

Abstract
Osteoglophonic dysplasia (OD) is an extremely rare, skeletal dysplasia with an autosomal dominant mode of inheritance. Rhizomelic dwarfism, craniosynostosis, impacted teeth, hypodontia or anodontia, and multiple nonossifying bone lesions are the salient features of this condition. We report a 14-year-old girl with clinical and radiological features consistent with OD. She presented with disproportionate short stature, craniosynostosis, a prominent supraorbital ridge, delayed teeth eruption, hypodontia, and multiple nonossifying bone lesions in the femur, tibia, and fibula. She had hypophosphatemia, which is a known association in this dysplasia. She also had advanced bone age, which is an unreported feature of this dysplasia. This condition is caused by activating mutations in FGFR1 . A missense mutation was detected in the FGFR1 , NM_001174067 ( FGFR1 _v001):c.1115G > A [p.(Cys372Tyr)] confirming the diagnosis; this is the first mutation-proven case to be reported from India.
Keywords: craniosynostosis, hypodontia, FGFR1, nonossifying fibroma, brachydactyly
Introduction
Osteoglophonic dysplasia (OD) (MIM 166250) is an extremely rare, autosomal dominant disorder. There are only 15 reported cases in the literature. This is the first mutation-proven case to be reported from India. It is characterized by craniosynostosis, rhizomelic dwarfism, hypodontia, and nonossifying bone lesions.
Case Report
A 14-year-old girl born to a nonconsanguineous couple was brought for evaluation of short stature. She had normal intelligence and had attained menarche at 13 years of age. Anthropometric measurements showed a height of 131 cm, weight of 36 kg, and occipitofrontal circumference of 51 cm (all < 3rd centile). She had rhizomelic short stature with an upper segment to lower segment (US:LS) ratio of 1.07:1. She had turribrachycephaly, hypertelorism, prominent supraorbital ridge, flat nasal bridge, bilateral low-set, posteriorly rotated pinnae, high-arched palate, ( Fig. 1A, B ) rhizomelic shortening of the limbs, brachydactyly of hands and feet, stubby fingers and toes ( Fig. 1D, E ), and varus deformity of both lower tibia. She had only eight teeth at the age of 14 years with severe gum hypertrophy ( Fig. 1C ). She had prematurely lost her primary dentition by 3 years of age, with delayed eruption of the secondary dentition.
Fig. 1.
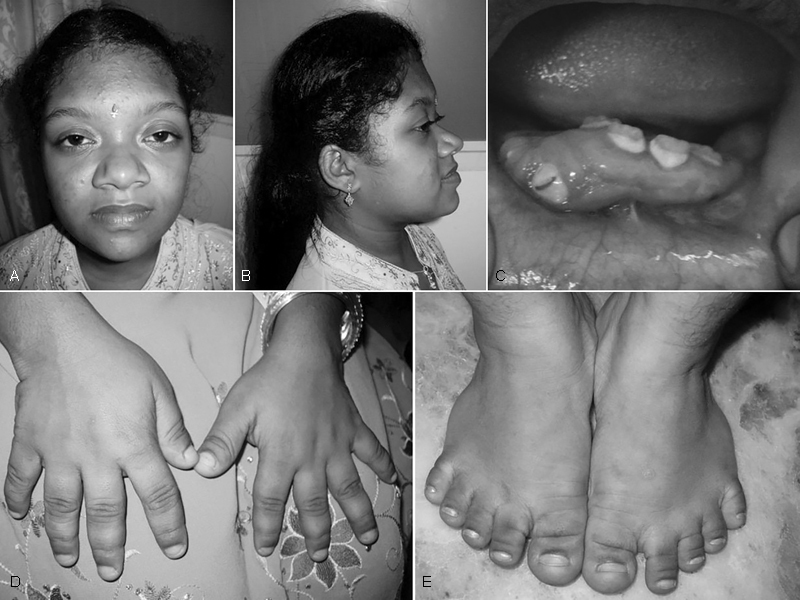
( A ) Photograph of the proband showing a high forehead, hypertelorism, prominent supraorbital ridge, flat nasal bridge, and prognathism. ( B ) Profile photo of the proband showing turribrachycephaly, low-set, posteriorly rotated pinna, and mid-facial hypoplasia. ( C ) Intraoral picture showing gum hypertrophy with embedded teeth and oligodontia. ( D ) Brachydactyly with stubby fingers. ( E ) Brachydactyly with stubby toes.
