

Abstract
The efficient production, folding, and secretion of proteins is critical for cancer cell survival. However, cancer cells thrive under stress conditions that damage proteins, so many cancer cells overexpress molecular chaperones that facilitate protein folding and target misfolded proteins for degradation via the ubiquitin-proteasome or autophagy pathway. Stress response pathway induction is also important for cancer cell survival. Indeed, validated targets for anti-cancer treatments include molecular chaperones, components of the unfolded protein response, the ubiquitin-proteasome system, and autophagy. We will focus on links between breast cancer and these processes, as well as the development of drug resistance, relapse, and treatment.
Breast cancer subtypes and cellular protein quality control pathways
Breast cancer is a complex and heterogeneous disease that remains the most prevalent cancer diagnosed in women and is responsible for the greatest proportion of cancer-related deaths in women [1]. Breast cancers are divided into different subtypes depending on the expression of hormone receptors, including estrogen receptor (ER), progesterone receptor (PR), and epidermal growth factor receptor 2 (HER2 or erbB2) [2, 3]. Luminal breast cancers are characterized by ER overexpression and fall into the luminal A or B class, in which, respectively, either both ER and PR are overexpressed or ER is overexpressed and HER2 may also be overexpressed. HER2-positive breast cancer, in which HER2 is overexpressed, represents another subtype, and can be diagnosed at a younger age compared to the luminal A and luminal B cancers. Finally, triple negative breast cancers (TNBC) are ER-, PR-, and HER2-negative [2, 3].
ER-positive breast cancer groups are especially prevalent and mainly afflict postmenopausal women because luminal cells become more sensitive to estrogen (17β-estradiol or E2) levels as a result of hormonal fluctuations [4]. Activation of the ER signaling cascade stimulates cell division, tumor growth, and metastasis. Therefore, ER-positive patients are initially treated with anti-estrogen therapies [5] (Table 1). Tamoxifen was one of the first FDA approved drugs used to treat these patients, is a non-steroid inhibitor of the receptor, and blocks downstream signaling [6, 7]. However, in many tamoxifen treated patients, ER activation was still detected, highlighting the demand for improved compounds and new targets [8]. In fact, since tamoxifen was approved, different ER-targeted drugs were introduced that downregulate the receptor, induce receptor degradation, or attenuate ER signaling (Table 1) [9–12]. Nevertheless, endocrine-treatment resistance remains one of the leading causes of breast cancer mortality [12, 13].
Table 1.
Examples of drugs used in ER-positive breast cancer treatment
| Drug name | Notes | Developmental stage | References |
|---|---|---|---|
| Fulvestrant (Faslodex) | Induces ER degradation, nonsteroidal selective estrogen receptor degrader (SERD) | FDA approved | [288–292] |
| Tamoxifen | Nonsteroidal selective estrogen receptor modulator (SERM) | FDA approved | [6, 288, 293–295] |
| Raloxifene hydrochloride | Nonsteroidal selective estrogen receptor modulator (SERM) | FDA approved | [296–301] |
| Toremifene (Fareston) | Nonsteroidal selective estrogen receptor modulator (SERM) | FDA approved | [301, 302] |
| ARN-810 | Nonsteroidal selective estrogen receptor degrader (SERD) | Clinical trial | [303, 304] |
| AZD9496 | Nonsteroidal selective estrogen receptor degrader (SERD) | Clinical trial | [305, 306] |
| RAD1901 | Nonsteroidal selective estrogen receptor degrader (SERD) | Clinical trial | [307–309] |
| Letrozole (Femara) | Nonsteroidal inhibitor of estrogen synthesis (aromatase inhibitor) | FDA approved | [310–313] |
| Anastrozole (Arimidex) | Nonsteroidal inhibitor of estrogen synthesis (aromatase inhibitor) | FDA approved | [314–317] |
