

Abstract
Background
Restriction-site associated DNA sequencing (RADseq) technology was recently employed to identify a large number of single nucleotide polymorphisms (SNP) for linkage mapping of a North American and Eastern Asian Populus species. However, there is also the need for high-density genetic linkage maps for the European aspen (P. tremula) as a tool for further mapping of quantitative trait loci (QTLs) and marker-assisted selection of the Populus species native to Europe.
Results
We established a hybrid F1 population from the cross of two aspen parental genotypes diverged in their phenological and morphological traits. We performed RADseq of 122 F1 progenies and two parents yielding 15,732 high-quality SNPs that were successfully identified using the reference genome of P. trichocarpa. 2055 SNPs were employed for the construction of maternal and paternal linkage maps. The maternal linkage map was assembled with 1000 SNPs, containing 19 linkage groups and spanning 3054.9 cM of the genome, with an average distance of 3.05 cM between adjacent markers. The paternal map consisted of 1055 SNPs and the same number of linkage groups with a total length of 3090.56 cM and average interval distance of 2.93 cM. The linkage maps were employed for QTL mapping of one-year-old seedlings height variation. The most significant QTL (LOD = 5.73) was localized to LG5 (96.94 cM) of the male linkage map, explaining 18% of the phenotypic variation.
Conclusions
The set of 15,732 SNPs polymorphic in aspen and high-density genetic linkage maps constructed for the P. tremula intra-specific cross will provide a valuable source for QTL mapping and identification of candidate genes facilitating marker-assisted selection in European aspen.
Electronic supplementary material
The online version of this article (10.1186/s12870-017-1127-y) contains supplementary material, which is available to authorized users.
Keywords: European aspen, Intra-specific cross, Full-sibling population, RADseq, High resolution linkage map, SNP, Growth-related traits, QTL mapping
Background
Populus species are the fast-growing trees appreciated worldwide due to their rapid growth and quality of wood, which is amenable to a variety of mechanical and chemical processing. They are of particular importance for the pulp and paper industries and the fuel and energy sectors, providing valuable raw materials in a short time. In Europe, the most frost-resistant and productive Populus species that has adapted to unfertile and acidic soils is aspen (Populus tremula L.) [1]. Despite its value and advantages, aspen has a major drawback - massive trunk damage by heart rot caused by the fungi Phellinus tremulae (Bond.) Bond. et. Boriss. Aspen clones with the highest growth rate (i.e. natural triploids) have been reportedly less affected by the heart rot pathogen [1, 2]. Therefore, the priority tasks of the aspen breeding programs in Russia have been growth rate, resistance to diseases and winter hardiness, which in turn, are closely related to the phenology traits [3].
The timing of bud flush and bud set are important adaptive traits which represent critical trade-offs between survival and growth [4]. Too early bud flush or late bud set increases the risk of frost damage, whereas too early bud set might be disadvantageous in the competition for resources with other trees during the short growing season characteristic of northern latitudes [5]. The knowledge about the genetic architecture of the adaptive and grow-related traits will allow effective breeding of the most valuable commercial aspen clones.
Breeding in woody perennial species is a long-term process where the availability and application of genomic tools would have obvious benefits. Several remarkable genomic resources for Populus breeding were recently made available, e.g. the assembled genome sequence of a P. trichocarpa [6], a 34 K SNP array available for genotyping of natural populations of Populus trichocarpa and other Populus species [7], and a number of genetic linkage maps of different species in the genus (reviewed in [8]). The linkage maps allowed identification of QTLs related to valuable traits followed by candidate gene identification and developing gene-specific markers that should facilitate breeding in long-lived perennial plants like Populus. For example, the linkage maps developed for full-sib F1 progenies of interspecific crosses between P. tremula x P. tremuloides allowed identification of QTLs related to sex determination in aspen [9]. Consequently, the sex-linked genomic region was reviewed considering gene loci located in congruent genome region of the sequenced P. trichocarpa. These genes were bioinformatically evaluated for their potential as candidate genes for sex determination in aspen, suggesting as a target Potri.019G047300 (‘TOZ19’). A PCR-based marker was subsequently developed for determination of the sex of non-flowering aspen individuals or seedlings [10].
Technologies such as next-generation sequencing (NGS) provide new inspiring opportunities to obtain genome-wide genetic markers for high resolution linkage map construction. Recently, Restriction-site Associated DNA Sequencing (RADseq) was employed to identify a large number of SNPs for linkage mapping of F1 progenies derived from an interspecific cross of North American P. deltoides and P. simonii, a native tree species widely distributed in northern China. 2545 high quality SNP markers allowed construction of two parent-specific linkage maps with an average distance of ~3 cM between adjacent markers, facilitating extensive QTL mapping studies in poplar [11]. Using the same F1 hybrid population of P. deltoides × P. simonii Mousavi et al. [12] demonstrated a strategy for constructing dense genetic linkage maps in hybrid forest trees by combining RADseq and whole-genome sequencing technologies.
So far only inter-specific Populus crosses were assessed for the high-resolution linkage mapping, although some Populus species with huge distribution area would provide the large diversity of phenotypic traits that is comparable with variability observed among the different species [3]. In Europe two native Populus species have a sufficiently wide geographical distribution to be considered for studies of natural variation: black poplar (P. nigra) and aspen (P. tremula) [5].