Laboratory investigations revealed serum phosphorus 2.5 mg/dL (2.7–4.5 mg/dL), serum calcium 9.5 mg/dL (8.6–10 mg/dL), and serum alkaline phosphatase 173 IU/L (100–320 IU/L). Skull X-rays revealed turribrachycephaly with extensive copper beaten appearance in the frontal region, enlarged sella, and flattening of forehead ( Fig. 2C ). She had midfacial hypoplasia, prognathism, and multiple, unerupted impacted teeth ( Fig. 2D ). Chest radiograph revealed a bell-shaped thorax with down-slanting broad ribs ( Fig. 3F ). Hand radiographs showed severe brachydactyly with very short metacarpals and phalanges, predominantly the middle and distal phalanges; the proximal and middle phalanges showed coning. The carpal bone age corresponded to 16 years ( Fig. 2A ), and the epiphyses of metacarpals, phalanges, and distal radioulnar joint had fused already ( Fig. 3D ). Foot radiography showed a corresponding appearance ( Fig. 2B ). Spine radiograph showed a mild flattening of the thoracic vertebrae with a mild lower anterior beaking of the L5 vertebra with posterior scalloping from L1–L5 ( Fig. 3E ). Radiograph of the lower limbs revealed short long bones, and a short femoral neck with coxa vara ( Fig. 3A ). Multiple epiphyseal lucencies were seen in the medial aspect of the lower femur, the upper tibia, and fibula, which were consistent with nonossifying fibromas (NOFs) ( Fig. 3B, C ). Tibia vara was evident in the lower half, and the fibula was longer than the tibia with a wavy outline. Orthopantomogram showed multiple impacted permanent tooth buds in the maxilla and mandible ( Fig. 2E ). In view of these clinical features and multiple metaphyseal lucencies around the knee, a clinical diagnosis of OD was considered.
Fig. 2.
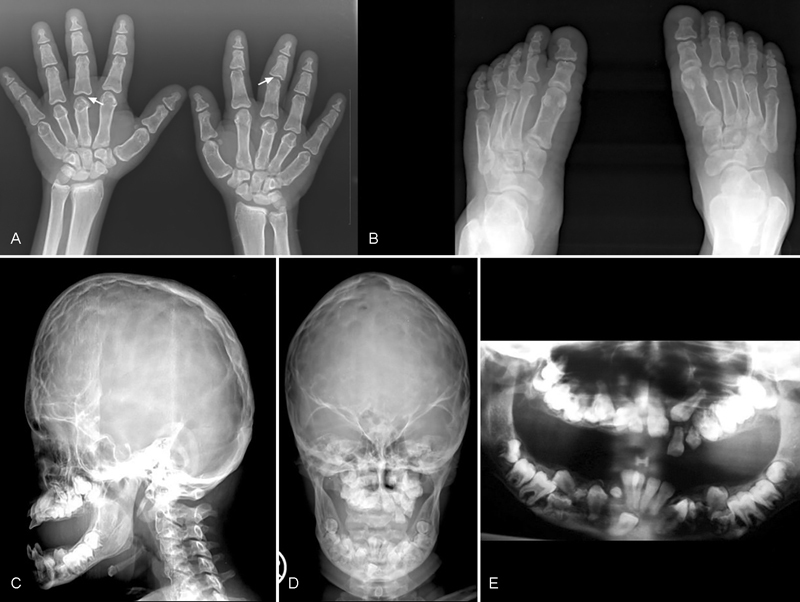
( A ) Radiograph of the hand showing severe brachydactyly with short metacarpals and middle and distal phalanges (arrows). Note coning of the proximal and middle phalanges. ( B ) Radiograph of the feet showing brachydactyly of the metatarsals and phalanges with shortening of the middle and terminal phalanges of all toes. ( C ) Radiograph of the lateral view of skull showing turribrachycephaly with prominent “copper beaten” appearance of the frontal region, enlarged sella, flat forehead, midfacial hypoplasia, and prognathism. ( D ) Radiograph of the anteroposterior view of skull showing “copper beaten' appearance and multiple unerupted teeth in the maxilla and mandible. ( E ) Orthopantomogram showing multiple impacted permanent tooth buds in mandible and maxilla.