| Exemestane (Aromasin) | Nonsteroidal inhibitor of estrogen synthesis (aromatase inhibitor) | FDA approved | [318–321] |
| BEZ235 | Dual inhibitor of PI3K and mTOR | Clinical trial | [322–325] |
| SAR245409 | PI3K inhibitor | Clinical trial | [285, 313] |
| Taselisib | Class I PI3K alpha inhibitor | Clinical trial | [326–328] |
| Buparlisib | PI3K inhibitor, competes for the ATP binding | Clinical trial | [63, 326] |
| Venetoclax (ABT-199) | Bcl-2 inhibitor | Clinical trial | [329, 330] |
| Everolimus (Afinitor) | Inhibitor of mTORC1 used both in luminal A and in HER2-positive tumors | FDA approved | [277–279, 283] |
| Temsiroliums | Inhibitors of mTORC1 used in luminal A, TNBC, and HER2-positive tumors | Clinical trial | [331] |
| Vorinostat (SAHA) | Global inhibitor of HDAC | Clinical trial | [332–334] |
| Entinostat | Inhibitor of HDAC1 and HDAC3 | Clinical trial | [335, 336] |
| Panobinostat | Specific inhibitor of HDAC | Clinical trial | [337–339] |
| Rapamycin | mTOR inhibitor | HER- and ER-positive breast cancer cells | [340–344] |
| SNIPER(ER) | PROTAC-mediated ER degradation in breast cancer cells | ER-positive breast cancer cells | [98, 345] |
| BHPI | Modulator of ER-dependent UPR response | ER-positive breast cancer cells | [168, 187, 188, 346] |
| MAb159 | Monoclonal antibody against BiP | ER-positive breast cancer cells | [222] |
| Plumbagin | BiP downregulator, induces BIK levels | ER-positive breast cancer cells | [220] |
| Epigallocatechin gallate (EGCG) | Inhibits cellular oxidation and DNA methyltransferase to block EGFR and HER2 activation and can induce UPR response by binding BiP | TNBC, ER- and HER2-positive breast cancer cells | [347–352] |
| Resveratrol | Activates SIRT-1 and inhibits TNF-induced activation of NFkB. Used in combination with bortezomib, reduces cell viability through autophagy inhibition | ER- and HER2-positive breast cancer cells | [353, 354] |
| Hydroxychloroquine | Autophagy inhibitor, suppresses lysosomal acidification | Clinical trial | [167, 225] |
The HER2 amplified class represents 15–20% of all the breast cancers and patients with this variant have benefited from significant clinical successes [3, 14]. HER2 plays a key role in cellular hemostasis and tissue development (such as during epithelial mammary gland organization [15]). There are four members of the HER tyrosine kinase family, HER, HER2, HER3, and HER4 (also known as EGFR, erbB2, erbB3, and erbB4, respectively), and maintenance of proper receptor numbers at the plasma membrane is critical for signal transduction [15, 16]. Upon ligand binding, the receptors homo- or hetero-dimerize, inducing an intracellular signaling cascade [17]. Receptor endocytosis also represents a key regulatory event [16]. While many ligands for EGRF, HER3, and HER4 are known, no HER2 ligand has been identified. Moreover, in contrast to the other family members, HER2 is the preferred dimerization partner of the other HERs, in particular HER3, which activates downstream PI3K/Akt signaling [18–21]. Preclinical studies supported a key function for HER3 in promoting the growth of HER2-positive breast cancer cells [22], and these patients are now prime candidates for trastuzumab (Herceptin, a monoclonal antibody against HER2), perstuzumab (Perjeta, a HER2 and HER3 dimerization inhibitor), and lapatinib (Tykerb, a tyrosine kinase inhibitor), as well as for other compounds directed against HER2 and/or downstream kinases (Table 2). Yet despite the apparent specificity of these drugs, some patients remain resistant to these treatments [23], mainly due to metastasis and receptor mutations, which reduces patient survival and increases tumor relapse [24]. Moreover, ~ 10% of metastatic luminal breast cancers metastasize to the brain, and in this case the only treatment options include chemotherapy, radiation, and/or surgery [12]. Therefore, the identification of markers during early stage therapy is also of fundamental importance.
Table 2.