The distribution area of P. tremula covers Western and Eastern Europe, the European part of Russia, the Crimea, the Caucasus, Siberia, the Far East, Kazakhstan, Central Asia, Mongolia, China, the northern part of the Korean peninsula and Algeria [1]. Inhabiting a wide range of environmental conditions, aspen exhibits variability in numerous morphological and physiological traits. To analyze the natural phenotypic and genetic variation, the Swedish Aspen (SwAsp) Collection was established to reveal genetic determinants underlying genotypic variation of traits important for adaptation [5]. Investigation of the aspen germplasm collection elucidated exceptionally high levels of phenotypic and genotypic variation in aspen and exposed high levels (>0.55) of broad-sense heritability of phenology traits such as bud set, bud burst, and onset of senescence. Remarkably, a significant correlation between bud set and growth was reported [13]. The availability of a comprehensive set of SNP markers with known position on the aspen linkage map would facilitate the future identification of genes controlling adaptive traits.
The objective of this study was to construct high-density genetic linkage maps for the European aspen (P. tremula) using a comprehensive set of SNP markers generated with RADseq data. Herein, double digest RADseq (ddRADseq) method [14] was used for the genotyping purposes. The ddRADseq technique was earlier reported as an efficient and low-cost method for de novo SNP discovery and genotyping of a non-model species compared to the existing RADseq approaches [14, 15]. The linkage maps would provide a tool for identifying quantitative trait loci (QTLs) underlying the variation of forestry-important biological traits such as fast growth, resistance to heart rot, wood quality, root-cropping ability, phenology and winter hardiness.
Methods
Mapping population
To construct a high resolution linkage map an F1 full-sib mapping population was developed from the intra-specific cross of two aspen genotypes (P. tremula). Both male and female trees grew in the same location in the St. Petersburg area at a distance of only 8 m from each other. The parental trees had comparable ages: 30–32 years for the female and 28–30 years for the male tree, based on the number of the annual rings estimated by taking a core sample. The height of the female tree was h = 19.8 m and the diameter at breast height was d = 0.27 m. For the male tree those measurements were estimated as h = 17.6 m and d = 0.23 m, correspondingly. Inhabiting the same environment, both parents had similar dendrometry records, except for phenological traits and winter hardiness. Ten and 6 days delay were recorded for the female tree compared to the male tree for generative bud flush and vegetative bud flush, respectively. As a consequence, serious frost damage of twigs was registered for the male parent, whereas no frost damage was recorded for the female tree.
Controlled crossing was performed in April 2016 in a greenhouse of St. Petersburg State Forest Technical University. Freshly cut twigs of male and female aspen about 1 m long were placed in a 10-l clean plastic jug with water. After 10 days of incubation at room temperature (20 °C), catkins emergence was observed for both male and female twigs. Hybridization was conducted by artificial pollination of female flowers using a small brush with pollen collected from male catkins.
Next, twigs with pollinated female catkins were bagged to exclude random airborne pollen. After 14 days female catkins produced small fruit splitting to release lots of tiny, cottony seeds. The seeds were immediately planted in small plastic containers (12x10x7cm) filled with wet peat substrate. Germination occurred within a couple of days after the planting (Additional file 1: Figure S1). After 2 weeks the seedlings were transferred into individual containers and placed in lining-out nursery (Additional file 1: Figure S2). In May 2016, the seedlings reached the height of approx. 50 cm and were re-planted into larger individual containers (9x9x11 cm). By September 2016 about 1100 containerized seedlings were ready for the field.
The seedling stock was then divided into three parts to establish the F1 full-sibling aspen plantations in three diverse environments. 192 seedlings were planted in September 2016 in a field 70 km southwest of Saint Petersburg (59°16′33″N, 30°08′32″E). This location is classified as a humid continental climate; distinct moderating influence of the Baltic Sea cyclones result in warm, humid and short summers and long, moderately cold wet winters. Another set of 192 seedlings was transported to West Siberia and planted in a field nursery in Surgut (61°15′00″N, 73°25′00″E). This environment features a continental subarctic climate with long and very cold winters (daily mean temperature in January is (20 °C) and short but relatively warm summers. The third part of the containerized seedlings was used to establish the hybrid aspen plantation in the South of Russia (Voronezh, 51°40′19″N, 39°11′03″E). The Saint Petersburg population of F1 hybrids was used to construct the high-density linkage map.
DNA isolation
DNA was extracted from dried leaves collected from parental trees and each of 122 one-year progeny seedlings. For each sample 0.15 g of dried leaf material were frozen in liquid nitrogen and grinded with Mixer Mill MM 400 (Retsch, Germany). Grinded samples were washed with fresh washing buffer: 100 mM Tris-HCl, pH 8.0; 50 mM EDTA; 1 M NaCl; 1% 2-mercaptoethanol; 1% polyvinylpyrrolidone (PVP) (k-30, SABC) [16]. DNA extraction was carried out using a DNA Extran-3 kit (Syntol, Moscow) according to the manufacture protocol. Extracted DNA was cleaned with Genomic DNA Clean & Concentrator kit (Zymo Research).
