraskevas KI S- Editor: Qi Y L- Editor: A E- Editor: Huang Y
Contributor Information
Ihtzaz Ahmed Malik, Institute of Anatomy and Cell Biology, University Medical Center, D-37075 Goettingen, Germany. i.malik@med.uni-goettingen.de.
Jörg Wilting, Institute of Anatomy and Cell Biology, University Medical Center, D-37075 Goettingen, Germany.
Giuliano Ramadori, Department of Gastroenterology and Endocrinology, University Medical Center, D-37075 Goettingen, Germany.
Naila Naz, Faculty of Life Sciences, The University of Manchester, Manchester M13 9PL, United Kingdom.
References
- 1.Frazer DM, Anderson GJ. Iron imports. I. Intestinal iron absorption and its regulation. Am J Physiol Gastrointest Liver Physiol. 2005;289:G631–G635. doi: 10.1152/ajpgi.00220.2005. [DOI] [PubMed] [Google Scholar]
- 2.Ganz T, Nemeth E. Iron metabolism: interactions with normal and disordered erythropoiesis. Cold Spring Harb Perspect Med. 2012;2:a011668. doi: 10.1101/cshperspect.a011668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson GJ, Frazer DM. Hepatic iron metabolism. Semin Liver Dis. 2005;25:420–432. doi: 10.1055/s-2005-923314. [DOI] [PubMed] [Google Scholar]
- 4.Cairo G, Bernuzzi F, Recalcati S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006;1:25–39. doi: 10.1007/BF02829934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muckenthaler MU, Rivella S, Hentze MW, Galy B. A Red Carpet for Iron Metabolism. Cell. 2017;168:344–361. doi: 10.1016/j.cell.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graham RM, Reutens GM, Herbison CE, Delima RD, Chua AC, Olynyk JK, Trinder D. Transferrin receptor 2 mediates uptake of transferrin-bound and non-transferrin-bound iron. J Hepatol. 2008;48:327–334. doi: 10.1016/j.jhep.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Cairo G, Recalcati S, Mantovani A, Locati M. Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol. 2011;32:241–247. doi: 10.1016/j.it.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Darshan D, Vanoaica L, Richman L, Beermann F, Kühn LC. Conditional deletion of ferritin H in mice induces loss of iron storage and liver damage. Hepatology. 2009;50:852–860. doi: 10.1002/hep.23058. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan J, Ward DM. The essential nature of iron usage and regulation. Curr Biol. 2013;23:R642–R646. doi: 10.1016/j.cub.2013.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramm GA, Ruddell RG. Iron homeostasis, hepatocellular injury, and fibrogenesis in hemochromatosis: the role of inflammation in a noninflammatory liver disease. Semin Liver Dis. 2010;30:271–287. doi: 10.1055/s-0030-1255356. [DOI] [PubMed] [Google Scholar]
- 11.Levi S, Yewdall SJ, Harrison PM, Santambrogio P, Cozzi A, Rovida E, Albertini A, Arosio P. Evidence of H- and L-chains have co-operative roles in the iron-uptake mechanism of human ferritin. Biochem J. 1992;288(Pt 2):591–596. doi: 10.1042/bj2880591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sammarco MC, Ditch S, Banerjee A, Grabczyk E. Ferritin L and H subunits are differentially regulated on a post-transcriptional level. J Biol Chem. 2008;283:4578–4587. doi: 10.1074/jbc.M703456200. [DOI] [PubMed] [Google Scholar]
- 13.Sargent PJ, Farnaud S, Evans RW. Structure/function overview of proteins involved in iron storage and transport. Curr Med Chem. 2005;12:2683–2693. doi: 10.2174/092986705774462969. [DOI] [PubMed] [Google Scholar]
- 14.Naz N, Moriconi F, Ahmad S, Amanzada A, Khan S, Mihm S, Ramadori G, Malik IA. Ferritin L is the sole serum ferritin constituent and a positive hepatic acute-phase protein. Shock. 2013;39:520–526. doi: 10.1097/SHK.0b013e31829266b9. [DOI] [PubMed] [Google Scholar]
- 15.Ahmad S, Sultan S, Naz N, Ahmad G, Alwahsh SM, Cameron S, Moriconi F, Ramadori G, Malik IA. Regulation of iron uptake in primary culture rat hepatocytes: the role of acute-phase cytokines. Shock. 2014;41:337–345. doi: 10.1097/SHK.0000000000000107. [DOI] [PubMed] [Google Scholar]