Fig. 3.
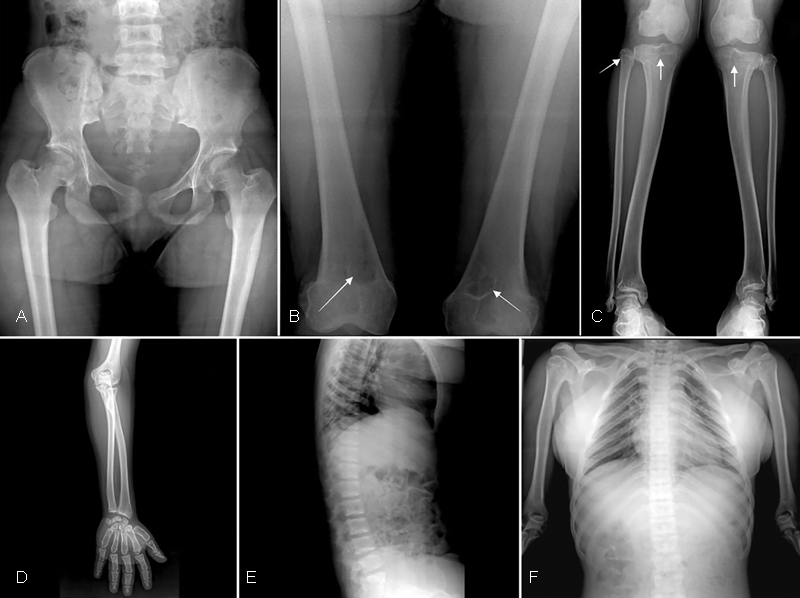
( A ) Radiograph of the pelvis showing coxa vara with short femoral neck. ( B ) Radiograph of the lower femur showing multiple lucencies in the medial aspect consistent with nonossifying fibromas (NOFs, arrows). ( C ) Wavy tibia and fibula with epiphyseal lucencies in the upper tibia (bilateral) and right fibula. ( D ) Radiograph of the forearm and hand showing advanced bone age with complete fusion of distal radioulnar joint corresponding to bone age of 16 years. ( E ) Lateral view of thoracolumbar spine showing posterior scalloping from L1–L5 and mild lower anterior beaking of L5 vertebra. ( F ) Chest radiograph showing bell-shaped thorax with down-slanting broad ribs. Also, medial broadening of the clavicle is observed.
Genetic analysis revealed a heterozygous NM_001174067 (FGFR1_v001):c.1115G > A [p.(Cys372Tyr)] missense mutation in exon 10 in the FGFR1 ( Fig. 4A, B ). This mutation is predicted to be localized in the extracellular portion of the receptor, adjacent to the single transmembrane domain.
Fig. 4.