Examples of drugs used in HER2 breast cancer treatment
| Drug name | Notes | Developmental stage | References |
|---|---|---|---|
| Trastuzumab (Herceptin) | Monoclonal antibody against HER2 | FDA approved | [355–360] |
| Ado trastuzumab emtansine (T-DM1) | Bifunctional antibody-drug (trastuzumab linked to ematansine, DM1). Binds HER2 and inhibits microtubule assembly/disassembly | FDA approved | [361–364] |
| Eetumaxomab | Monoclonal antibody with CD3 and HER2 recognition sites | Clinical trial | [365, 366] |
| Pertuzumab (Perjeta) | Recombinant humanized antibody against domain II of HER2 | FDA approved | [284, 367, 368] |
| MM-111 | Antibody against HER2-HER3 dimers | Clinical trial | [369, 370] |
| Lapatinib (Tykerb) | Irreversible tyrosine kinase inhibitor in luminal B cancers | FDA approved | [371, 372] |
| Afatinib | Irreversible pan-HER tyrosine kinase inhibitor | Clinical trial | [373–375] |
| Canertinib | Irreversible tyrosine kinase inhibitor | Clinical trial | [19, 376, 377] |
| Neratinib | Irreversible pan-HER tyrosine kinase inhibitor, effective against EGFR, HER2, and HER4 | Clinical trial | [378–381] |
| Gefitinib | EGFR tyrosine kinase inhibitor | Clinical trial | [382, 383] |
| Erlotinib hydrochloride (Erlotinib) | Reversibly binds to the intracellular catalytic domain of EGFR; used also in TBNC cancers | Clinical trial | [383–385] |
| Sapitinib | Tyrosine kinase inhibitor effective in luminal B resistant cells | Clinical trial | [380, 386] |
| Sorafenib | Blocks the enzyme RAF kinase, inhibiting cancer cell proliferation and autophagy induction | Clinical trial | [63, 387–389] |
| Sildenafil citrate | Selectively inhibits cyclic guanosine monophosphate (cGMP)-specific type 5 phosphodiesterase | Clinical trial | [63] |
| MM-121 | Human monoclonal antibody against HER3 | Clinical trial | [267, 390–392] |
| MM-302 | Doxorubicin encapsulated within liposomes, and conjugated to a monoclonal antibody against HER2. Inhibits HER2 and topoisomerase II | Clinical trial | [393, 394] |
| ARRAY-380 | Reversible selective HER2 inhibitor | Clinical trial | [395] |
| TAK-285 | HER2-EGFR tyrosine kinase inhibitor | HER2-positive breast cancer cells | [396–399] |
| Everolimus (Afinitor) | Inhibitor of mTORC1 used both in luminal A and in HER2-positive tumors | FDA approved | [277–279, 283] |
| Temsiroliums | Inhibitor of mTORC1 used in luminal A, TNBC, and HER2-positive tumors | Clinical trial | [331] |
| GDC-0941 | PI3K inhibitor | Clinical trial | [400, 401] |
| SAR245408 | PI3K inhibitor | Clinical trial | [402, 403] |
| 17-AAG | Hsp90 inhibitor | Clinical trial | [404–406] |
| Retaspinmycin (IPI-504) | Hsp90 inhibitor | Clinical trial | [406–408] |
| Genetespib | Hsp90 inhibitor used in metastatic HER2 breast cancers | Clinical trial | [409–412] |
| Pazopanib | inhibitor of VEGFRs able to inhibit Hsp90 ATPase activity | Clinical trial | [63, 413, 414] |
| SNX-2112 | Hsp90 inhibitor, effective in HER2 and luminal B breast cancers | Clinical trial | [83, 280, 415] |
| Geldanamycin | Hsp90 inhibitor | HER2-positive breast cancer cells | [20, 109, 114–116] |
| KIN001-51 | HER3 binder, impairs dimerization | HER2-positive breast cancer cells | [416, 417] |
| TX1-85-1 | Induces HER3 degradation by covalent binding to a residue in the receptor | HER2-positive breast cancer cells | [100, 383] |
| TX2-121-1 | Derivate of TX1-85-1 linked to adamantane group. Induces HER3 degradation | HER2-positive breast cancer cells | [100, 383] |
| Patritumab (AMG 888) | Monoclonal antibody directed against the ligand-binding pocket of HER3 | Clinical trial | [418] |
| MEHD7945A | Monoclonal antibody directed against EGFR and HER3 | Clinical trial | [419, 420] |
| Pilaralisib | Pan-class I PI3K inhibitor | HER2-positive breast cancer cells | [224, 421] |
| liposomal paclitaxel | Inhibits tubulin assembly/disassembly | FDA approved | [224, 421, 422] |
| hydroxychloroquine | Autophagy inhibitor, suppresses lysosomal acidification | Clinical trial | [167, 225] |
| Eeyarestatins | p97 inhibitor | HER2-positive breast cancer cells | [81, 85] |
| NMS-873 | p97 inhibitor | HER2-positive breast cancer cells | [81] |
| HA15 | Inhibitor of BiP ATPase activity | HER2-positive breast cancer cells | [173] |
Over the past several years, alternative approaches to treat breast cancer have been pursued, focusing on the regulation of protein homeostasis (“proteostasis”) and stress response pathways [25–30]. These approaches include modulation of protein degradation pathways mediated by the proteasome and autophagy, and the regulation of cellular stress responses, with particular attention paid to the unfolded protein response (UPR).
