

Abstract
Activation of immunomodulatory pathways in response to invasive fungi can impair clearance and promote persistent infections. The Programmed Cell Death Protein-1 (PD-1) signaling pathway inhibits immune effector responses against tumors and immune checkpoint inhibitors that block this pathway are being increasingly used as cancer therapy. The objective of the current study was to investigate whether this pathway contributes to persistent fungal infection and to determine whether anti-PD-1 antibody treatment improves fungal clearance. Studies were performed using C57BL/6 mice infected with a moderately virulent strain of Cryptococcus neoformans (52D) which resulted in prolonged elevations in fungal burden and histopathologic evidence of chronic lung inflammation. Persistent infection was associated with increased and sustained expression of PD-1 on lung lymphocytes, including a mixed population of CD4+ T cells. In parallel, expression of the PD-1 ligands, PD-L1 and PD-L2, was similarly upregulated on specific subsets of resident and recruited lung dendritic cells and macrophages. Treatment of persistently-infected mice for four weeks by repetitive administration of neutralizing anti-PD-1 antibody significantly improved pulmonary fungal clearance. Treatment was well tolerated without evidence of morbidity. Immunophenotyping revealed that anti-PD-1 antibody treatment did not alter immune effector cell numbers or myeloid cell activation. Treatment did reduce gene expression of IL-5 and IL-10 by lung leukocytes and promoted sustained upregulation of OX40 by Th1 and Th17 cells. Collectively, this study demonstrates that PD-1 signaling promotes persistent cryptococcal lung infection and identifies this pathway as a potential target for novel immune-based treatments of chronic fungal disease.
Keywords: PD-1, T cells, dendritic cells, macrophages, lung, rodent, Cryptococcus
Introduction
Cryptococcus neoformans (C. neoformans) is an encapsulated fungus acquired by inhalation. Pulmonary infection with C. neoformans results in one of three generalized outcomes: clearance, persistence, or progression (1). Persistent and progressive infections are frequent in immunocompromised individuals; infection with C. neoformans constitutes the second most common fungal infection in organ transplant recipients (1). Further, out of an estimated 278,000 new cases of cryptococcosis annually, approximately 223,100 patients will develop cryptococcal meningitis with 81% of these cases resulting in death (2); thus cryptococcal disease is the second leading cause of AIDS-related mortality in HIV-positive individuals behind tuberculosis. In patients surviving initial infection, up to 15% of HIV-positive, C. neoformans-infected individuals relapse despite conventional anti-fungal treatments (3). Although uncommon, clinically significant infections have been reported in seemingly immunocompetent hosts (4–6). Antibiotic treatment of these infections is often toxic and lengthy (potentially life-long) (7). Thus, novel approaches that augment conventional therapy are needed.
Persistent cryptococcal lung infection, effectively modeled by intratracheal inoculation of C57BL/6 mice with a moderately virulent strain of C. neoformans (52D), is characterized by a mixed T helper (Th) polarization immunophenotype, which contains but does not eliminate the fungus (8–12). Persistent lung infection reflects an immune balance between concurrent induction of interferon gamma (IFNγ) and interleukin 17 (IL-17), which promote classical (M1) macrophage activation and fungal killing (13–16), with expression of IL-4, IL-13 and IL-10 which promote alternative (M2) macrophage activation and intracellular fungal survival (9, 15, 17). Our studies have shown that C. neoformans can evade host defenses by expression of virulence factors which impair Type 1 (T1) and favor non-protective Type 2 (T2) responses (18–20). To counteract this, we have successfully intervened with anti-IL-10 receptor antibody blockade, which augmented protective T1 immunity and improved fungal clearance in persistently-infected mice (17). Importantly, these studies demonstrated that exogenous immune modulation, even at late stages of established infection, can be therapeutically effective.
In the current study, we investigate whether the Programmed Cell Death Receptor-1 (PD-1) pathway contributes to persistent cryptococcal lung infection. PD-1 and its two ligands, PD-1 Ligand-1 (PD-L1) and PD-L2 comprise an important immunomodulatory pathway which under homeostatic conditions limits effector immune responses and promotes tolerance (reviewed in (21)). PD-1 is primarily expressed on lymphoid cells, especially activated T cells (22, 23). PD-L1 is expressed on a wide variety of antigen-presenting cells (APCs), T cells, and epithelial cells whereas PD-L2 expression is restricted primarily to antigen presenting cells (APCs) (22). Our interest in studying this pathway in the context of fungal persistence stems from studies demonstrating that PD-1 signaling impairs clearance of some viral (24–26) and bacterial pathogens (27, 28). Furthermore, numerous studies have shown that this pathway promotes tumor immune evasion which has led to the development of potent immune checkpoint inhibitors which effectively treat tumors in mice (29, 30) and humans (reviewed in (31, 32)).
Knowledge of the role of this pathway in cryptococcal infection is limited. A study by Guerrero et al demonstrated that infection with more virulent, mucoid colonies of C. neoformans is associated with increased expression of PD-L1 and PD-L2 by alveolar macrophages (33). We have recently shown that GM-CSF promotes PD-L2 expression by dendritic cells (DCs) in the lungs of mice with persistent cryptococcal lung infection (8). Neither study provided a detailed evaluation of this pathway throughout infection or assessed the therapeutic effects of PD-1 blockade. Results of the current study show that persistent cryptococcal lung infection induced broad and sustained upregulation of PD-1 and its ligands, PD-L1 and PD-L2, on specific lung lymphoid and myeloid cell subsets. Treatment of persistently-infected mice with a blocking anti-PD-1 antibody enhanced fungal clearance, likely through mechanisms that diminished T2 bias and enhanced T cell activation. Collectively, this study demonstrates that the PD-1 signaling pathway promotes persistent cryptococcal lung infection and that targeted blockade of this pathway is therapeutically beneficial and well-tolerated.
Materials and Methods
Mice
Female C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed under specific pathogen-free conditions in the Animal Care Facility at the VA Ann Arbor Healthcare System. All experiments were approved by the Veterans Administration Institutional Animal Care and Use Committee. Mice were 8–12 weeks of age at the time of infection with C. neoformans.
C. neoformans
C. neoformans strain 52D was obtained from the American Type Culture Collection (catalog #24067; Manassas, VA) and grown to a late logarithmic phase by incubation on a shaker at 37°C for 60–72 h in Sabouraud dextrose broth (1% Neopeptone, 2% dextrose; Difco, Detroit, MI). Cultures were centrifuged and the pellets washed with non-pyrogenic saline (Travenol, Deerfield, IL). Cells were counted in the presence of trypan blue using a hemocytometer and subsequently resuspended at 3.33 × 105 cells/mL in non-pyrogenic saline immediately prior to intratracheal inoculation.
Intratracheal inoculation of C. neoformans
Mice were anesthetized by intraperitoneal (IP) injection of ketamine (100 mg/kg; Hospira, Inc., Lake Forest, IL) and xylazine (6.8 mg/kg; Lloyd Laboratories, Shenandoah, IA). Following a midline neck incision, strap muscles were separated laterally to expose the trachea. Under direct vision, 104 C. neoformans (suspended in 30 μL non-pyrogenic saline) were injected into the trachea using a 30-gauge needle attached to a 1-mL syringe mounted on a repetitive pipette (Stepper; Tridak, Brookfield, CT). After inoculation, skin was closed using a cyanoacrylate adhesive.
Tissue Collection
Lungs were perfused in situ via the right ventricle using 8–10 mL of PBS until pulmonary vessels appeared clear. Lungs were excised, minced, and placed in a gentleMACS C tube (Miltenyi Biotec; San Diego, CA) containing 5 mL of digestion buffer (34). After a short, gentle agitation using a gentleMACS dissociator (Miltenyi Biotec), lung suspensions were incubated on a rotator at 37°C for 35 m. Lung suspensions were next vigorously homogenized using a gentleMACS dissociator and centrifuged. ACK Lysing Buffer (KD Medical; Columbia, MD) was used to lyse erythrocytes. Samples were then passed through 10-mL syringes rapidly 10–12 times to further break apart tissue and cell clumps. Next, samples were centrifuged in 20% Percoll gradients (Sigma-Aldrich; St. Louis, MO) to separate leukocytes from epithelial cells and debris. Thereafter, cells were resuspended in RPMI complete media (34) and counted on a hemocytometer in the presence of trypan blue.
RNA Isolation and Quantitative RT-PCR
Five million cells were pelleted in 1.5-mL microcentrifuge tubes and RNA was isolated using TRIzol (Ambion by Life Technologies; Carlsbad, CA). DNA contamination was removed using the Turbo DNA-free kit (Ambion by Life Technologies ; Carlsbad, CA) according to the manufacturer’s instructions. RNA was quantified and assessed for quality using a NanoDrop 2000 (Thermo Fisher Scientific; Wilmington, DE). Quantitative RT-PCR (qPCR) was performed using the QuantiTect SYBR Green RT-PCR Kit (Qiagen; Germantown, MD) according to the manufacturer’s instructions with a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific; Wilmington, DE). qPCR primers are listed in Supplemental Table I.
PD-1 Blocking Antibody
A low-endotoxin (<2EU/mg), azide-free purified antibody against murine PD-1 (clone RMP1-14) or rat IgG2a, κ low-endotoxin isotype control antibody (clone 2A3) (both from Bio X Cell; West Lebanon, NH) was administered at a dose of 200 μg antibody in 200 μL sterile PBS (~ 10 mg/kg) via IP injection twice per week based on the previously-established efficacy of this dosing strategy in other experimental models of fungal infection (35, 36). Treatment was initiated at 3 weeks post infection (WPI) and continued for either 2 or 4 weeks.