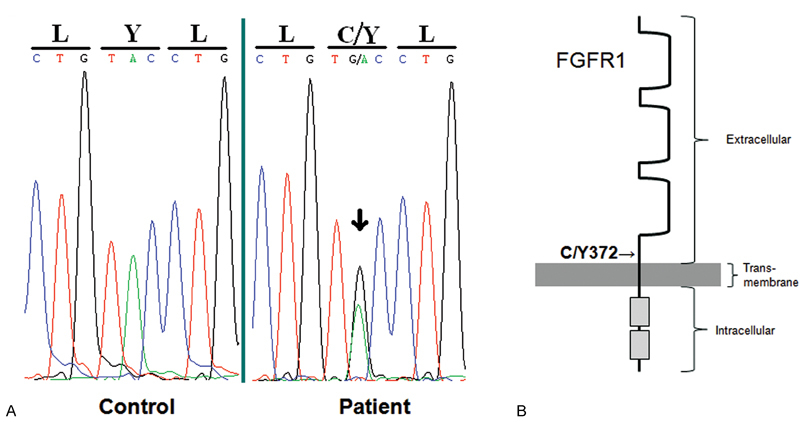
( A ) A heterozygous c.1115G > A [p.(Cys372Tyr)] missense mutation (arrow) was detected in the patient in FGFR1 exon 10 near the predicted transmembrane domain that was not detected in normal controls. ( B ) An FGFR1 monomer is shown with the predicted location of the Y372C mutation adjacent to the transmembrane domain.
Discussion
OD is extremely rare; its exact incidence is unknown. It was first reported by Fairbank in 1951. 1 The term “osteoglophonic” in Greek implies “hollowed out appearance of the bone.” 2 The autosomal dominant pattern of inheritance was highlighted by the father to son transmission reported in three families. 3 4 5 The proband had all the classical features of OD and her elder brother and parents were normal, suggesting de novo inheritance.
The salient craniofacial features of this condition include craniosynostosis, frontal bossing, hypertelorism, prognathism, and proptosis that are evident at birth. Anteverted nares, macroglossia, gum hypertrophy, and oligodontia are early findings of OD. Rhizomelic shortening, stubby fingers and toes, and extension defect of the elbow and knee joints become apparent with age. 6 The height is typically below the third centile (expected adult height 97 to 146 cm). 6 These patients have normal intelligence and a normal life span. 6 Death in infancy is attributed to complications arising from feeding difficulties, nasal obstruction, or respiratory problems. 6 7 Inguinal hernia and hypertrophic pyloric stenosis have been reported. 6
These patients often require multiple surgeries for the correction of craniosynostosis. 3 8 The typical ‘copper beaten’ appearance seen in the skull radiograph may regress by adulthood. 6 9 Platyspondyly with anterior beaking, posterior scalloping with cephalocaudal narrowing of the vertebral canal, and broad ribs are known features of this disorder. 6 9
The hallmark of OD is multiple nonossifying fibromas (MNOFs), manifesting as lucencies seen in the metaphyses of long bones, especially at the knee. 1 3 4 6 8 10 11 They increase in size and number until the attainment of skeletal maturation and gradually fill with advancing age causing abnormal modeling and expansion of metaphyses. 3 4 8 Rarely, fractures in these foci can result in pseudarthrosis. 7 Biopsy typically shows whorls of fibrous tissue. 6 In our case, these lesions were seen in the lower end of femur and upper ends of tibia and fibula.
The oral manifestations of OD are oligodontia or anodontia, gingival hypertrophy, and macroglossia. 9 12 Non-eruption of teeth could be due to crowding, inverted teeth, soft tissue impaction, or even odontogenic tumors and cysts. Histopathological examination of the gingival swelling in three cases with OD was consistent with giant cell granuloma. 5 9 12
OD is caused by activating mutations in highly conserved residues of FGFR1 , which is probably a negative regulator of growth of long bones. 5 The FGFR1 -Y372C mutation found in this case was previously observed in an OD family associated with hypophosphatemia. 5 Our patient too had hypophosphatemia. The patients may develop more severe bone disease owing to the constitutive activation of FGFR1 , which in turn leads to an upregulation of FGF23 in bone, thereby leading to renal phosphate wasting. Patients with more metaphyseal lytic lesions tend to develop more profound hypophosphatemia potentially due to elevated expression of FGF23 . 13
The fibroblast growth factors and their receptor, FGFR 1-4 have key roles in skeletal development. FGFR1 and FGFR2 mutations cause syndromes associated with craniosynostosis (Apert, Crouzon, and Pfeiffer syndromes), whereas FGFR3 mutations lead to achondroplasia, hypochondroplasia, and thanatophoric dysplasia. OD shares features of craniosynostosis syndromes and dwarfing syndromes but in addition has distinctive nonossifying fibromas (NOFs). 5
Funding
None.