The ubiquitin-proteasome pathway utilizes a cascade of E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases [31–34]. Once a protein substrate has acquired at least four ubiquitin species, the substrate is delivered to the 26S proteasome, which houses three unique protease activities to destroy protein substrates. Protein ubiquitylation can be reverted by a family of proteins called deubiquitylating enzymes (DUBs) that are involved in removing mono- and poly-ubiquitin chains from proteins, thereby changing the fates of their targets and maintaining the pool of free ubiquitin [35–37]. In contrast, autophagy is a process in which damaged proteins are encapsulated and degraded in double-membrane structures, called autophagolysosomes [38–42]. During canonical autophagy (formally called macroautophagy), an isolation membrane encloses a portion of the cytoplasm containing misfolded proteins, protein aggregates, and even organelles to form a vesicle called the autophagosome. The contents of the autophagosomes are degraded upon subsequent fusion with lysosomes [2, 38–41, 43, 44].
The proteasome pathway has been effectively targeted in select cancers, primarily because protein folding is an inherently slow, energy expensive, and inefficient pathway. Therefore, each cellular compartment is equipped with a variety of molecular chaperones and folding enzymes that coordinate protein folding; however, if protein folding is delayed or compromised, aberrant proteins are instead targeted to the ubiquitin-proteasome system [45–48]. Proteasome inhibitors such as carfilzomib (Kyprolis) and bortezomib (Velcade) are especially effective for the treatment of multiple myeloma [49, 50] because myeloma cells produce high levels of misfolded or unassembled immunoglobulin subunits [51–55]. Also, due to their higher proliferation rate, cancer cells require increased levels of ATP for enhanced protein production and there is a heightened demand on cellular protein folding pathways. Cancer cells can also become hypoxic, hypoglycemic, and acidic, which perturbs cellular hemostasis and—in particular—secretory protein folding in the endoplasmic reticulum [28, 56, 57]. Moreover, most cancer cells contain DNA duplications, deletions, inversions, and translocations, as well as altered chromosome numbers (aneuploidy) [58], which alters the stoichiometries of protein complexes. Consequently, many proteins become “orphaned” and misfold [59, 60]. To overcome the ensuing onslaught of these misfolded proteins, cancer cells also overexpress multiple chaperones to maintain cellular homeostasis [33, 48, 61–63].
In the remainder of this review, we will discuss the links between protein folding, degradation, and transport and breast cancer survival with an emphasis on the different mechanisms involved in controlling secretory protein folding. The highlighted mechanisms include the endoplasmic reticulum-associated degradation (ERAD) pathway, the unfolded protein response (UPR), and the autophagy pathway (Fig. 1). Finally, we will emphasize critical areas for further research that may improve breast cancer treatments.
Fig. 1.