- 16.Ahmad S, Moriconi F, Naz N, Sultan S, Sheikh N, Ramadori G, Malik IA. Ferritin L and Ferritin H are differentially located within hepatic and extra hepatic organs under physiological and acute phase conditions. Int J Clin Exp Pathol. 2013;6:622–629. [PMC free article] [PubMed] [Google Scholar]
- 17.Sheikh N, Dudas J, Ramadori G. Changes of gene expression of iron regulatory proteins during turpentine oil-induced acute-phase response in the rat. Lab Invest. 2007;87:713–725. doi: 10.1038/labinvest.3700553. [DOI] [PubMed] [Google Scholar]
- 18.Ahmad G, Sial GZ, Ramadori P, Dudas J, Batusic DS, Ramadori G. Changes of hepatic lactoferrin gene expression in two mouse models of the acute phase reaction. Int J Biochem Cell Biol. 2011;43:1822–1832. doi: 10.1016/j.biocel.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Sultan S, Pascucci M, Ahmad S, Malik IA, Bianchi A, Ramadori P, Ahmad G, Ramadori G. LIPOCALIN-2 is a major acute-phase protein in a rat and mouse model of sterile abscess. Shock. 2012;37:191–196. doi: 10.1097/SHK.0b013e31823918c2. [DOI] [PubMed] [Google Scholar]
- 20.Naz N, Malik IA, Sheikh N, Ahmad S, Khan S, Blaschke M, Schultze F, Ramadori G. Ferroportin-1 is a ‚nuclear‘-negative acute-phase protein in rat liver: a comparison with other iron-transport proteins. Lab Invest. 2012;92:842–856. doi: 10.1038/labinvest.2012.52. [DOI] [PubMed] [Google Scholar]
- 21.Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48:1420–1429. doi: 10.1002/hep.22486. [DOI] [PubMed] [Google Scholar]
- 22.Pietrangelo A. Iron in NASH, chronic liver diseases and HCC: how much iron is too much? J Hepatol. 2009;50:249–251. doi: 10.1016/j.jhep.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 23.Amanzada A, Schneider S, Moriconi F, Lindhorst A, Suermann T, van Thiel DH, Mihm S, Ramadori G. Early anemia and rapid virological response improve the predictive efficiency of IL28B-genotype for treatment outcome to antiviral combination therapy in patients infected with chronic HCV genotype 1. J Med Virol. 2012;84:1208–1216. doi: 10.1002/jmv.23323. [DOI] [PubMed] [Google Scholar]
- 24.Amanzada A, Goralczyk AD, Moriconi F, van Thiel DH, Ramadori G, Mihm S. Vitamin D status and serum ferritin concentration in chronic hepatitis C virus type 1 infection. J Med Virol. 2013;85:1534–1541. doi: 10.1002/jmv.23632. [DOI] [PubMed] [Google Scholar]
- 25.Kowdley KV, Belt P, Wilson LA, Yeh MM, Neuschwander-Tetri BA, Chalasani N, Sanyal AJ, Nelson JE; NASH Clinical Research Network. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2012;55:77–85. doi: 10.1002/hep.24706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amanzada A, Moriconi F, Mansuroglu T, Cameron S, Ramadori G, Malik IA. Induction of chemokines and cytokines before neutrophils and macrophage recruitment in different regions of rat liver after TAA administration. Lab Invest. 2014;94:235–247. doi: 10.1038/labinvest.2013.134. [DOI] [PubMed] [Google Scholar]
- 27.Malik IA, Moriconi F, Sheikh N, Naz N, Khan S, Dudas J, Mansuroglu T, Hess CF, Rave-Fränk M, Christiansen H, et al. Single-dose gamma-irradiation induces up-regulation of chemokine gene expression and recruitment of granulocytes into the portal area but not into other regions of rat hepatic tissue. Am J Pathol. 2010;176:1801–1815. doi: 10.2353/ajpath.2010.090505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal Biochem. 2004;331:370–375. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 29.Rahman TM, Hodgson HJ. The effects of early and late administration of inhibitors of inducible nitric oxide synthase in a thioacetamide-induced model of acute hepatic failure in the rat. J Hepatol. 2003;38:583–590. doi: 10.1016/s0168-8278(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 30.Chen TM, Subeq YM, Lee RP, Chiou TW, Hsu BG. Single dose intravenous thioacetamide administration as a model of acute liver damage in rats. Int J Exp Pathol. 