Colony Forming Units (CFU) Assay
To assess fungal burden, 10 μL aliquots of lung digests or brain or spleen homogenates were plated on Sabouraud dextrose agar plates in duplicate serial 10-fold dilutions. Plates were incubated at room temperature for 45–48 hours. At the end of incubation, colonies were counted and CFU/organ was calculated.
Effect of isotype control and anti PD-1 antibodies on fungal growth in vitro
To assess the effect of antibodies on the growth of C. neoformans in vitro, 106 C. neoformans ere cultured in SBD media at 37°C with either 0.8 μg/mL (low) or 8.0 μg/mL (high) anti-PD-1 or rat IgG2a isotype control antibody followed by counting viable C. neoformans on a hemocytometer at 24 and 48 hours.
Lung Histology
Lungs were fixed by inflation with a 1:1 mixture of PBS and Optimum Cutting Temperature (OCT) compound followed by freezing on dry ice. After freezing, 5 μm sections were cut and stained with hematoxylin and eosin (H&E) for histological analysis.
Antibodies for Flow Cytometry
Antibodies targeting the following murine proteins were purchased from BioLegend (San Diego, CA): PD-L1 (clone 10F.9G2), I-A/I-E (M5/114.15.2), CD45 (30-F11), CD19 (6D5), CD11b (M1/70), PD-1 (29F.1A12), TCRβ chain (H57-597), CD8a (53-6.7), PD-L2 (TY25), CD80 (16-10A1), CD4 (GK1.5), Ly6G (1A8), Ly6C (HK1.4), T-bet (4B10), ICOS (C398.4A), OX40 (OX-86), TCRγ/δ (GL3) and CD16/32 (clone 93). Antibodies targeting the following murine proteins were purchased from BD Biosciences (Franklin Lake, NJ): Siglec F (E50-2440), CD24 (M1/69), and CD3ε (145-2C11). Antibodies targeting the following murine proteins were purchased from eBioscience (Asheville, NC): iNOS (CXNFT), FoxP3 (FJK-16s), Ki67 (SolA15), CD11c (N418), Gata3 (TWAJ), and RORγt (B2D). A polyclonal antibody targeting murine arginase was purchased from R&D Systems. Isotype control antibodies were used according to manufacturer’s instructions in all experiments. Antibodies were conjugated to the following fluorophores: Allophycocyanin (APC), Alexa Fluor 700, APC-cyanine 7 (APC-Cy7), fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin-chlorophyll-protein (PerCP)-Cy5.5, PE-eFluor 610, PE-Cy5, PE-Cy7, Brilliant Violet (BV) 421, and BV 650.
Cell Staining and Flow Cytometry
Cell surface staining, including blockade of Fc receptors, and sample analysis by flow cytometry were performed as described previously (34). For staining of intracellular and nuclear proteins, cells were incubated with a viability dye (Zombie Aqua Fixable Viability dye; BioLegend) for 20 min at room temperature in the dark. Cells were next washed in PBS then incubated with an Fc receptor-blocking antibody for 10 min at 4°C. After washing, cells were incubated and washed using a transcription factor staining kit (FoxP3/transcription factor staining kit; eBioscience) per manufacturer’s instructions. Thereafter, cells were incubated with antibodies raised against cytoplasmic and nuclear antigens (in permeabilization buffer) for 30 min at room temperature in the dark. Cells were then washed twice with permeabilization buffer and subsequently stored in 2% paraformaldehyde in PBS overnight. Flow cytometry was performed on either a Becton Dickinson (BD) LSR II or a BD LSR Fortessa (BD Biosciences), utilizing 12 PMT channels. After gating out debris and doublets, a total of 100,000 events were acquired from each sample using FACSDiva data acquisition software. Data were analyzed using FlowJo software (Treestar; Ashland, OR). Specific leukocyte populations were identified using gating strategies described in the Results Sections and Figure Legends. To calculate the total number of cells in each population of interest in each sample, the corresponding percentage was multiplied by the total number of CD45+ cells in that sample. The latter value was calculated for each sample as the product of the percentage of CD45+ cells and the original hemocytometer count of total cells identified within that sample.
Statistical Analysis
All data are presented as mean ± SEM of 4–5 (Figures 1–3) or 4–9 (Figures 4, 6–9, S1, S2) mice per group. Multiple cohort data were analyzed by ANOVA with post-hoc Fisher’s LSD test for multiple comparisons. Two cohort comparisons were performed by Student’s T test. P<0.05 was considered significant in all cases.
Figure 1.
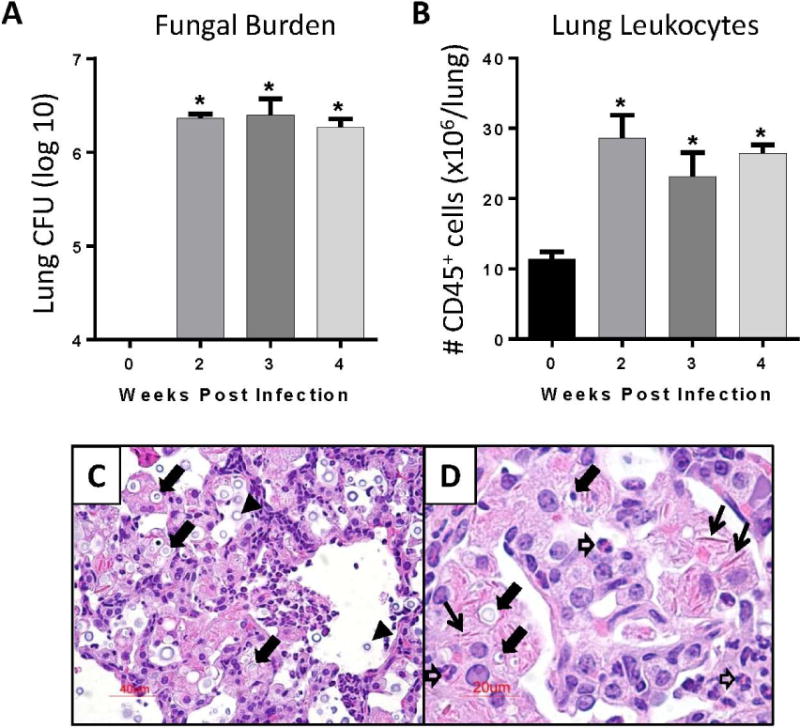
Induction of persistent cryptococcal lung infection in C57BL/6 mice. (A - D) C57BL/6 mice were infected by the intratracheal route with C. neoformans strain 52D. At day 0 (uninfected) and 2, 3, and 4 weeks post infection (WPI), lungs were harvested for analysis. (A) Fungal lung burden was assessed using CFU assays. (B) Number of CD45+ lung leukocytes was assessed using flow cytometric analysis. (C, D) Representative lung sections (H&E stain) from infected mice at 4 WPI at 200 X (C) and 400 X (D) magnifications. Note the presence of: numerous intracellular cryptococci within macrophages (closed block arrows), extracellular cryptococci in the alveolar space (black arrowheads), eosinophils (open block arrows), and extracellular crystals (thin black arrows). For A, B: n=4–5 mice assayed individually per time-point; *p<0.05 by ANOVA with Fisher’s LSD post hoc test vs. Day 0 (uninfected).
Figure 3.