Construction and sequencing of ddRAD libraries
Six ten double-digested RADseq (ddRADseq) libraries were constructed for the two parental genotypes (8 library replicates for each parent) and their 122 progenies (1 library per individual) according to the protocol described by Plekhanova et al. [17]. Concentration of DNA for each individual sample was normalized to 10 ng/ul and digested for 1 h at 37°C with 0.7 units of HindIII (NEB, USA) in a 20-ul reaction volume. Digestion was stopped by incubating the reaction mixture at 65°C for 20 min. Next, the “barcode” adapter was ligated to the end generated by HindIII, allowing pooling of the samples. The ligation reaction was carried out at 22°C for 1 h in a volume of 50 ul with 1.6 units of T4 ligase (NEB, USA). T4 ligase inactivation was reached by heating at 65°C for 10 min. Next, 10 ul for each sample were pooled and simultaneously purified with an Agencourt AMPure XP kit. The second restriction digestion was performed with 0.7 units of NlaIII (NEB, USA) in a 20-ul reaction volume at 37°C for 1 h with subsequent inactivation at 65°C for 20 min. The second “common” adapter was ligated to the overhanging end of NlaIII. The reaction mixture volume (50 ul) was dispersed into 14 aliquots and PCR was performed on the 14 aliquots to amplify the DNA fragments. PCR mix (50 ul) contained 1× HF buffer, 25 uM of each primer, 12.5 uM dNTPs and 1 unit of Phusion® High-Fidelity DNA Polymerase (NEB, USA). PCR conditions were: 98°C for 30 s, 14 cycles of 98°C for 10 s, 65°C for 30 s, 72°C for 15 s, then 72°C for 2 min. All 14 PCR reactions were pooled and purified with Agencourt AMPure XP kit. Concentration and size distribution of the prepared libraries were checked by automated electrophoresis with 2100 Bioanalyzer (Agilent Technologies, USA). Fragments were sequenced as 150 base reads on an Illumina HiSeq2500 at the University of Southern California Genome and Cytometry Core. Illumina data is available at NCBI under the BioProject PRJNA395596.
SNP calling
Illumina reads were mapped to the P. trichocarpa reference genome [6] using BWA MEM [18] under default mapping parameters. Polymorphisms were called using the GATK pipeline [19] which considers indel realignment and base quality score recalibration, and calls variants across all samples simultaneously through the HaplotypeCaller program in GATK. Variants were filtered using standard hard filtering parameters according to GATK Best Practices recommendation [20, 21]. More precisely, GBS data was filtered to only retain SNP calls with Mapping Quality (MQ) > 37 and Quality by Depth (QD) > 24. Both metrics take into consideration the quality of the mapping and genotype calls to ensure that only those with highest confidence were used. The SNPs were also filtered to retain those with MQRankSum < |2.0|, which ensure that there was no difference in the Mapping Quality scores for both alleles. This filtering retained those that pass all three criteria. Lastly, VCFtools software [22] was used to implement the following inclusion criteria: minor allele frequency (MAF) more than 3%, genotype call-rate more than 90%, and Hardy-Weinberg Equilibrium (HWE) exact P-value more than 10−5.
Linkage map construction
A RADseq linkage map of aspen was constructed using the pseudo-testcross mapping strategy [23], where a mapping population is developed by hybridizing two unrelated highly heterozygous parents to produce a set of F1 progeny [15]. With this strategy the maternal and paternal linkage maps were constructed separately using linkage phase inferred between adjacent markers.
A linkage map was constructed using JoinMap 3.0 [24]. The linkage group assignments were made under the logarithm of odds (LOD) score limit of 5.0. The regression mapping algorithm and Kosambi’s mapping function were used for map construction with the following settings: Rec = 0.4, LOD = 1.0, Jump = 5. The resulting linkage maps were drawn by JoinMap 3.0 output.
QTL mapping
Composite Interval Mapping (CIM) analysis was implemented using Windows QTL Cartographer v.2.5 [25] with a 2-cM walk speed and a type-I error rate of 5%. Intervals of five background markers with a window width of 10 cM were analyzed to control the QTL background effects. The Kosambi mapping function was chosen for recalculation of maps on genotype data. The LOD significance threshold was determined by a permutation test with 1000 permutations at a significance level of P < 0.05. The QTL results were drawn using a custom Perl script, which is available upon request.
Results
Construction and sequencing of ddRAD libraries
A total of 138 ddRAD libraries were constructed from the two parents (each in 8 replicates) and their 122 F1 offsprings. The 138 ddRAD libraries were sequenced to generate 236.56 million reads, comprising approximately 33.83 Gb of sequencing data (Additional file 2: Figure S3). The female and male parental data sets contained 20.17 million reads (comprising 2.88 Gb) and 13.08 million reads (1.87 Gb), respectively. From the 122 offspring, 204.108 million reads containing 29.07 Gb of data were produced for SNP detection. 82.62% and 81.89% of reads were successfully mapped to the P. trichocarpa reference genome for the female and male parent, respectively. For the 122 progenies the average percentage of mapped reads was 77.51%.
SNP genotyping
GATK identified 16,234 SNPs to further analysis after passing through filtering criteria (see Methods). Using these SNPs, 122 progenies were screened for their individual ancestry by maximum likelihood estimation using the ADMIXTURE software [26]. The number of ancestral populations was K = 2, corresponding to 8 reps of each female and male parent (Additional file 3: Figure S4). 7 out of 122 F1 offsprings showed unclear ancestry and were excluded from the further analysis, whereas 115 progenies perfectly demonstrated their hybrid nature and were used for the linkage map construction.
Next, chi-square tests were performed for each of 16,234 SNPs to check for correspondence to Mendelian segregation ratios 1:1, 1:2:1 and 1:1:1:1 within the 122 F1 progenies. Those SNPs deviating seriously from the Mendelian ratios (p > 0.05) were excluded from the analysis. There were SNPs showing no polymorphism between the 2 P. tremula parents, but polymorphic when compared to the P. trichocarpa reference. Those SNPs were considered as potentially valuable markers and were included in the final list of 15,732 robust SNPs useful for genotyping purposes in aspen (Additional file 4).
High-resolution genetic map construction
To construct the female map SNPs from the 15,732 robust SNPs list were selected if they i) were maternal heterozygous (ab) and paternal homozygous (aa), ii) showed segregation ratio 1:1 among the progeny, iii) showed clear polymorphism when 8 reps of maternal genotype and 8 reps of paternal genotype were compared, iiii) had less than 10% of missing data across the entire F1 population. Based on these criteria, 1000 SNPs were chosen for female map construction. Correspondingly, 1055 SNPs that were paternal heterozygous (ab) and maternal homozygous (aa) passing through the above mentioned criteria were selected to develop the male linkage map (Table 1).