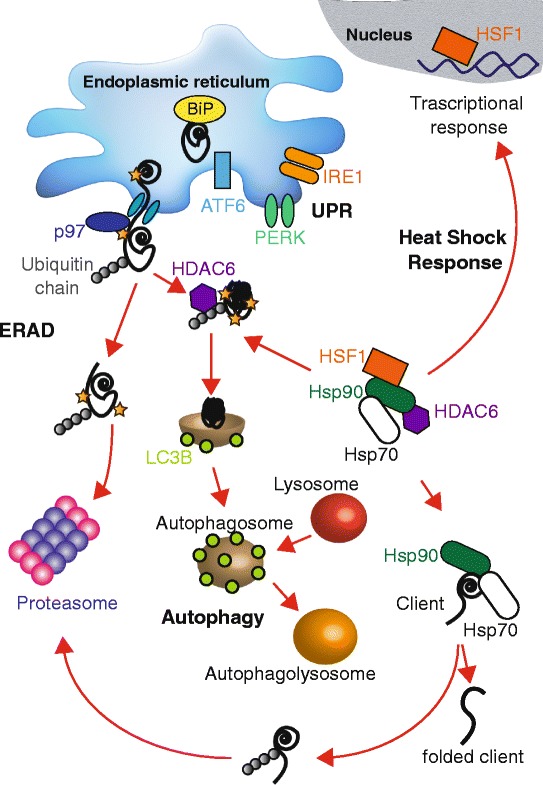
Schematic representation of secretory protein folding and quality control, the unfolded protein response pathway, and the heat shock response. In the endoplasmic reticulum, unfolded proteins (black lines) can be recognized and bound by chaperones, such as BiP (yellow circles). The increase in the concentration of BiP-unfolded protein complexes in the endoplasmic reticulum favors induction of the unfolded protein response (UPR). The UPR is regulated by ATF6 (light blue rectangle), PERK (green dimers), and IRE1 (orange dimers), which reside in the endoplasmic reticulum membrane. Upon activation, the UPR can increase cellular folding capacity by increasing chaperone synthesis, inducing endoplasmic reticulum expansion, and increasing the concentration of components of the endoplasmic reticulum associated degradation (ERAD) machinery. During ERAD, unfolded proteins in the endoplasmic reticulum are recognized, ubiquitylated by E3 ubiquitin ligases, and retrotranslocated via the action of p97 (blue circle), an AAA-ATPase, to the cytosol where they are degraded by the proteasome. Misfolded, aggregation-prone proteins, protein aggresomes, and damaged organelles can alternatively be targeted for autophagy via encapsulation in double membrane vesicles known as autophagosomes (light brown vesicles). LC3BII is an established marker of cellular autophagy and is associated with autophagosome membranes (light green circles), and proteins can be directed to autophagy degradation via HDAC6 (purple hexagon). Upon fusion with lysosomes (red vesicles), the material incorporated in the autophagolysosome is degraded (orange vesicles). In the absence of stress, HSF1, HSP90, HSP70, and HDAC6 can form a complex in the cytoplasm. During stress (for example, an increase in the concentration of unfolded protein or heat), HSF1 (orange rectangle) can translocate to the nucleus and induce the transcription of other proteins, like chaperones, to increase the cellular folding capacity. This is known as the heat shock response. At the same time, Hsp90 and Hsp70 (green and white rounded rectangles, respectively) are involved in cytoplasmic protein folding, dictating the fate of their clients. If the client fails to attain its final conformation, it will be ubiquitylated and degraded by the proteasome
Proteasome-mediated degradation as a possible therapeutic target
Approximately one-third of all proteins in eukaryotes are targeted to the endoplasmic reticulum [45, 46, 64]. Nevertheless, proteins entering this compartment—which represents the first step in the secretory pathway—may misfold due to stochastic errors in the folding pathway or as a result of cellular stress. Cellular attempts to temper protein toxicity in the secretory pathway are based on two different mechanisms: first, the UPR can be induced, which increases the chaperone reservoir in the endoplasmic reticulum and induces endoplasmic reticulum expansion, and second, misfolded proteins can be targeted for degradation to either the lysosome (via autophagy) or to the proteasome [31, 65, 66]. The process that targets unfolded proteins in the endoplasmic reticulum to the proteasome is known as endoplasmic reticulum-associated degradation, or ERAD.