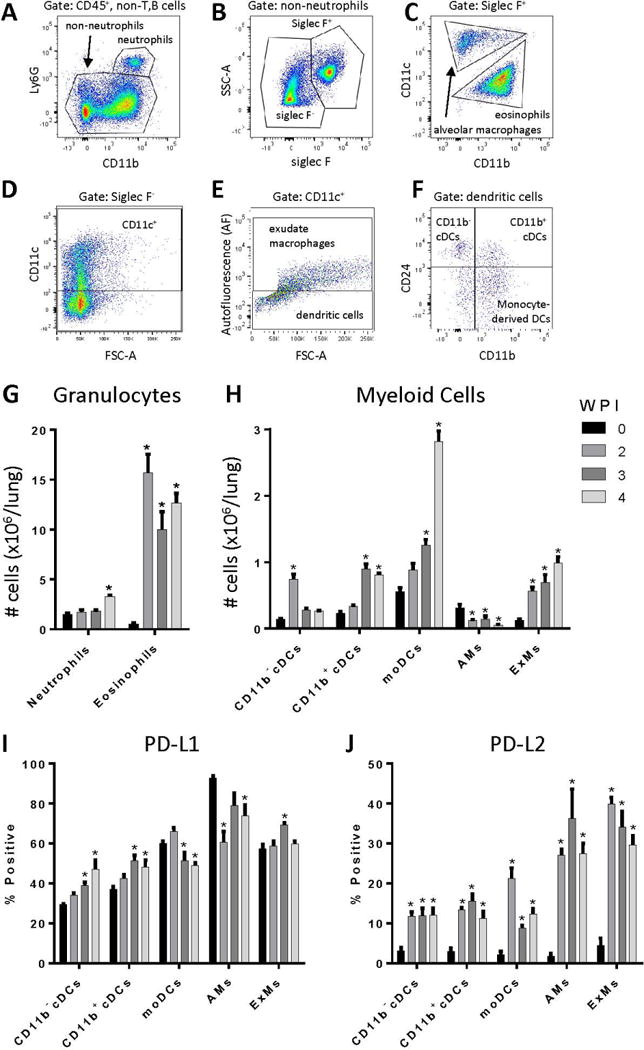
Persistent cryptococcal lung infection causes accumulation of numerous myeloid cell populations and increased dendritic cell and macrophage PD-L1 and PD-L2 expression. Single cell suspensions of lung cells were subjected to 12-color flow cytometry for analysis of myeloid cell populations (A-F). Representative gating on cells obtained at 3 WPI is shown. Following selection of viable leukocytes (not shown), T and B cells were excluded by expression of CD19 or TCRβ (not shown). Next, neutrophils were characterized based on expression of CD11b and Ly6G (A). Non-neutrophils were then separated into Siglec F+ and Siglec F− populations (B). Within the Siglec F+ population, alveolar macrophages were identified as CD11b−CD11c+, whereas eosinophils were defined as CD11b+CD11c− (C). Siglec F− cells were gated on CD11c+ cells (D). The Siglec F−CD11c+ population was divided into exudate macrophages (ExMs) and dendritic cells (DCs); exudate macrophages were characterized as autofluorescence (AF)+ whereas dendritic cells were AF− (E). Dendritic cells were divided into three subgroups based on expression of CD24 and CD11b (F): CD11b− cDCs (CD24+CD11b−; i.e., CD103+ DCs), CD11b+ cDCs (CD24+CD11b+), and monocyte-derived DCs (moDCs; CD24−CD11b+). (G-H) Application of this gating scheme facilitated the enumeration of: (G) granulocyte subsets and (H) myeloid cell subsets including CD11b− and CD11b+ conventional DCs (cDCs), monocyte-derived DCs (moDCs), alveolar macrophages (AMs), and exudate macrophages (ExMs). Expression of PD-L1 and PD-L2 on myeloid cells was assessed by flow cytometry. Relative expression of PD-L1 (I) and PD-L2 (J) on dendritic cells and macrophages is shown. For G-J: n=4–5 mice assayed individually per time-point; *=p<0.05 by ANOVA with Fisher’s LSD post hoc test vs. Day 0 (uninfected).
Figure 4.
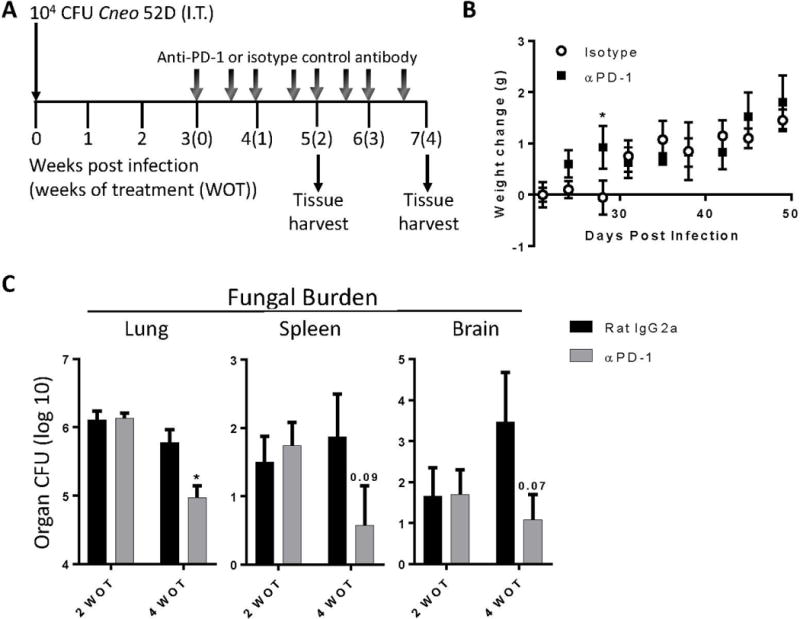
Anti-PD1 antibody treatment promotes fungal clearance in mice with cryptococcal lung infection. (A) C57BL/6 mice were infected I.T. with C. neoformans strain 52D. Beginning at 3 WPI, mice were administered 200 μg of either neutralizing anti-PD-1 antibody (RMP1-14) or isotype-matched control antibody (2A3) twice per week for 2–4 weeks of treatment (WOT). (B) Weight change in cohorts of treated mice. (C) Fungal burden in lungs, spleens, and brains of treated cohorts of mice. For B, C: n=4–9/cohort assayed individually in two separate experiments; *=p<0.05 by unpaired Student t test.
Figure 9.
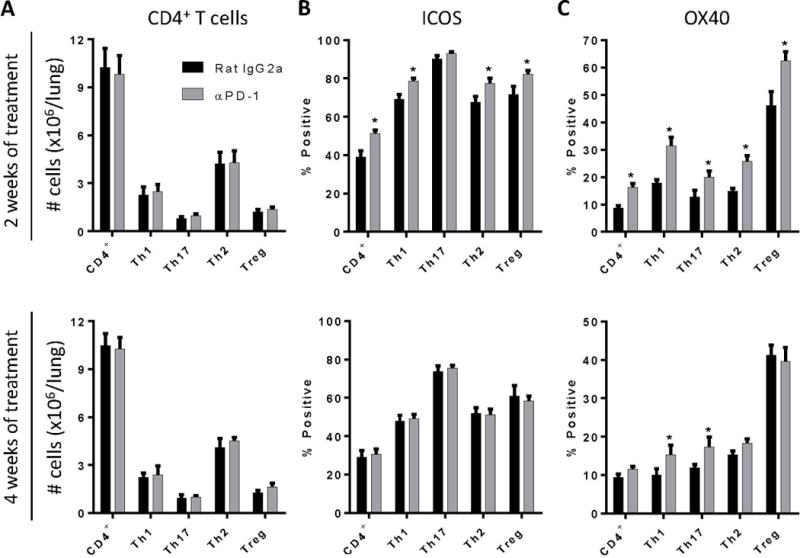
Anti-PD-1 antibody treatment promotes sustained expression of ICOS and OX40 on Th1 and Th17 cells. Infected C57BL/6 mice treated with either neutralizing anti-PD-1 or control antibody were evaluated (by flow cytometric analysis) at 2 WOT (top panels) or 4 WOT (bottom panels) for: (A) total numbers of CD4+ T cells and CD4+ T cell subsets and their expression of T cell activation markers including (B) ICOS, and (C) OX40. For A-C: n=4–9/cohort assayed individually in two separate experiments; *=p<0.05 by unpaired Student t test.
Figure 6.
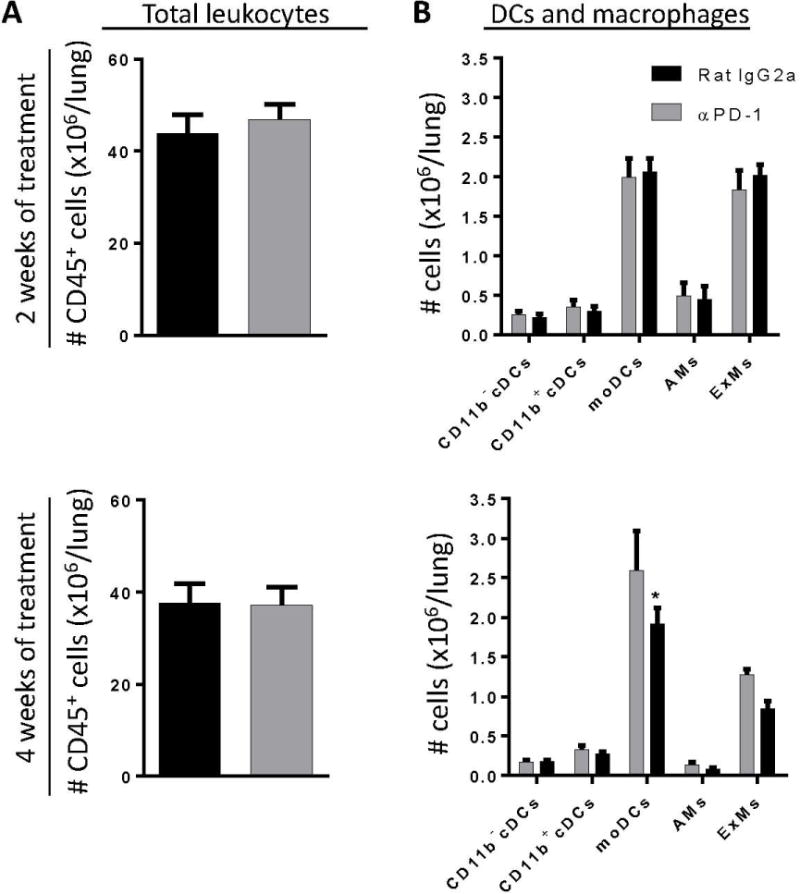
Anti-PD-1 antibody treatment does not substantially alter lung myeloid cell accumulation in the lungs of mice with cryptococcal lung infection. Infected C57BL/6 mice treated with either neutralizing anti-PD-1 antibody or control antibody were evaluated (by flow cytometric analysis) at 2 WOT (top panels) or 4 WOT (bottom panels) for numbers of (A) total lung CD45+ leukocytes, and (B) subsets of lung DC and macrophages. For A, B: n=4–9/cohort assayed individually in two separate experiments; *p<0.05 by unpaired Student t test.