Table 1.
Number of SNPs used for developing of the female and male P. tremula linkage maps
| Female genotypea | Male genotype | SNPs in parents | SNPs used for mapping |
|---|---|---|---|
| ab | aa | 2429 | 1000 |
| aa | ab | 2426 | 1055 |
| ab | ab | 2229 | 0 |
| aa | bb | 8584 | 0 |
| ab | ac | 64 | 0 |
| Total | 15,732 | 2055 | |
aa, b, c denote up to four possible alleles from two parents at an SNP site
Due to the high level of synteny and collinearity between the linkage maps of different Populus species and physical map of P. trichocarpa, [11, 12, 27] we divided the SNPs into 19 preliminary groups according to their distribution across the 19 chromosomes of the reference genome of P. trichocarpa. SNP markers were then assembled separately for each of 19 chromosomes for the male and female map using default parameters of JoinMap 3.0. For the best mapping order of the SNP markers on a chromosome and precise estimation of the genetic distances, F1 offsprings showing more than 10% of missing data for a particular linkage group were excluded from the mapping procedure. Therefore, the number of F1 individuals involved in SNP mapping for each chromosome varied between 99 to 115.
For the resulting maternal map 1000 SNPs were assigned to 19 linkage groups at the LOD threshold of 10.0 comprising a total genetic distance of 3054.99 cM. The length of single LG ranged from 106.01 cM (LG9) to 300.24 cM (LG1), with an average interlocus distance of 3.05 cM (Table 2). The corresponding paternal map represented a total length of 3090.56 cM and ranged from 107.3 cM (LG9) to 361.07 cM (LG1), with an average distance between neighboring markers of 2.93. There was a good consistency between the two parental maps in terms of length for the 19 linkage groups with the total correlation coefficient 0.91. The highest similarity was detected for LG2 and LG9, the deepest discordance was revealed for LG3. The two parental linkage maps are presented by Figs. 1 and 2; the detailed information on genetic distance and linkage phase between adjacent SNP markers is listed in Additional files 5 and 6.
Table 2.
Number of mapped SNPs and genetic distances of P. tremula female and male linkage groups (LG)
| P. tremula (female) | P. tremula (male) | Chromosome size (Mb)a | ||||
|---|---|---|---|---|---|---|
| Group | SNP number | Length (cM) | Group | SNP number | Length (cM) | |
| LG1 | 104 | 300.24 | LG1 | 133 | 361.07 | 50.5 |
| LG2 | 88 | 220.13 | LG2 | 73 | 222.56 | 25.26 |
| LG3 | 56 | 126.87 | LG3 | 48 | 175.8 | 21.82 |
| LG4 | 63 | 172.75 | LG4 | 46 | 191.32 | 24.27 |
| LG5 | 70 | 205.57 | LG5 | 74 | 196.78 | 25.89 |
| LG6 | 80 | 222.72 | LG6 | 87 | 208.34 | 27.91 |
| LG7 | 53 | 139.98 | LG7 | 39 | 124.54 | 15.61 |
| LG8 | 48 | 159.65 | LG8 | 52 | 171.18 | 19.47 |
| LG9 | 28 | 106.01 | LG9 | 57 | 107.3 | 12.95 |
| LG10 | 73 | 213.06 | LG10 | 65 | 174.74 | 22.58 |
| LG11 | 37 | 132.98 | LG11 | 46 | 139.9 | 18.5 |
| LG12 | 40 | 132.84 | LG12 | 34 | 107.64 | 15.76 |
| LG13 | 31 | 115.16 | LG13 | 47 | 120.65 | 16.32 |
| LG14 | 53 | 151.63 | LG14 | 64 | 165.61 | 18.92 |
| LG15 | 45 | 129.96 | LG15 | 45 | 117.82 | 15.28 |
| LG16 | 43 | 131.81 | LG16 | 33 | 109.14 | 14.49 |
| LG17 | 30 | 147.87 | LG17 | 37 | 127.76 | 16.08 |
| LG18 | 31 | 133.65 | LG18 | 43 | 127.5 | 16.96 |
| LG19 | 27 | 112.11 | LG19 | 32 | 140.91 | 15.94 |
| Total | 1000 | 3054.99 | 1055 | 3090.56 | ||
aGenome size refers to the reference genome of P. trichocarpa [6]
Fig. 1.

Maternal linkage map constructed for P. tremula intra-specific cross using the JoinMap 3.0. software. Genetic distances (cM) between neighboring SNPs are given on the left side of the linkage groups. SNP marker names are denoted by the names and positions of chromosomes of the reference genome P. trichocarpa
Fig. 2.

Paternal linkage map constructed for P. tremula intra-specific cross using the JoinMap 3.0. software. Genetic distances (cM) between neighboring SNPs are given on the left side of the linkage groups. SNP marker names are denoted by the names and positions of chromosomes of the reference genome P. trichocarpa
We also estimated syntenic relationships between the maternal and paternal linkage maps of P. tremula to the physical map of P. trichocarpa (Fig. 3). The order of SNP markers in the aspen parental genetic maps and the P. trichocarpa genome was highly consistent, showing high levels of synteny between the genomes of the 2 Populus species. However, for some linkage groups e.g. Chr 1,11,12,15,17,18 (Fig. 3) a block of SNPs was detected whose genetic distances were not congruent to the respective SNP positions in the chromosomes of P. trichocarpa. Since both maternal and paternal maps showed the same mode of the discordancy relatively to the reference genome, it suggests the presence of chromosome inversions, such as those found on Chr 1, 11, 15, 18.