ERAD is a multistep pathway, and many of the components required for this event are induced by the UPR [66, 67]. ERAD substrates are selected by a pool of chaperones and lectins and, in the case of soluble luminal substrates, are partially transported across the membrane to expose their cytoplasmic domains, which allows for ubiquitylation [34, 36, 68, 69]. The acquisition of a poly-ubiquitin chain recruits the AAA+ ATPase p97, which “retrotranslocates” ERAD substrates from the endoplasmic reticulum [70–73]. Once retrotranslocated, ubiquitylated substrates are degraded by the 26S proteasome. p97 is not only involved in ERAD substrate retrotranslocation but also modulates protein trafficking in the secretory pathway, mitochondrial-associated protein degradation, the degradation of aberrant ribosome-associated proteins, chromatin remodeling, and autophagy, each of which is also associated with cancer [74–80]. Recently, HER2-positive breast cancer cells were shown to have elevated levels of p97, suggesting in one study that ERAD contributes to cancer cell survival [81]. Based on these data and the importance of p97 during the disposal of misfolded proteins, p97 inhibitors are being examined for possible therapeutic applications in cancer cells. The eeyrestatins (Eer I and II) were the first generation of p97 inhibitors and in several studies were shown to reduce ERAD efficiency [82–85]. HER2-positive cells are more sensitive to both eeyarestatin and NMS-873, a refined p97 inhibitor, compared to other breast cancer cells that did not overexpress HER2 [81]. Recently a new p97 inhibitor was characterized (CB-5083) as a potential anti-cancer drug both in multiple myeloma and in various solid tumors, including lung and colorectal carcinoma [86]. Thus far, clinical trials using this compound in breast cancer patients have not yet begun.
Human epidermal growth factor receptors, or HER family members, are selected for endosomal sorting and lysosomal degradation from the plasma membrane, and as noted above the regulation of receptor numbers at the plasma membrane is crucial for cell survival [87, 88]. However, the levels of HER3 that ultimately reside at the plasma membrane are also regulated in the endoplasmic reticulum by ERAD in a p97-dependent manner. In fact, HER3 is ubiquitylated by an E3 ubiquitin ligase known as Nrdp1 in the endoplasmic reticulum [88, 89]. Nrdp1 also ubiquitylates HER3 at the plasma membrane, which is required for endocytosis and lysosomal sorting/degradation [88, 90, 91]. Consistent with the importance of properly regulating the levels of HER3-containing heterodimers, Nrdp1 is suppressed in 57% of breast cancer tissues [92–94]. This E3 ubiquitin ligase also interacts with HER4 in breast cancer cells [95]. One study suggested that, after endoplasmic reticulum stress and UPR induction, Nrdp1 becomes trapped in tubular structures, which impairs HER3 degradation [96]. These data highlight how ERAD, plasma membrane protein degradation, and stress may be linked to signal transduction pathways and proteostasis in cancer cells.
Based on its central role in protein quality control, compromising other early steps in the proteasome-dependent degradation pathway might deplete cancer cells of their oncogenic signaling potential and provide another therapeutic route. To this end, PROTACs were developed, which are heterobifunctional molecules that contain a binding motif for the target of interest and for an E3 ubiquitin ligase. Addition of the molecule induces selective degradation of a target via the proteasome or, less frequently, via chaperone-mediated autophagy [8, 97]. For example, a PROTAC was used to downregulate ER levels in breast cancer cells. Here, the PROTAC was composed of a methyl-ester of bestatin, which binds the cellular inhibitor of apoptosis protein-1 (IAP) and is linked to 4-hydroxy tamoxifen (4-OHT), which binds ER [98]. Under normal conditions, estrogen binding to ER leads to receptor translocation to the nucleus where it activates transcription [99]. However, the PROTAC induced cIAP1-mediated ubiquitylation and proteasomal degradation of ER in MCF7 cells, triggering necrotic cell death [98]. PROTAC technology also successfully decreased HER3 levels in breast cancer cells [100]. In the immediate future, it will be exciting to observe the effects of PROTACs in other cancer models and in animal models. Even with recent successes, the delivery and bioavailability of PROTACs remain the largest hurdles prior to clinical trials.