Results
Persistent cryptococcal lung infection is associated with increased PD-1 expression on polarized CD4+ T cells
The first objective of the current study was to determine whether the PD-1 signaling pathway is upregulated in response to cryptococcal lung infection. To establish persistent cryptococcal lung infection, we inoculated C57BL/6 mice with C. neoformans strain 52D via the intratracheal route. At 0, 2, 3 and 4 WPI, pulmonary fungal burden was quantified by CFU assays, and CD45+ lung leukocytes were enumerated by flow cytometric analysis. Fungal burden increased to approximately 106–107 CFU by 2 WPI and then remained at the same level throughout the duration of the study (Figure 1A) consistent with other studies performed using this model system (10, 17, 37–40). Persistent infection was associated with a sustained accumulation of lung leukocytes (Figure 1B). Histologic evaluation of lung sections obtained from mice at 4 WPI (Figure 1C, D) identified characteristic features of persistent infection including the presence of numerous cryptococci, many of them located intracellularly within large macrophages. Loose alveolar infiltrates containing extracellular crystals, eosinophils, and small mononuclear cells with lymphocyte morphology were readily observed.
We used flow cytometric analysis of lung leukocytes obtained at 2, 3, and 4 WPI to assess whether lymphocytes present in these immune infiltrates expressed PD-1 (Figure 2A–I). Our results demonstrated that persistent cryptococcal lung infection is associated with a robust and sustained 2–3 fold increase in the total numbers of CD4+ T cells in the lung at 2, 3, and 4 WPI. CD8+ T cell numbers were increased only at 4 WPI, whereas the relatively small numbers of δγ T cells briefly increased at 2 and 3 WPI but returned to baseline thereafter. A non-significant trend towards increased accumulation of B cells was also observed. We found that PD-1 expression was significantly increased on CD4+, CD8+, and δγ T cells in the lungs of persistently-infected mice as early as 2 WPI relative to expression on T cells in uninfected mice (Figure 2J). PD-1 expression remained elevated on CD4+ and CD8+ T cells at 3 and 4 WPI whereas expression of PD-1 on γδ T cells returned to baseline by 4 WPI.
Figure 2.
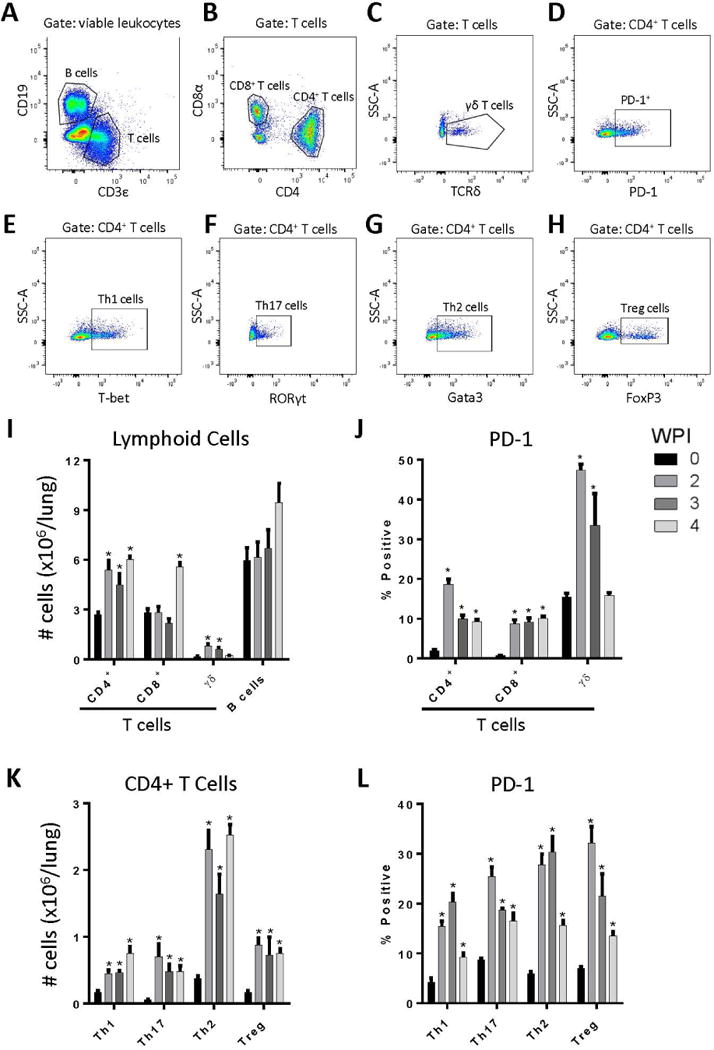
Persistent cryptococcal lung infection promotes lymphoid cell accumulation and increased T cell PD-1 expression. Following infection of mice with C. neoformans strain 52D, lungs were collected and processed for flow cytometry analysis at 0, 2, 3, and 4 WPI. Lung single cell suspensions were subjected to 12-color flow cytometry for analysis of lymphoid cell populations and PD-1 expression (A-H). Representative gating on cells obtained at 3 WPI is shown. Following selection of viable leukocytes based on expression of CD45 and a viability dye (not shown), T and B cells were characterized based on expression of CD19 (B cells; A) or CD3ε (T cells; A). Next, subsets of T cells were characterized by expression of CD8α (CD8+ T cells; B), CD4 (CD4+ T cells; B), or TCRδ (γδ T cells; C). Representative PD-1 staining pattern for CD4+ T cells is shown (D). Subsets of CD4+ T cells were characterized based on expression of hallmark transcription factors T-bet (Th1 cells; E), RORγt (Th17 cells; F), Gata3 (Th2 cells; G), and FoxP3 (Treg cells; H). (I-L) Application of this gating scheme facilitated: (I) the enumeration of specific lymphocyte subsets; (J) PD-1 expression on lymphocytes subsets; (K) enumeration of CD4+ T cell subsets; and (L) PD-1 expression on CD4+ T cell subsets. For I-L: n=4–5 mice assayed individually per time-point; *=p<0.05 by ANOVA with Fisher’s LSD post hoc test vs. Day 0 (uninfected).
To evaluate PD-1 expression on specific Th subsets, we utilized intracellular staining of the nuclear transcription factors T-bet, RORγt, GATA3, and FoxP3 to identify Th1, Th17, Th2, and T regulatory (Treg) cells respectively (refer to Figure 2A–H). Using this approach, our data demonstrated that the numbers of Th1 and Th17 cells increased 2- to 3-fold relative to uninfected mice, whereas the number of Th2 cells and Treg cells increased 4 to 5-fold (Figure 2K). In uninfected mice, PD-1 expression on each Th subset was less than 10% (Figure 2L). In response to infection, PD-1 expression increased to between 20 and 30% on each Th subset and remained elevated relative to uninfected mice at all time-points studied. Collectively, our findings demonstrate increased PD-1 expression on CD4+, CD8+, and γδ T cells accumulating in the lungs of mice that develop persistent cryptococcal infection. Moreover, increased PD-1 expression was not limited to any one subset of CD4+ T cells; rather, PD-1 expression was comparably increased and sustained on Th1, Th17, Th2, and Treg cells (Figure 2L) suggesting that the preferential enhancement of Th2 responses observed in this model is not attributable to differential expression of PD-1.
Persistent cryptococcal lung infection is associated with increased expression of PD-L1 and PD-L2 on specific subsets of lung dendritic cells and macrophages
We evaluated whether increased PD-1 expression on T cell subsets in the lung was associated with a concomitant increase in the expression of PD-1 ligands, PD-L1 and PD-L2, on lung myeloid cells. Specific subsets of lung dendritic cells and macrophages were identified within lung leukocyte populations obtained at 0, 2, 3, and 4 WPI using flow cytometric analysis and a detailed gating scheme (refer to (8, 41, 42) and Figure 3A–F). Results demonstrated a significant 2-fold increase in the number of lung neutrophils present at 4 WPI, and a significant 10- to 15-fold increase in the number of eosinophils present at all time-points post-infection (Figure 3G). Amongst DC populations (Figure 3H), CD11b− cDC numbers were increased 4-fold at 2 WPI followed by a return to baseline by 3 WPI, whereas the number of CD11b+ cDCs were unchanged at 2 WPI but were increased 3-fold at 3 and 4 WPI (Figure 3H). In contrast, numbers of non-resident monocyte-derived DCs increased almost 6-fold by 4 WPI at which point they outnumbered CD11b− DCs and CD11b+ DCs by a 2:1 ratio. Amongst macrophage populations, numbers of resident alveolar macrophages decreased following infection, whereas we observed a progressive 5-fold expansion in recruited exudate macrophages by 4 WPI (Figure 3H). In response to infection, lung macrophages demonstrated increased intracellular staining for the M2 activation marker, arginase, but not the M1 activation marker, inducible nitric oxide synthase (iNOS) consistent with an alternatively-activated phenotype (data not shown).
In uninfected mice, PD-L1 was expressed on approximately 30% - 60% of lung dendritic cells and 60% - 90% of macrophages (Figure 3I). In response to infection, PD-L1 expression was modestly increased on CD11b− cDCs, CD11b+ cDCs, and exudate macrophages whereas expression decreased slightly on moDCs and alveolar macrophages (Figure 3I). In contrast, PD-L2 was expressed on less than 5% of all myeloid cells in uninfected lungs but its expression was markedly increased and sustained on all myeloid cell populations examined at 2, 3, and 4 WPI (Figure 3J); increased PD-L2 expression was most prominent on alveolar and exudate macrophages, with 30–40% of these populations expressing PD-L2 in response to persistent infection. Thus, our data reveal increased and sustained lung myeloid cell expression of PD-L1 and PD-L2 in mice with persistent cryptococcal infection.