Fig. 3.
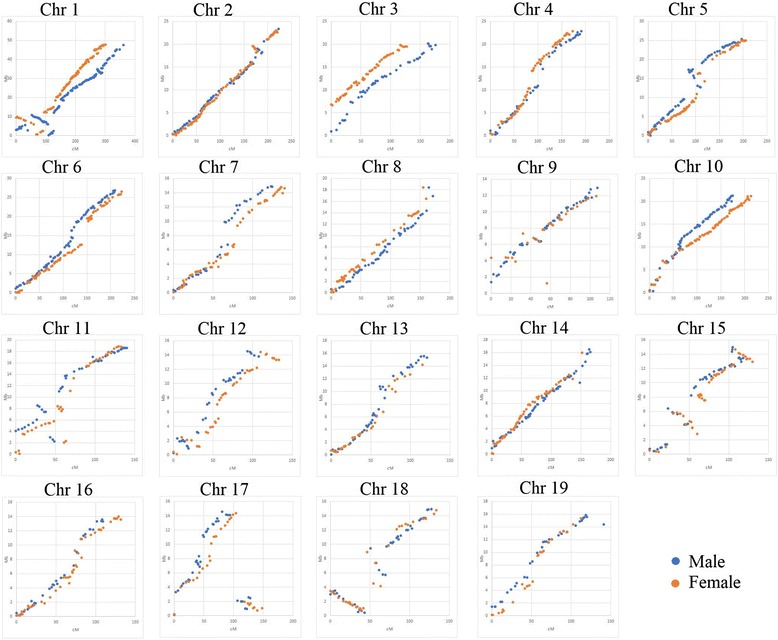
Collinearity between maternal (red) and paternal (blue) linkage maps of P. tremula and physical map of P. trichocarpa. Genetic position of SNPs on the linkage maps of P. tremula are given in cM on the X-axis, while corresponding positions of the SNPs on physical map of P. trichocarpa are shown in Mb on the Y-axis
Recently published high resolution linkage maps of P. deltoides and P. simonii allowed estimating of correlation between the genetic length of the 19 chromosomes of both the species and physical chromosome size of P. trichocarpa [11]. In the present study we developed the high density genetic maps for P. tremula. Thus, it was worthwhile to estimate the correlation between the chromosome sizes of all four Populus species (Table 3). The highest correlation (0.98) was detected between the genetic map of the P. tremula male parent and the physical map of P. trichocarpa, suggesting that the linkage map is quite accurate in marker ordering. The correlation coefficients between genetic lengths of linkage groups on the male P. tremula map and in the maps of P. deltoides and P. simonii were nearly identical (0.927 and 0.929 respectively).
Table 3.
The correlation between genetic length of 19 linkage groups on the maternal and paternal P. tremula genetic maps and physical length of P. trichocarpa chromosomes
| P. tremula maternal map | P. tremula paternal map | P. trichocarpa a | P. deltoides b | P. simonii | |
|---|---|---|---|---|---|
| P. tremula maternal map | 1.000000 | ||||
| P. tremula paternal map | 0.912140 | 1.000000 | |||
| P. trichocarpa | 0.911496 | 0.980897 | 1.000000 | ||
| P. deltoides | 0.853154 | 0.926683 | 0.914898 | 1.000000 | |
| P. simonii | 0.899007 | 0.928977 | 0.921623 | 0.939383 | 1.000000 |
Employing of the constructed high resolution genetic map for QTL mapping of growth-related traits in aspen
The F1 mapping population deriving from the intra-specific cross of two P. tremula genotypes was developed to monitor phenotypic trait variation in aspen under different environmental conditions. To aid in that that, experimental planting of F1 progenies were established in three different geographical locations facilitating further QTL mapping studies. Since in 2017 only one-year-old seedlings of the F1 population were available for phenotype assessment, we estimated the plant height variation of the seedlings before they were planted in the field (Additional file 7: Figure S5). Among the hybrids of the mapping population, the height of seedlings varied extensively from 15 cm to 127 cm, following a normal distribution (p = 0.5450) (Additional file 7: Figure S6). The estimated significant threshold for the seedling height QTLs was determined using a permutation test as LOD = 3.4 (P < 0.05). The most significant QTL (LOD = 5.73) was localized to LG5 (96.94 cM) of the male linkage map, explaining 18% of the phenotypic variation (Fig. 4). The nearest co-segregated markers were NC_008471.2_15136737; NC_008471.2_13107624 and NC_008471.2_12784748. Another significant QTL related to one-year-seedling height variation was identified on LG16 of the female map at 70.84 cM in an interval between SNPs NC_008482.2_6139958 and NC_008482.2_6542789. Its contribution to the phenotypic variation was estimated as 15%.
Fig. 4.