Targeting the Hsp70 and Hsp90 molecular chaperones in breast cancer
The Hsp70 and Hsp90 heat shock proteins are among the most abundant and important house-keeping chaperones and are overexpressed in many cancers [61, 101–103]. These chaperones bind and release protein substrates via a complex ATP-regulated cycle and play central roles in multi-protein assemblies that mediate each event during protein folding, degradation, and activation (Fig. 1). Based on the fact that cancers amass misfolded proteins, and because they are over-expressed in some breast cancers, Hsp70 and Hsp90 have been targeted in breast cancer [104–107].
Hsp90 helps fold several oncogenic proteins, such as BRAF, HER2, AKT, and CREF [108–110], and elevated Hsp90 correlates with decreased survival in breast cancer patients [111]. Therefore, inhibition or knock down of Hsp90 is a viable target for cancer therapy, especially in HER2-positive breast cancers. Normally, HER2 is degraded after ligand-induced endocytosis [112, 113] and after binding select drugs [19].
A class of compounds developed to treat HER2-positive breast cancers includes derivatives of the benzoquinoid ansamycin antibiotic geldanamycin (GA; Table 2), which bind and inactivate Hsp90 and, in turn, induce HER2 degradation [20, 114, 115]. Hsp90 interacts with HER2 at the plasma membrane [109], and GA treatment leads to rapid HER2 downregulation by mediating ubiquitiylation and degradation [19, 115, 116]. It is unclear whether Hsp90 stabilizes HER2-containing heterodimers or if Hsp90-HER2 binding favors faster recycling to the plasma membrane in breast cancer cells. However, lapatinib, a tyrosine kinase inhibitor used to treat HER2-positive breast cancers, binds inactive HER2 and inhibits Hsp90–HER2 association to the same extent as GA and another analog, 17-AAG (Table 2). These data suggest that Hsp90 may be directly involved in dimer stabilization. On the contrary, lapatinib treatment does not induce HER2 degradation after ubiquitylation, which can occur via the action of another E3 ligase, CHIP; therefore, Hsp90 prevents CHIP recruitment [117, 118]. Recently, it was reported that kinase inhibitors, such as sorafenib (Nexavarand) and pazopanib (Votrient) (Table 2), were linked to the inhibition of Hsp90 activity in HER2-positive breast cancer cells [63]. Together, these results suggest that modulating HER tyrosine kinase activity, in conjunction with Hsp90, may become a valuable, new therapeutic strategy in breast cancer.
Even though promising pre-clinical data have emerged in HER2-positive breast cancers after treatment with Hsp90 inhibitors, in clinical trials no clear efficacy was detected. In fact, Hsp90 inhibition usually induces compensatory effects, such as increased expression of other heat shock proteins (Hsp70 and Hsp27) and activation of an anti-apoptotic response [61, 119–122]. In particular, upon Hsp90 inhibition, overexpression of Hsp27 was reported to modulate the oxidative stress response in cancer cells, and higher levels of Hsp70 were also detected, which helps block apoptosis induction and reduces the efficacy of Hsp90 inhibitors [120, 121]. Moreover, in patients Hsp90 inhibitor exposure is limited due to poor drug solubility and liver toxicity [25, 123].
One cellular Hsp90 partner is HSF1, a transcription factor involved in the cytoplasmic stress response (Fig. 1) [124, 125]. In the absence of stress, Hsp90 and other chaperones form a complex with HSF1 [125, 126]. However, Hsp90 inhibitors alter the composition of the HSF1-containing complex, which releases HSF1 and stimulates HSF1-activated transcription of heat shock proteins [125, 127]. As might be expected, then, HSF1 is associated with cancer progression [128]. To date, specific inhibitors of HSF1 have not been identified, but this represents an active area of research.