Anti-PD-1 antibody treatment improves fungal clearance in mice with persistent cryptococcal lung infection
Numerous studies have shown that blockade of the PD-1 signaling pathway can promote enhanced immune responses against cancers by altering important immunomodulatory mechanisms active in established tumor microenvironments (29–31, 43, 44). Having identified that PD-1 and its ligands were upregulated on immune effector cells in the lungs of mice with persistent cryptococcal lung infection, our next objective was to determine whether antibody-mediated blockade of this pathway could improve fungal clearance in this model. We administered neutralizing anti-PD-1 antibody or isotype control antibody twice per week at a dose of 200 μg/mouse (see Materials and Methods and Figure 4A). Treatment was initiated at 3 WPI and continued for 2 or 4 weeks at which times fungal burden in the lung, spleen, and brain was assessed. During the period of antibody administration, no significant weight loss (Figure 4B) or alterations in animal behavior scores (data not shown) were observed between cohorts of mice treated with anti-PD-1 versus control antibody. Our results showed that 2 weeks of treatment (WOT) with anti-PD-1 antibody did not significantly alter fungal burden in the lungs, brains, or spleens of infected mice relative to isotype control treated mice (Figure 4C). However, mice treated for 4 weeks with anti-PD-1 antibody displayed significant reductions in pulmonary fungal burden relative to isotype control treated mice (Figure 4C); non-significant trends toward reduced fungal burden in the brains and spleens of anti-PD-1 antibody treated mice were also observed.
We considered whether antibody administration might directly alter fungal growth. Data from our prior study investigating the effects of antibody-mediate IL-10 blockade demonstrated comparable pulmonary fungal burden at 5 WPI in mice receiving isotype control antibody relative to infected but untreated mice (17). In addition, data from the current study shows that mice treated with isotype control antibody remained persistently-infected (pulmonary fungal burdens near 6 log CFU; Figure 4C). Furthermore, we observed no alteration in fungal growth in vitro when C. neoformans was cultured for 24 or 48 hours in the presence of two different doses (0.8 μg/mL or 8.0 μg/mL) of either isotype control (IgG2A) or anti-PD-1 antibodies (relative to C. neoformans cultured with no antibody added; data not shown). This collective data suggests that the antibodies administered to mice in this study did not directly alter fungal growth. Thus, given the additional animal use required and the logistical complexity involved, additional cohorts of infected but untreated mice were not included in our in-vivo studies assessing the effects of antibody-mediated PD-1 blockade.
We assessed microanatomic features of PD-1 blockade by histologic evaluation using lung sections obtained from infected mice treated for 4 weeks with either isotype control antibody or anti-PD-1 antibody (Figure 5). In control mice, we observed numerous intracellular and extracellular cryptococci located in patchy alveolar infiltrates comprised of numerous mononuclear cells, eosinophils, and larger cells with abundant cytoplasm consistent with macrophage morphology (Figure 5A, B). In mice treated with anti-PD-1 antibody, the overall number of lung leukocytes appeared comparable to that observed in isotype control antibody-treated mice; however, the number of cryptococci identified, especially those located extracellularly, appeared markedly reduced as did the number of foamy macrophages (Figure 5C, D). Collectively, our data demonstrate that anti-PD-1 antibody treatment in mice with established cryptococcal lung infection promotes fungal clearance in the lung and may reduce systemic fungal dissemination.
Figure 5.
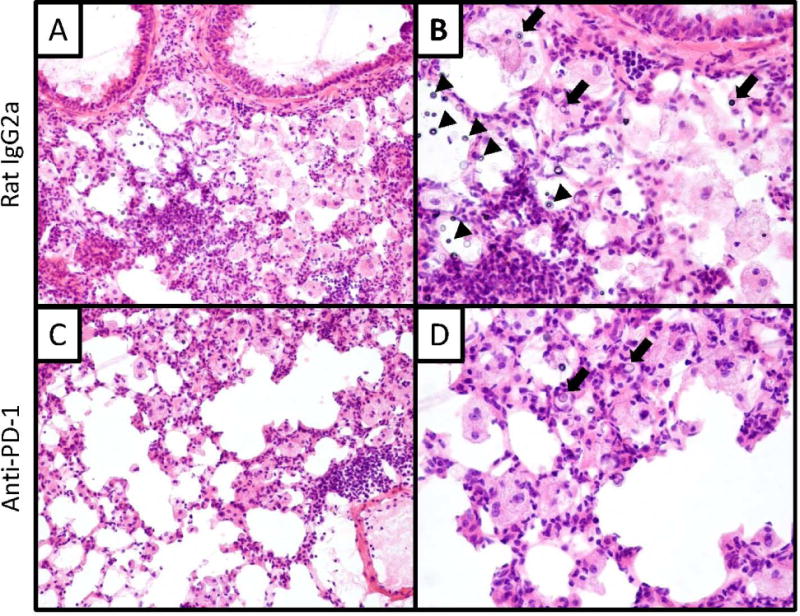
Effect of anti-PD-1 antibody treatment on lung histopathology. Representative lung sections obtained from mice treated (at 3 WPI) with control (A,B) and anti-PD-1 antibody (C,D) for 4 weeks. Sections were H&E stained and examined by light microscopy at 200x (A,C) or 400x (B,D) magnification. Sections at low magnification (A,C) show similar overall immune cell infiltration between treatments. Sections obtained from the isotype-treated group at high magnification (B) show numerous cryptococci located both intracellularly (closed block arrows) and extracellularly (black arrowheads). Sections obtained from the anti-PD-1 treated group at high magnification (D) reveal fewer cryptococci and a diminished number of foamy macrophages (relative to control treated mice).
Anti-PD-1 antibody treatment does not alter the accumulation or activation profile of lung myeloid cells in mice with persistent cryptococcal lung infection
Our next objective was to investigate cellular and molecular pathways altered by PD- blockade. We first sought to determine if differences in fungal burden between anti-PD-1 and control antibody treated mice were associated with changes in the accumulation of myeloid cells in the lung. Our results show that anti-PD-1 antibody treatment did not alter the total number of accumulating lung leukocytes (Figure 6A) or the number of any specific subset of accumulating DCs or macrophages (Figure 6B) in the lung after 2 or 4 WOT (top and bottom panels, respectively) as compared to control treated mice with the exception of a small reduction in the number of moDCs after 4 WOT (Figure 6B, bottom panel).
Since minimal differences in lung myeloid cell numbers were identified following treatment with anti-PD-1 antibody, we determined whether PD-1 blockade altered the activation profile of lung myeloid cells in treated mice. Our findings demonstrated that PD-1 blockade did not alter gene expression of either iNOS or arg1 by total lung leukocytes (Figure 7A, B) or intracellular staining for iNOS or arginase within any lung dendritic cell or macrophage subset studied after either 2 or 4 WOT (Figure 7C, D). Treatment with anti-PD-1 antibody also did not affect DC or macrophage cell surface expression of two other molecules associated with classical activation, MHC Class II and CD80, after either 2 or 4 WOT (data not shown). Note that for all myeloid cell subsets, intracellular staining for arginase was considerably greater than that of iNOS consistent with a predominantly alternatively-activated (M2) phenotype. Taken together, these data demonstrate that changes in pulmonary fungal burden in anti-PD-1 treated mice are not associated with substantial differences in the accumulation or activation of lung dendritic cells or macrophages.
Figure 7.
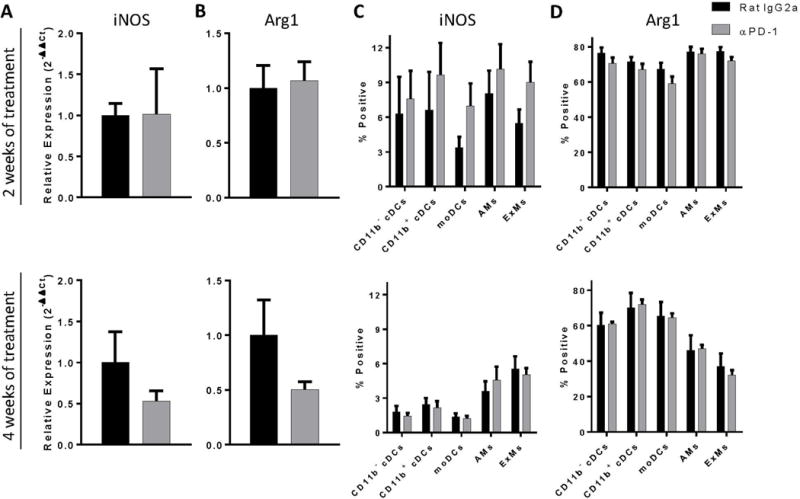
Anti-PD-1 antibody treatment does not substantially alter lung myeloid cell activation in the lungs of mice with cryptococcal lung infection. Infected C57BL/6 mice treated with either neutralizing anti-PD-1 or control antibody were evaluated for iNOS and arginase expression by (A) qPCR and (B) flow cytometric analysis using lung leukocytes obtained at 2 WOT (top panels) or 4 WOT (bottom panels). For A, B: n=4–9/cohort assayed individually in two separate experiments; *p<0.05 by unpaired Student t test.