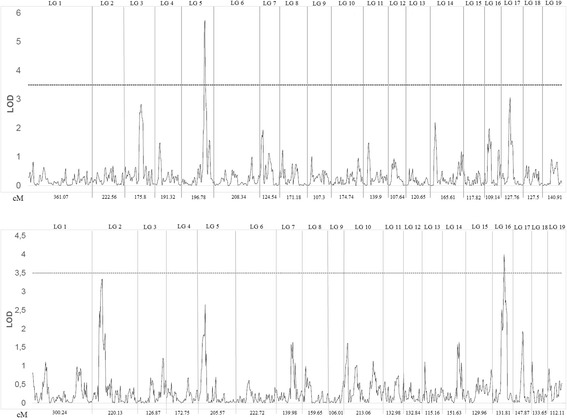
Mapping of QTLs affecting the height variation of one-year-old seedlings derived from artificial cross of two P. tremula genotypes. Upper scan: paternal linkage map. Lower scan: maternal linkage map. Length of 19 linkage groups (cM) are given below. LOD significance threshold were determined on the basis of 1000 permutations at a significance level of P < 0.05
Discussion
In the present study we first report the high-resolution linkage map for European aspen (P. tremula). This is the most widespread Populus species in the Northern hemisphere and harbors substantial genetic diversity both within populations and across the species range. Levels of nucleotide polymorphism for many nuclear genes of P. tremula were reported to be 2- to 10-fold higher than those in other woody, long-lived perennial plants, such as Pinus and Cryptomeria, and 2 to 5-fold higher than those in Betula pendula. Remarkably, the estimated population subdivision is low, thus, a substantial fraction of the aspen species-wide diversity can be sampled from single populations [28].
Due to the rapid decay of LD in P. tremula, it was previosuly assumed that the number of markers needed to ensure adequate genome coverage for QTL mapping purposes may be too high to be feasible in this species. On other hand, the rapid decay of LD suggests that it may be possible to map functional variation to very fine scales in P. tremula [28]. The limitation in the number of markers was overcome with the development of NGS technologies (e.g. RADseq [29]), so currently one could take the advantage of the low LD in aspen to achieve fine mapping and candidate genes identification for P. tremula.
There are numerous potential targets for the QTL fine mapping in aspen. The huge natural intraspecific genetic variation of P. tremula is reflected in the phenotypic diversity and constantly attracts the attention of forest researchers. At least 10 P. tremula varieties have been described in the last century just by the shape of leaves (var. monticola, parvifolia, grandifolia, rotundifolia, minor, oxyodonta, stricta, laevigata, pendula, supine) and two varieties have been reported based on bark color difference: the very ordinary light-colored P. tremula var. vulgaris and the dark-colored P. tremula var. fisca that occurs occasionally in the middle and southern regions of Russia [30]. Phenological races were also designated as intraspecific taxa: aspen with late bud flush (P. tremula L. var. tardifolia N. Rubtz.) was described from Trans-Ili Alatau (Tien Shan) [31] and reported for Belarus [32]. The late bud flushing variety showed 10–15 days delay compared to an ordinary early aspen. Revising the aspen intra-specific diversity A.Tzarev [3] reported 3% of occurrence of the late bud flushing variety in the forest-steppe regions of Russia (Voronezh area). There, bud flush delay varied from 6 to 14 days depending on the weather conditions in spring, with the late form requiring effective temperatures 1.7–2-fold higher than the early form of aspen. Remarkably, the inspection of plantations of early and late-bud flushing varieties over 30 years old showed that the percentage of trees affected by heart rot is less in plantations of the late form in approximately 1.5 times than in plantations of early aspen [3]. Special attention was traditionally paid to aspen clones with natural resistance to heart rot. Beginning from the nineteenth century small fragmentary natural populations of healthy aspen trees were regularly described within forests dramatically damaged by Phellinus pathogen in the Central part of Russia. Similarly, groups of 50–60 trees in otherwise infected aspen forests of Northwest Russia produced coppice shoots at late age (80–90 years), with no any visible signs of fungi destruction. It was intuitively concluded that resistance to the Phellinus pathogen in aspen is an inherited trait suitable for breeding [1].
In this paper we report a set of 15,732 SNP markers that are polymorphic between two genotypes of P. tremula. This is only a subset of the nucleotide polymorphism in aspen and does not include those regions in P.tremula without homology to the P. trichocarpa reference genome. About 20% of reads were not mapped to the reference, thus, part of the polymorphism in aspen is still left unexplored. 30% of the SNPs that mapped to the P. trichocarpa reference genome produced heterozygous genotypes in the maternal and paternal aspen trees assessed in this study. This is in good concordance with previous reports showing 29.2% of loci in P. trichocarpa accessions carrying heterozygous genotypes [7]. The developed set of 15,732 SNP markers would contribute to further association mapping studies in aspen.
Two thousand fifty five SNP markers were successfully employed for the construction of high-resolution maternal and paternal linkage maps for an F1 full-siblings P. tremula population. The constructed map comprised 19 linkage groups based on 1055 SNPs for the male map and 1000 SNPs for the female map. The estimated lengths of 3054.99 and 3090.56 cM for the two parental genetic maps were slightly shorter in comparison with the total genetic lengths reported earlier for the high density map of P. deltoides (4057 cM) and P. simonii (3.809 cM) [11]. On other hand, these values were larger than those reported in P. adenopoda (2443.2 cM) and P. alba (2719.5) [27] based on SSR and SRAP markers. The genetic lengths of the aspen linkage groups in our study were, however, consistent with the chromosome size of P. trichocarpa (Tables 1, 2).
Despite the highly conserved synteny and collinearity observed between SNP order on the chromosomes of P. tremula and P. trichocarpa, our results suggest the presence of several chromosome inversions between the two taxonomically remoted Populus species. P. tremula belongs to the section Populus (syn. Leuce Duby), whereas balsam poplars P. trichocarpa and P. simonii are from the same section Tacamahaca Spach. Even for P. simonii genetic maps, several linkage groups exhibited inverse orders of some SNPs relative to the P. trichocarpa genome [11]. Moreover, for nearly all linkage groups of P. deltoides and P. simonii one or more local regions have been reported where SNP order was inconsistent with reference genome positions [12]. We hypothesize that for some linkage groups of P. tremula (e.g. Chr 1, 11, 15, 18) chromosome inversions could be exposed relatively to the SNP marker order on P. trichocarpa chromosomes. An inversion occurs when a chromosome breaks at two points and the segment bounded by the breakpoints is reinserted in the reversed orientation [33]. A key evolutionary effect of inversions is that they suppress recombination as heterozygotes. As a result, inversions may contribute to reproductive isolation, local adaptation and speciation in plants [34]. The comparison of the whole genome sequencing of P. tremula with the reference genome of P. trichocarpa would make it possible to determine whether the observed inconsistency in the order of SNP markers on two genomes is due to the inversion of chromosomes, rather than the distortion of the genetic map.