2008;89:223–231. doi: 10.1111/j.1365-2613.2008.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malik IA, Naz N, Sheikh N, Khan S, Moriconi F, Blaschke M, Ramadori G. Comparison of changes in gene expression of transferrin receptor-1 and other iron-regulatory proteins in rat liver and brain during acute-phase response. Cell Tissue Res. 2011;344:299–312. doi: 10.1007/s00441-011-1152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cairo G, Tacchini L, Schiaffonati L, Rappocciolo E, Ventura E, Pietrangelo A. Translational regulation of ferritin synthesis in rat liver. Effects of chronic dietary iron overload. Biochem J. 1989;264:925–928. doi: 10.1042/bj2640925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cairo G, Rappocciolo E, Tacchini L, Schiaffonati L. Expression of the genes for the ferritin H and L subunits in rat liver and heart. Evidence for tissue-specific regulations at pre- and post-translational levels. Biochem J. 1991;275(Pt 3):813–816. doi: 10.1042/bj2750813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyron-Holtz EG, Moshe-Belizowski S, Cohen LA. A possible role for secreted ferritin in tissue iron distribution. J Neural Transm (Vienna) 2011;118:337–347. doi: 10.1007/s00702-011-0582-0. [DOI] [PubMed] [Google Scholar]
- 35.Surguladze N, Patton S, Cozzi A, Fried MG, Connor JR. Characterization of nuclear ferritin and mechanism of translocation. Biochem J. 2005;388:731–740. doi: 10.1042/BJ20041853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai CX, Linsenmayer TF. Nuclear translocation of ferritin in corneal epithelial cells. J Cell Sci. 2001;114:2327–2334. doi: 10.1242/jcs.114.12.2327. [DOI] [PubMed] [Google Scholar]
- 37.Smith AG, Carthew P, Francis JE, Edwards RE, Dinsdale D. Characterization and accumulation of ferritin in hepatocyte nuclei of mice with iron overload. Hepatology. 1990;12:1399–1405. doi: 10.1002/hep.1840120622. [DOI] [PubMed] [Google Scholar]
- 38.Thompson KJ, Fried MG, Ye Z, Boyer P, Connor JR. Regulation, mechanisms and proposed function of ferritin translocation to cell nuclei. J Cell Sci. 2002;115:2165–2177. doi: 10.1242/jcs.115.10.2165. [DOI] [PubMed] [Google Scholar]
- 39.Izawa T, Murakami H, Wijesundera KK, Golbar HM, Kuwamura M, Yamate J. Inflammatory regulation of iron metabolism during thioacetamide-induced acute liver injury in rats. Exp Toxicol Pathol. 2014;66:155–162. doi: 10.1016/j.etp.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Kuramochi M, Izawa T, Pervin M, Bondoc A, Kuwamura M, Yamate J. The kinetics of damage-associated molecular patterns (DAMPs) and toll-like receptors during thioacetamide-induced acute liver injury in rats. Exp Toxicol Pathol. 2016;68:471–477. doi: 10.1016/j.etp.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Golbar HM, Izawa T, Wijesundera KK, Bondoc A, Tennakoon AH, Kuwamura M, Yamate J. Depletion of Hepatic Macrophages Aggravates Liver Lesions Induced in Rats by Thioacetamide (TAA) Toxicol Pathol. 2016;44:246–258. doi: 10.1177/0192623315621191. [DOI] [PubMed] [Google Scholar]
- 42.Neubauer K, Lindhorst A, Tron K, Ramadori G, Saile B. Decrease of PECAM-1-gene-expression induced by proinflammatory cytokines IFN-gamma and IFN-alpha is reversed by TGF-beta in sinusoidal endothelial cells and hepatic mononuclear phagocytes. BMC Physiol. 2008;8:9. doi: 10.1186/1472-6793-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woolbright BL, Jaeschke H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol. 2017;66:836–848. doi: 10.1016/j.jhep.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moriconi F, Ahmad G, Ramadori P, Malik I, Sheikh N, Merli M, Riggio O, Dudas J, Ramadori G. Phagocytosis of gadolinium chloride or zymosan induces simultaneous upregulation of hepcidin- and downregulation of hemojuvelin- and Fpn-1-gene expression in murine liver. Lab Invest. 2009;89:1252–1260. doi: 10.1038/labinvest.2009.92. [DOI] [PubMed] [Google Scholar]