Anti-PD-1 antibody treatment reduces IL-5 and IL-10 gene expression by lung leukocytes in mice with persistent cryptococcal lung infection
We next investigated whether anti-PD-1 antibody treatment altered gene expression of immunostimulatory cytokines (IFNγ and IL-17a) and immunomodulatory cytokines (IL-5 and IL-10) known to influence fungal clearance (9, 11, 12, 14, 16, 17). Our data showed no significant differences in gene expression of IFNγ or IL-17a by total lung leukocytes between treatment groups at either 2 or 4 WOT (Supplemental Figure 1). In contrast, expression of IL-5 was significantly reduced in leukocytes of anti-PD-1 versus control antibody-treated mice following 2 WOT and trended toward a reduction after 4 WOT (Figure 8A). Gene expression of IL-10 was significantly reduced following anti-PD-1 antibody treatment after 4 WOT (Figure 8B). Thus, anti-PD-1 antibody treatment reduces gene expression of two cytokines associated with fungal persistence.
Figure 8.
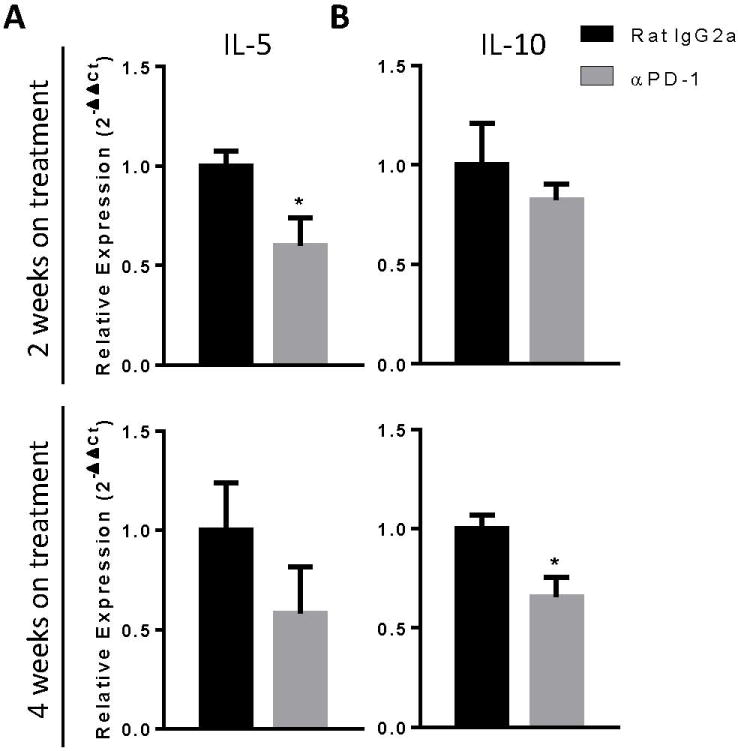
Anti-PD-1 antibody treatment decreases IL-5 and IL-10 gene expression in mice with cryptococcal lung infection. Infected C57BL/6 mice treated with either neutralizing anti-PD-1 or control antibody were evaluated for (A) IL-5 and (B) IL-10 gene expression by qPCR analysis using lung leukocytes obtained at 2 WOT (top panels) or 4 WOT (bottom panels). n=4-9/cohort assayed individually in two separate experiments; *=p<0.05 by unpaired Student t test.
Anti-PD-1 treatment does not alter accumulation of polarized CD4+ T cells in the lungs of mice with persistent cryptococcal lung infection but does enhance sustained expression of OX40 by Th1 and Th17 cells
Our final objective was to determine whether PD-1 blockade altered accumulation and activation of lymphoid cell subpopulations in the lungs of mice with persistent cryptococcal lung infection. We observed that treatment with anti-PD-1 antibody did not alter the number of B cells, total T cells, CD8+ T cells, or γδ T cells relative to control mice (Supplemental Figure 2). Moreover, PD-1 blockade did not significantly affect accumulation of any specific subset of polarized CD4+ T helper cell, including Th1, Th17, Th2, or Treg cells, at either of the two time-points studied (Figure 9A).
Since PD-1 blockade did not change the number of T cells or CD4+ Th subsets, we investigated whether treatment with anti-PD-1 antibody altered T cell activation status. We examined the expression of ICOS, known to be important in secondary stimulation of activated T cells (45), and OX40, which promotes memory T cell responses and has previously been shown to enhance fungal clearance in C. neoformans-infected mice when treated with a receptor agonist (46). Our data showed that 2 weeks of treatment with anti-PD-1 antibody significantly increased the expression of both ICOS and OX40 on all subsets of CD4+ T cells (except for ICOS expression on Th17 cells; Figure 9B and 9C, top panels). After 4 WOT, ICOS expression had decreased back to baseline whereas OX40 expression remained upregulated on Th1 and Th17 cells but not on Th2 or Treg cells (Figure 9C, bottom panels). These collective results suggest that improved fungal clearance in response to targeted PD-1 signaling blockade in mice with persistent cryptococcal lung infection is mediated, at least in part, through beneficial effects on CD4+ T cell activation, including the sustained upregulation of OX40 on Th1 and Th17 cells.
Discussion
PD-1 signaling has proven beneficial by modulating immune effector responses and preventing autoimmunity. In contrast, our current findings contribute to an emerging body of evidence demonstrating that PD-1 signaling can contribute to immune evasion by tumors and certain invading pathogens. Using a murine model of persistent cryptococcal lung infection, our current study provides the following novel observations: 1) PD-1 expression is both increased and sustained on broad subsets of T cells in the lungs; 2) expression of the PD-1 ligands, PD-L1 and PD-L2, is enhanced on specific subsets of lung dendritic cells and macrophages; and 3) targeted PD-1 blockade improves fungal clearance. Our additional studies suggest that the beneficial effects of anti-PD-1 antibody treatment may be mediated through reductions of detrimental IL-5 and IL-10 cytokines and increased activation of Th1 and Th17 cells. Collectively, these data provide strong evidence that the PD-1 signaling pathway promotes persistence of cryptococcal lung infection and opens new avenues for the development of novel immune-based treatments for the treatment of chronic fungal diseases.
Our thorough kinetic evaluation of the expression of PD-1 and its ligands (PD-L1 and PD-L2) demonstrates that persistent cryptococcal lung infection is associated with sustained expression of this immunomodulatory pathway. The data shows that PD-1 expression was increased on multiple lymphocyte subsets including all four major subsets of polarized CD4+ Th cells residing in the lungs of infected mice. This finding argues that the relative skewing of the adaptive immune response towards a non-protective Th2/Treg bias in this model is not attributable to selectively limited or enhanced expression of PD-1 on any one (or more) Th subset. We similarly identified broad and sustained expression of PD-L1 and PD-L2 by numerous subsets of lung DCs and macrophages although the expression tended to be highest on lung macrophages. These observations confirm and extend those of Guerrero et al who demonstrated increased expression of PD-L1 and PD-L2 on alveolar macrophages in the lungs of C57BL/6 mice infected with C. neoformans strain 52D (33). Interestingly, although that study only assessed PD-L1 and PD-L2 expression on alveolar macrophages, they showed that increased expression of both ligands was associated with a more virulent, mucoid strain of the organism providing further evidence that fungal virulence factors may exploit this pathway to promote immune evasion. Our results also show that of the two ligands, PD-L2 is more dynamically responsive to changes in the infected lung microenvironment whereas the degree of PD-L1 upregulation is less prominent. This finding may be explained by the prominence of Th2 responses observed in this model as Loke et al (47) has previously shown that PD-L1 is widely expressed by peritoneal macrophages at baseline with increased expression in response to LPS, IFNγ, and IL-4 whereas PD-L2 expression is initially more restricted and is more specifically increased in response to a key Type 2 cytokine, IL-4.
Our observation that treatment of persistently-infected mice for 4 weeks with anti-PD-1 blocking antibody significantly improved fungal clearance within the lung provides direct evidence that the PD-1 signaling pathway actively opposes fungal clearance. The effect was not immediate, as 2 weeks of treatment did not improve fungal clearance although treatment may have begun to enhance specific immune effector mechanisms at this time point (see below). Our decision to treat with anti-PD-1 antibody was informed by our observation that both PD-L1 and PD-L2 are prominently expressed once persistent infection is established; thus we sought to effectively block input from both of these ligands. In addition to significantly reducing the fungal burden in the lung, treatment was also associated with a trend toward reductions in fungal burden in the spleen and brain suggesting that enhanced fungal containment in the lung reduced systemic dissemination of the organism. Alternatively, it’s possible that PD-1 blockade enhanced immunity in secondary infection sites including the CNS. The presence of organisms in the lung and brain shows that the current treatment protocol did not achieve sterilizing immunity and we suspect some mice in the anti-PD-1 antibody treated group might eventually die, perhaps of meningitis. Future studies are warranted to investigate whether reduced dissemination and/or enhanced immunity are the cause for reduced fungal burdens in the spleens and brains of anti-PD-1 treated mice.