Conclusions
In the present study we describe a set of 15,732 SNPs polymorphic within P. tremula, a plant that has the widest-known geographic distributions of any tree species. The SNPs were obtained by RADseq genotyping of two parental aspen trees and their 122 F1 progenies followed by mapping of 20.17 million Illumina reads to the P. trichocarpa reference genome. 2055 of the SNP markers were successfully employed for the construction of high-resolution maternal and paternal linkage maps of the intraspecific aspen cross. The constructed linkage maps were successfully employed for mapping of QTLs related to plant height variation in the full sibling family. The high resolution linkage map and the set of SNP markers will provide a valuable source for QTL mapping and association studies in P. tremula facilitating marker-assisted selection in European aspen.
Additional files
Germination of hybrid seeds (F1) obtained via artificial crossing of two parental aspen genotypes. Figure S2. Two-weeks-old F1 hybrid aspen seedlings placed in lining-out nursery. (PPTX 1375 kb)
Information about 138 sequenced ddRAD libraries and reads successfully mapped to the P. trichocarpa reference genome. (PDF 391 kb)
{kind=link}
Individual ancestry of 122 F1 aspen offspring estimated by maximum likelihood method based on 16,234 SNPs with ADMIXTURE software. (JPEG 1846 kb)
List of 15,732 robust SNPs available for aspen genotyping. (TXT 8857 kb)
Detailed information on genetic distances and linkage phase between adjacent SNP markers in the maternal linkage map constructed for P. tremula intra-specific cross. (XLSX 427 kb)
Detailed information on genetic distances and linkage phase between adjacent SNP markers in the paternal linkage map constructed for P. tremula intra-specific cross. (xlsx, 428 kb) (XLSX 463 kb)
Evaluation of one-year-old seedling height variation before the plants were planted in the field. Figure S6. Chi-square goodness of it test for Normality of seedling height values among the 122 F1 progenies derived from P. tremula intra-specific cross. (PPTX 939 kb)
Acknowledgments
We are grateful to Dr. Georg von Wühlisch for the initiation of the Russian-German collaborative MaRussiA project. We thank Dr. Dmitry Danilov for his help with the field work during the establishment of the hybrid aspen plantation in Saint Petersburg’s area (Druzhnaja Gorka).
Funding
This work was supported by the Ministry of Food and Agriculture of Germany (MaRussiA project, 2015–2016) and by scientific project no. 37.1521.2014/K of the Ministry of Science and Education of the Russian Federation. SVN collaboration with VIR was supported from Zumberg Foundation USC grant. The publication costs were funded by the MaRussiA project.
Availability of data and materials
Supporting data are included as additional files.
About this supplement
This article has been published as part of BMC Plant Biology Volume 17 Supplement 1, 2017: Selected articles from PlantGen 2017. The full contents of the supplement are available online at https://bmcplantbiol.biomedcentral.com/articles/supplements/volume-17-supplement-1.
Authors’ contributions
AVZ created the F1 population, established the full-siblings’ plantations, coordinated the collection of phenotyping data. PSU built the linkage maps, performed synteny analysis. PSU and MVL prepared the RADseq libraries. MVL made phenotyping, performed QTL analysis. PLC analyzed the Illumina reads, made SNP calling, performed admixture analysis. SVN contributed to the design of the study and granted the access to Illumina sequencing. EKP conceived the study and along with PLC wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study does not contain any research requiring ethical consent or approval.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12870-017-1127-y) contains supplementary material, which is available to authorized users.
References
- 1.Yablokov AS. Upbringing and breeding of healthy aspen. Leningrad: Goslesbumizdat; 1949. [Google Scholar]
- 2.Garipov NR. Selection and propagation of triploid aspen (Populus tremula L.) with the application of methods of molecular genetics and biotechnology in the Republic of Tatarstan. 2014. http://dlib.rsl.ru/01005552669. Accessed 16 Oct 2017.