Conflict of Interest None.
Authors' Contributions
S.K. prepared the manuscript and performed detailed literature search. K.E.W. performed molecular studies and interpretation, and wrote the discussion on molecular aspect. D.Y. performed the literature search and helped in the manuscript preparation. A.G. critically evaluated the manuscript and contributed suggestions. S.N. conceived the idea of drafting this paper and performed the final drafting, and will act as the guarantor of the manuscript.
References
- 1.Fairbank T. Edinburgh: Churchill Livingstone; 1951. An Atlas of General Affections of Skeleton; pp. 181–183. [Google Scholar]
- 2.Beighton P, Cremin B J, Kozlowski K. Osteoglophonic dwarfism. Pediatr Radiol. 1980;10(01):46–50. doi: 10.1007/BF01644343. [DOI] [PubMed] [Google Scholar]
- 3.Kelley R I, Borns P F, Nichols D, Zackai E H. Osteoglophonic dwarfism in two generations. J Med Genet. 1983;20(06):436–440. doi: 10.1136/jmg.20.6.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reynolds J F, Cohen M M, Jr, Michels V V, Ardinger H H, Kelly T E. Osteoglophonic dysplasia: natural history and confirmation of autosomal dominant inheritance. Proc Greenwood Genet Centre. 1988;7:167–168. [Google Scholar]
- 5.White K E, Cabral J M, Davis S I et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005;76(02):361–367. doi: 10.1086/427956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beighton P. Osteoglophonic dysplasia. J Med Genet. 1989;26(09):572–576. doi: 10.1136/jmg.26.9.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sklower Brooks S, Kassner G, Qazi Q, Keogh M J, Gorlin R J. Osteoglophonic dysplasia: review and further delineation of the syndrome. Am J Med Genet. 1996;66(02):154–162. doi: 10.1002/(SICI)1096-8628(19961211)66:2<154::AID-AJMG6>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 8.Azouz E M, Kozlowski K. Osteoglophonic dysplasia: appearance and progression of multiple nonossifying fibromata. Pediatr Radiol. 1997;27(01):75–78. doi: 10.1007/s002470050069. [DOI] [PubMed] [Google Scholar]
- 9.Shankar V N, Ajila V, Kumar G. Osteoglophonic dysplasia: a case report. J Oral Sci. 2010;52(01):167–171. doi: 10.2334/josnusd.52.167. [DOI] [PubMed] [Google Scholar]
- 10.Keats T E, Smith T H, Sweet D E. Craniofacial dysotosis with fibrous metaphyseal deffects. Am J Roentgenol Radium Ther Nucl Med. 1975;124(02):271–275. doi: 10.2214/ajr.124.2.271. [DOI] [PubMed] [Google Scholar]
- 11.Santos H, Campos P, Alves R, Torrado A. Osteoglophonic dysplasia: a new case. Eur J Pediatr. 1988;147(05):547–549. doi: 10.1007/BF00441988. [DOI] [PubMed] [Google Scholar]
- 12.Roberts T S, Stephen L, Beighton P. Osteoglophonic dysplasia: dental and orthodontic implications. Orthod Craniofac Res. 2006;9(03):153–156. doi: 10.1111/j.1601-6343.2006.00369.x. [DOI] [PubMed] [Google Scholar]
- 13.Bastepe M, Jüppner H. Inherited hypophosphatemic disorders in children and the evolving mechanisms of phosphate regulation. Rev Endocr Metab Disord. 2008;9(02):171–180. doi: 10.1007/s11154-008-9075-3. [DOI] [PubMed] [Google Scholar]