The improvement in fungal clearance we observe in mice with established cryptococcal lung infection treated with anti PD-1 antibody support and extend the findings of two prior studies showing that PD-1 deficiency or blockade improved fungal clearance and (or) survival of infection with H. capsulatum or C. albicans (35, 36). In both these studies, the investigators assessed the effects of PD-1 blockade throughout the entire duration of the infections which were both acute and lethal. Here we show that treatment with anti-PD-1 antibody is effective, even when PD-1 blockade is delayed until after persistent infection has been established. Thus, the results of our study suggest that PD-1 blockade might be effective at treating patients already infected with a fungal pathogen.
Anti-PD-1 antibody treatment did not result in substantial weight loss, abnormal animal behavior, or increase the total number of lung leukocytes (or any leukocyte subset). Thus, there was no evidence that PD-1 blockade in mice with fungal infection caused pneumonitis or any other form of immune reconstitution syndrome (48, 49) in a manner detrimental to the mice. Although we had correctly hypothesized that treatment would improve fungal clearance, it was possible that treatment might have enhanced susceptibility to infection; yet this did not occur. Given the widespread use of checkpoint inhibitors in patients with cancer, this data provides a measure of reassurance that the inadvertent treatment of patients with unrecognized underlying fungal infection is unlikely to cause harm.
Our data provide several key insights into the cellular and molecular mechanisms which might explain the improved fungal clearance observed in response to anti-PD-1 antibody treatment. Notably, PD-1 blockade had a minimal effect on the number of immune effector cells in the lung. This finding differed from our study performed using an anti-IL-10 receptor blocking antibody in which we attributed improved fungal clearance to its ability to increase the number of several effector cells including moDCs, exudate macrophages, and Th1 and Th17 cells (17). However, our findings do suggest some inter-relationship between the PD-1 and IL-10 immunomodulatory pathways as we found evidence that anti-PD-1 antibody treatment diminished IL-10 gene expression. IL-5 gene expression was also reduced by PD-1 blockade. Since both IL-10 and IL-5 are associated with non-protective Th2 responses (9, 11) it is likely that some of the beneficial effects of anti-PD-1 antibody treatment were mediated through reductions in unfavorable T2 cytokine responses. As we have previously shown that inhibiting IL-10 signaling promotes fungal clearance (17), combined intervention toward both PD-1 and cytokine signaling pathways provide an intriguing avenue of possible future studies.
Treatment with anti-PD-1 antibody enhanced T cell expression of ICOS and OX40. Specifically, our findings demonstrate that two weeks of anti-PD-1 treatment increased expression of ICOS on Th1, Th2, and Treg cells but these effects were transient and not observed following 4 weeks of treatment. In contrast, whereas OX40 expression was increased on all polarized CD4+ T cell populations following 2 weeks of anti-PD-1 treatment, its expression remained elevated on Th1 and Th17 cells, but not on Th2 and Treg cells. Both molecules are associated with T cell activation (50, 51) and OX40 promotes memory T cell formation; thus, sustained activation of Th1 and Th17 cells as observed following 4 WOT may contribute to the beneficial effects of PD-1 blockade observed in this study. Support for this hypothesis is provided by a study by Humphreys et al which showed that engagement of OX40, utilizing an OX40L fusion protein, reduced pulmonary eosinophilia and promoted fungal clearance when evaluated in the identical model of murine cryptococcal lung infection used in the present study (46). Thus, it might prove particularly informative to perform future studies seeking to determine whether combination therapy with PD-1 blockade and agonistic OX40L stimulation might further enhance clearance relative to either intervention alone. It would be of additional interest to determine whether the PD-1 expressing T cells are antigen-specific and to assess whether the upregulation in OX40L in response to PD-1 blockade is limited to this T cell subset.
In summary, this study advances our understanding of molecular mechanisms that actively contribute to pathogen immune evasion and promote persistent infection. We specifically show that the PD-1 signaling pathway critically contributes to the pathogenesis of cryptococcosis and our findings provide important proof of concept that checkpoint inhibitors may be incorporated into highly novel, immune-based therapies for the treatment of chronic fungal lung infections. These results motivate future studies seeking to further refine the appropriate dose and duration of therapy and to assess the exciting possibility that anti-PD-1 antibody treatment in combination with conventional antibiotics or IL-10 signaling blockade might yield additive or synergistic results.
Supplementary Material
Acknowledgments
We would like to acknowledge the enthusiastic input of Morgan Gordon who provided additional scientific support of this project.
Supported by a Merit Review Award (J.J.O.) from the Biomedical Laboratory Research and Development Service, Department of Veterans Affairs and an NIH T32 Training Grant (J.A.R., trainee).
The abbreviations used are
- AM
alveolar macrophage
- ExM
exudate macrophage
- DC
dendritic cell
- cDC
conventional dendritic cell
- moDC
monocyte-derived dendritic cell
- CFU
colony forming unit
- DPI
days post infection
- WPI
weeks post infection
- WOT
weeks of treatment
- IL
interleukin
- IFNγ
interferon gamma
- ICOS
inducible costimulatory of T cells
- PD-1
programmed cell death protein-1
- PD-L1/2
programmed cell death protein-1 ligand-1/2
- C. neoformans
Cryptococcus neoformans
- Th
T helper
- qPCR
quantitative reverse-transcriptase polymerase chain reaction
- CD
cluster of differentiation
- LPS
lipopolysaccharide
- IP
intraperitoneal
- APC
allophycocyanin
- PE
phycoerythrin
- PerCP
peridinin chlorophyll protein
- Cy
cyanine
- FITC
fluorescein isothiocyanate
- BV
brilliant violet
Footnotes
Portions of this work have been presented previously at the International Conference of the American Thoracic Society, Washington, D.C., May 23, 2017.
References
- 1.Li SS, Mody CH. Cryptococcus. Proceedings of the American Thoracic Society. 2010;7:186–196. doi: 10.1513/pats.200907-063AL. [DOI] [PubMed] [Google Scholar]
- 2.Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, Denning DW, Loyse A, Boulware DR. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. The Lancet Infectious diseases. 2017;17:873–881. doi: 10.1016/S1473-3099(17)30243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bozzette SA, Larsen RA, Chiu J, Leal MA, Jacobsen J, Rothman P, Robinson P, Gilbert G, McCutchan JA, Tilles J, et al. A placebo-controlled trial of maintenance therapy with fluconazole after treatment of cryptococcal meningitis in the acquired immunodeficiency syndrome. California Collaborative Treatment Group. The New England journal of medicine. 1991;324:580–584. doi: 10.1056/NEJM199102283240902. [DOI] [PubMed] [Google Scholar]
- 4.Chen S, Sorrell T, Nimmo G, Speed B, Currie B, Ellis D, Marriott D, Pfeiffer T, Parr D, Byth K. Epidemiology and host- and variety-dependent characteristics of infection due to Cryptococcus neoformans in Australia and New Zealand. Australasian Cryptococcal Study Group. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2000;31:499–508. doi: 10.1086/313992. [DOI] [PubMed] [Google Scholar]
- 5.Choi YH, Ngamskulrungroj P, Varma A, Sionov E, Hwang SM, Carriconde F, Meyer W, Litvintseva AP, Lee WG, Shin JH, Kim EC, Lee KW, Choi TY, Lee YS, Kwon-Chung KJ. Prevalence of the VNIc genotype of Cryptococcus neoformans in non-HIV-associated cryptococcosis in the Republic of Korea. FEMS yeast research. 2010;10:769–778. doi: 10.1111/j.1567-1364.2010.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitchell TG, Perfect JR. Cryptococcosis in the era of AIDS–100 years after the discovery of Cryptococcus neoformans. Clinical microbiology reviews. 1995;8:515–548. doi: 10.1128/cmr.8.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu LY, Ng ES, Koh LP. Common and emerging fungal pulmonary infections. Infectious disease clinics of North America. 2010;24:557–577. doi: 10.1016/j.idc.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Chen GH, Teitz-Tennenbaum S, Neal LM, Murdock BJ, Malachowski AN, Dils AJ, Olszewski MA, Osterholzer JJ. Local GM-CSF-Dependent Differentiation and Activation of Pulmonary Dendritic Cells and Macrophages Protect against Progressive Cryptococcal Lung Infection in Mice. J Immunol. 2016;196:1810–1821. doi: 10.4049/jimmunol.1501512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernandez Y, Arora S, Erb-Downward JR, McDonald RA, Toews GB, Huffnagle GB. Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. J Immunol. 2005;174:1027–1036. doi: 10.4049/jimmunol.174.2.1027. [DOI] [PubMed] [Google Scholar]
- 10.Hoag KA, Street NE, Huffnagle GB, Lipscomb MF. Early cytokine production in pulmonary Cryptococcus neoformans infections distinguishes susceptible and resistant mice. American journal of respiratory cell and molecular biology. 1995;13:487–495. doi: 10.1165/ajrcmb.13.4.7546779. [DOI] [PubMed] [Google Scholar]
- 11.Huffnagle GB, Boyd MB, Street NE, Lipscomb MF. IL-5 is required for eosinophil recruitment, crystal deposition, and mononuclear cell recruitment during a pulmonary Cryptococcus neoformans infection in genetically susceptible mice (C57BL/6) J Immunol. 1998;160:2393–2400. [PubMed] [Google Scholar]