- 3.Tzarev AP. ‘Sortology’ of poplar. Voronezh: Voronezh State University; 1985. [Google Scholar]
- 4.Frewen BE, Chen TH, Howe GT, Davis J, Rohde A, Boerjan W, Bradshaw HD. Quantitative trait loci and candidate gene mapping of bud set and bud flush in Populus. Genetics. 2000;154(2):837–845. doi: 10.1093/genetics/154.2.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luquez V, Hall D, Albrectsen BR, Karlsson J, Ingvarsson P, Jansson S. (2008). Natural phenological variation in aspen (Populus tremula): the SwAsp collection. Tree Genetics Genomes. 2008;4(2):279–292. doi: 10.1007/s11295-007-0108-y. [DOI] [Google Scholar]
- 6.Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 7.Geraldes A, Difazio SP, Slavov GT, Ranjan P, Muchero W, Hannemann J, et al. A 34K SNP genotyping array for Populus Trichocarpa: design, application to the study of natural populations and transferability to other Populus species. Mol Ecol Resour. 2013;13(2):306–323. doi: 10.1111/1755-0998.12056. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Sun X, Tan B, Zhang B, Xu LA, Huang M, Wang M. A genetic linkage map of Populus adenopoda maxim. × P. alba L. hybrid based on SSR and SRAP markers. Euphytica. 2010;173(2):193–205. doi: 10.1007/s10681-009-0085-3. [DOI] [Google Scholar]
- 9.Pakull B, Groppe K, Mecucci F, Gaudet M, Sabatti M, Fladung M. Genetic mapping of linkage group XIX and identification of sex-linked SSR markers in a Populus tremula× Populus tremuloides cross. Can J For Res. 2011;41(2):245–253. doi: 10.1139/X10-206. [DOI] [Google Scholar]
- 10.Pakull B, Kersten B, Lüneburg J, Fladung M. A simple PCR-based marker to determine sex in aspen. Plant Biol. 2015;17(1):256–261. doi: 10.1111/plb.12217. [DOI] [PubMed] [Google Scholar]
- 11.Tong C, Li H, Wang Y, Li X, Ou J, Wang D, et al. Construction of high-density linkage maps of Populus deltoids × P. simonii using restriction-site associated DNA sequencing. PLoS One. 2016;11(3):e0150692. doi: 10.1371/journal.pone.0150692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mousavi M, Tong C, Liu F, Tao S, Wu J, Li H, Shi J. De novo SNP discovery and genetic linkage mapping in poplar using restriction site associated DNA and whole-genome sequencing technologies. BMC Genomics. 2016;17(1):656. doi: 10.1186/s12864-016-3003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michelson IH, Ingvarsson PK, Robinson KM, Edlund E, Eriksson ME, Nilsson O, Jansson S. Autumn senescence in aspen is not triggered by day length. Physiol Plantarum. 2017. doi:10.1111/ppl.12593. [DOI] [PubMed]
- 14.Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7(5):e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun C, Niu Y, Ye X, Dong J, Hu W, Zeng Q, et al. Construction of a high-density linkage map and mapping of sex determination and growth-related loci in the mandarin fish (Siniperca chuatsi) BMC Genomics. 2017;18(1):446. doi: 10.1186/s12864-017-3830-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li JT, Yang J, Chen DC, Zhang XL, Tang ZS. An optimized mini-preparation method to obtain high-quality genomic DNA from mature leaves of sunflower. Genet Mol Res. 2007;6(4):1064–1071. [PubMed] [Google Scholar]
- 17.Plekhanova E, Vishnyakova MA, Bulyntsev S, et al. Genomic and phenotypic analysis of Vavilov’s historic landraces reveals the impact of environment and genomic islands of agronomic traits. Scientific Reports. 2017;7:4816. doi:10.1038/s41598-017-05087-5. [DOI] [PMC free article] [PubMed]
- 18.Li H, Durbin R. Fast and accurate long-read alignment with burrows–wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;11:10. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grattapaglia D, Sederoff R. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics. 1994;137(4):1121–1137. doi: 10.1093/genetics/137.4.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Ooijen, Johan W, Voorrips RE. "JoinMap® 3.0, Software for the calculation of genetic linkage maps." Plant Research International, Wageningen, The Netherlands. (2001);1–51.
- 25.Wang S, Basten CJ, and Zeng Z-B. Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. 2012. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm. Accessed 01 Aug 2012.
- 26.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Zhang B, Sun X, Tan B, Xu LA, Huang M, et al. Comparative genome mapping among Populus adenopoda, P. alba, P. deltoides, P. euramericana and P. trichocarpa. Genes Genet Syst. 2011;86(4):257–268. doi: 10.1266/ggs.86.257. [DOI] [PubMed] [Google Scholar]
- 28.Ingvarsson PK. Nucleotide polymorphism and linkage disequilibrium within and among natural populations of European aspen (Populus tremula L., Salicaceae) Genetics. 2005;169(2):945–953. doi: 10.1534/genetics.104.034959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3(10):e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kunitskii B. Botanical and silvicultural characteristics of aspen with notes on its use. Yearbook of St.Petersburg Forest Institute. 1888;2:51–71.
- 31.Rubtzov NI. A note about a new form of aspen from the Trans-Ili Alatau. Botanical Journal of the USSR. 1938;23(1):23–26.
- 32.Kruglikov GG. On a new form of aspen. Forestry. 1938;6(12):34–42.
- 33.Kirkpatrick M. How and why chromosome inversions evolve. PLoS Biol. 2010;8(9):e1000501. doi: 10.1371/journal.pbio.1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowry DB, Willis JH. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biol. 2010;8:e1000500. doi: 10.1371/journal.pbio.1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Germination of hybrid seeds (F1) obtained via artificial crossing of two parental aspen genotypes. Figure S2. Two-weeks-old F1 hybrid aspen seedlings placed in lining-out nursery. (PPTX 1375 kb)
Information about 138 sequenced ddRAD libraries and reads successfully mapped to the P. trichocarpa reference genome. (PDF 391 kb)
Individual ancestry of 122 F1 aspen offspring estimated by maximum likelihood method based on 16,234 SNPs with ADMIXTURE software. (JPEG 1846 kb)
List of 15,732 robust SNPs available for aspen genotyping. (TXT 8857 kb)
Detailed information on genetic distances and linkage phase between adjacent SNP markers in the maternal linkage map constructed for P. tremula intra-specific cross. (XLSX 427 kb)
Detailed information on genetic distances and linkage phase between adjacent SNP markers in the paternal linkage map constructed for P. tremula intra-specific cross. (xlsx, 428 kb) (XLSX 463 kb)
Evaluation of one-year-old seedling height variation before the plants were planted in the field. Figure S6. Chi-square goodness of it test for Normality of seedling height values among the 122 F1 progenies derived from P. tremula intra-specific cross. (PPTX 939 kb)
Data Availability Statement
Supporting data are included as additional files.