- 12.Jain AV, Zhang Y, Fields WB, McNamara DA, Choe MY, Chen GH, Erb-Downward J, Osterholzer JJ, Toews GB, Huffnagle GB, Olszewski MA. Th2 but not Th1 immune bias results in altered lung functions in a murine model of pulmonary Cryptococcus neoformans infection. Infection and immunity. 2009;77:5389–5399. doi: 10.1128/IAI.00809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen GH, McDonald RA, Wells JC, Huffnagle GB, Lukacs NW, Toews GB. The gamma interferon receptor is required for the protective pulmonary inflammatory response to Cryptococcus neoformans. Infection and immunity. 2005;73:1788–1796. doi: 10.1128/IAI.73.3.1788-1796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arora S, Hernandez Y, Erb-Downward JR, McDonald RA, Toews GB, Huffnagle GB. Role of IFN-gamma in regulating T2 immunity and the development of alternatively activated macrophages during allergic bronchopulmonary mycosis. J Immunol. 2005;174:6346–6356. doi: 10.4049/jimmunol.174.10.6346. [DOI] [PubMed] [Google Scholar]
- 15.Arora S, Olszewski MA, Tsang TM, McDonald RA, Toews GB, Huffnagle GB. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infection and immunity. 2011;79:1915–1926. doi: 10.1128/IAI.01270-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murdock BJ, Huffnagle GB, Olszewski MA, Osterholzer JJ. Interleukin-17A enhances host defense against cryptococcal lung infection through effects mediated by leukocyte recruitment, activation, and gamma interferon production. Infection and immunity. 2014;82:937–948. doi: 10.1128/IAI.01477-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murdock BJ, Teitz-Tennenbaum S, Chen GH, Dils AJ, Malachowski AN, Curtis JL, Olszewski MA, Osterholzer JJ. Early or late IL-10 blockade enhances Th1 and Th17 effector responses and promotes fungal clearance in mice with cryptococcal lung infection. J Immunol. 2014;193:4107–4116. doi: 10.4049/jimmunol.1400650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osterholzer JJ, Surana R, Milam JE, Montano GT, Chen GH, Sonstein J, Curtis JL, Huffnagle GB, Toews GB, Olszewski MA. Cryptococcal urease promotes the accumulation of immature dendritic cells and a non-protective T2 immune response within the lung. Am J Pathol. 2009;174:932–943. doi: 10.2353/ajpath.2009.080673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olszewski MA, Zhang Y, Huffnagle GB. Mechanisms of cryptococcal virulence and persistence. Future microbiology. 2010;5:1269–1288. doi: 10.2217/fmb.10.93. [DOI] [PubMed] [Google Scholar]
- 20.Qiu Y, Davis MJ, Dayrit JK, Hadd Z, Meister DL, Osterholzer JJ, Williamson PR, Olszewski MA. Immune modulation mediated by cryptococcal laccase promotes pulmonary growth and brain dissemination of virulent Cryptococcus neoformans in mice. PloS one. 2012;7:e47853. doi: 10.1371/journal.pone.0047853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunological reviews. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, Azuma M, Yagita H. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169:5538–5545. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 23.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. The EMBO journal. 1992;11:3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufmann DE, Walker BD. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J Immunol. 2009;182:5891–5897. doi: 10.4049/jimmunol.0803771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eichbaum Q. PD-1 signaling in HIV and chronic viral infection--potential for therapeutic intervention? Current medicinal chemistry. 2011;18:3971–3980. doi: 10.2174/092986711796957239. [DOI] [PubMed] [Google Scholar]
- 26.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 27.Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol. 2011;186:1598–1607. doi: 10.4049/jimmunol.1003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rowe JH, Johanns TM, Ertelt JM, Way SS. PDL-1 blockade impedes T cell expansion and protective immunity primed by attenuated Listeria monocytogenes. J Immunol. 2008;180:7553–7557. doi: 10.4049/jimmunol.180.11.7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. International immunology. 2005;17:133–144. doi: 10.1093/intimm/dxh194. [DOI] [PubMed] [Google Scholar]
- 30.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, Tamada K, Chen L. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer research. 2005;65:1089–1096. [PubMed] [Google Scholar]
- 31.Janakiram M, Pareek V, Cheng H, Narasimhulu DM, Zang X. Immune checkpoint blockade in human cancer therapy: lung cancer and hematologic malignancies. Immunotherapy. 2016;8:809–819. doi: 10.2217/imt-2016-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shrimali RK, Janik JE, Abu-Eid R, Mkrtichyan M, Khleif SN. Programmed death-1 & its ligands: promising targets for cancer immunotherapy. Immunotherapy. 2015;7:777–792. doi: 10.2217/imt.15.49. [DOI] [PubMed] [Google Scholar]
- 33.Guerrero A, Jain N, Wang X, Fries BC. Cryptococcus neoformans variants generated by phenotypic switching differ in virulence through effects on macrophage activation. Infection and immunity. 2010;78:1049–1057. doi: 10.1128/IAI.01049-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osterholzer JJ, Curtis JL, Polak T, Ames T, Chen GH, McDonald R, Huffnagle GB, Toews GB. CCR2 mediates conventional dendritic cell recruitment and the formation of bronchovascular mononuclear cell infiltrates in the lungs of mice infected with Cryptococcus neoformans. J Immunol. 2008;181:610–620. doi: 10.4049/jimmunol.181.1.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazar-Molnar E, Gacser A, Freeman GJ, Almo SC, Nathenson SG, Nosanchuk JD. The PD-1/PD-L costimulatory pathway critically affects host resistance to the pathogenic fungus Histoplasma capsulatum. Proc Natl Acad Sci U S A. 2008;105:2658–2663. doi: 10.1073/pnas.0711918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, Unsinger J, Korman AJ, Green JM, Hotchkiss RS. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Critical care. 2013;17:R85. doi: 10.1186/cc12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen GH, McNamara DA, Hernandez Y, Huffnagle GB, Toews GB, Olszewski MA. Inheritance of immune polarization patterns is linked to resistance versus susceptibility to Cryptococcus neoformans in a mouse model. Infection and immunity. 2008;76:2379–2391. doi: 10.1128/IAI.01143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen GH, Olszewski MA, McDonald RA, Wells JC, Paine R, 3rd, Huffnagle GB, Toews GB. Role of granulocyte macrophage colony-stimulating factor in host defense against pulmonary Cryptococcus neoformans infection during murine allergic bronchopulmonary mycosis. Am J Pathol. 2007;170:1028–1040. doi: 10.2353/ajpath.2007.060595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arora S, McDonald RA, Toews GB, Huffnagle GB. Effect of a CD4-depleting antibody on the development of Cryptococcus neoformans-induced allergic bronchopulmonary mycosis in mice. Infection and immunity. 2006;74:4339–4348. doi: 10.1128/IAI.01989-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huffnagle GB, Toews GB, Burdick MD, Boyd MB, McAllister KS, McDonald RA, Kunkel SL, Strieter RM. Afferent phase production of TNF-alpha is required for the development of protective T cell immunity to Cryptococcus neoformans. J Immunol. 1996;157:4529–4536. [PubMed] [Google Scholar]
- 41.Osterholzer JJ, Chen GH, Olszewski MA, Curtis JL, Huffnagle GB, Toews GB. Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6Chigh monocytes. J Immunol. 2009;183:8044–8053. doi: 10.4049/jimmunol.0902823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. American journal of respiratory cell and molecular biology. 2013;49:503–510. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okudaira K, Hokari R, Tsuzuki Y, Okada Y, Komoto S, Watanabe C, Kurihara C, Kawaguchi A, Nagao S, Azuma M, Yagita H, Miura S. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. International journal of oncology. 2009;35:741–749. doi: 10.3892/ijo_00000387. [DOI] [PubMed] [Google Scholar]
- 44.Harvey RD. Immunologic and clinical effects of targeting PD-1 in lung cancer. Clinical pharmacology and therapeutics. 2014;96:214–223. doi: 10.1038/clpt.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rudd CE, Schneider H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nature reviews Immunology. 2003;3:544–556. doi: 10.1038/nri1131. [DOI] [PubMed] [Google Scholar]
- 46.Humphreys IR, Edwards L, Walzl G, Rae AJ, Dougan G, Hill S, Hussell T. OX40 ligation on activated T cells enhances the control of Cryptococcus neoformans and reduces pulmonary eosinophilia. J Immunol. 2003;170:6125–6132. doi: 10.4049/jimmunol.170.12.6125. [DOI] [PubMed] [Google Scholar]
- 47.Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gundacker ND, Jordan SJ, Jones BA, Drwiega JC, Pappas PG. Acute Cryptococcal Immune Reconstitution Inflammatory Syndrome in a Patient on Natalizumab. Open forum infectious diseases. 2016;3:ofw038. doi: 10.1093/ofid/ofw038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hashimoto H, Hatakeyama S, Yotsuyanagi H. Development of cryptococcal immune reconstitution inflammatory syndrome 41 months after the initiation of antiretroviral therapy in an AIDS patient. AIDS research and therapy. 2015;12:33. doi: 10.1186/s12981-015-0075-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prell RA, Evans DE, Thalhofer C, Shi T, Funatake C, Weinberg AD. OX40-mediated memory T cell generation is TNF receptor-associated factor 2 dependent. J Immunol. 2003;171:5997–6005. doi: 10.4049/jimmunol.171.11.5997. [DOI] [PubMed] [Google Scholar]
- 51.Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, Boucher LM, Bouchard D, Chan VS, Duncan G, Odermatt B, Ho A, Itie A, Horan T, Whoriskey JS, Pawson T, Penninger JM, Ohashi PS, Mak TW. ICOS is essential for effective T-helper-cell responses. Nature. 2001;409:105–109. doi: 10.1038/35051113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.