Cytosolic Hsp70 as well an ER lumenal Hsp70 homolog, BiP (or GRP78; see below), also play important roles in breast cancer, as well as in many other cancers. Hsp70 upregulation has been detected in different types of cancers, including endometrial cancer, osteosarcoma, and renal carcinoma [129, 130], and higher levels of Hsp70 are associated with metastasis and resistance to chemotherapy in glioblastoma and breast, endometrial, and cervical cancers [45, 131–133]. Hsp70 overexpression correlates with TNBC metastasis in murine models and human breast cancer patients [24], but knock down of cytosolic Hsc70 (a constitutive Hsp70) or Hsp70 (the inducible Hsp70) in breast cancer lines exhibits distinct responses, probably due to the relative expression levels of the two chaperones [61, 134]. However, dual knock down of the chaperones increased Hsp90 client ubiquitylation, that is, HER2, in ovarian cancer, suggesting a role for Hsp70 inhibitors in the treatment of HER2-positive breast cancer. Moreover, Hsp70 inhibition induces caspase-3 and PARP cleavage as well as sustained apoptosis in breast cancer cells [135]. The proposed mechanism by which Hsp70 inhibition triggers cancer cell death involves lysosomal membrane permeabilization [136–138]. Notably, Hsp70 binds bis-monoacylglycerol-phosphate, a membrane-bound anionic phospholipid that predominantly localizes to the inner lysosomal membrane, which maintains lysosomal membrane stability and cell survival. In rhabdomyosarcoma, on the other hand, cancer cell death mediated by Hsp70 inhibition was linked to UPR induction [139].
The unfolded protein response in breast cancer: a protective pathway and a potential therapeutic target
As described above, the UPR is an adaptive mechanism that restores endoplasmic reticulum homeostasis [28, 30, 65]. There are three UPR branches that are coordinately activated in mammalian cells: the inositol-requiring enzyme 1 (IRE1), PKR-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) branches [28, 140] (Fig. 1). Under normal conditions, PERK, IRE1, and ATF6 are maintained in an inactive form in the endoplasmic reticulum membrane by binding to BiP (also known as glucose-regulated protein 78 (GRP78)), which also recognizes unfolded secretory proteins with exposed hydrophobic patches and helps target misfolded proteins for proteasomal degradation via the ERAD pathway [141–143]. As described later, BiP is also an important therapeutic target in cancer. During endoplasmic reticulum stress, BiP is released from PERK, IRE1, and ATF6 and binds misfolded proteins that begin to accumulate [144]. As a result, PERK phosphorylates eIF2α, inhibiting protein translation and leading to the expression of activating transcription factor 4 (ATF4) [145]. Higher levels of ATF4 in turn increase the levels of CHOP, a pro-apoptotic transcription factor (also known as DDIT3) and growth arrest and DNA damage-inducible protein 34 (GADD34). Both ATF4 and CHOP also upregulate the transcription of autophagy-related genes (ATGs) [146] and, after a prolonged response, induce cell death [147, 148]. In contrast, ATF6 is transported to the Golgi during endoplasmic reticulum stress and is cleaved to liberate a soluble, active transcription factor that induces the production of chaperones and redox related proteins, including BiP, GRP94, PDIA4, and PDIA6 [149–151]. Finally, the release of BiP from IRE1 favors receptor oligomerization and autophosphorylation. In addition to its kinase activity, IRE1 is an endonuclease that catalyzes splicing of the X-box binding protein 1 (XBP1) message. Once the message is spliced and translated, the resulting Xbp1 transcription factor activates the synthesis of chaperones, ERAD components, and inflammatory responsive genes [151–153].
UPR induction can be either protective or deleterious for cell survival, depending on the cancer. In fact, the UPR can even act as a pro-tumorigenic signal, increasing tumor cell protein folding capacity and drug resistance [154]. However, prolonged endoplasmic reticulum stress activates cell death pathways, such as mitochondrial-associated apoptosis [155, 156] and CHOP-dependent cell death (see above) [144, 157, 158]. Examples of these disparate functions include the fact that high levels of Xbp1 are present in some breast cancer tissues, and a greater amount of the spliced XBP1-message correlates with poor prognosis in TNBCs [159, 160]. It is also known that HIF1α, the hypoxia-inducing factor 1α, is hyperactive in TNBCs [161, 162]. A genome-wide map of the Xbp1 regulatory network in TNBCs suggested that Xbp1 tumorigenicity is assisted by the formation of a complex with HIF1α [162]. Moreover, an analysis of independent cohorts of patients with TNBC revealed that a spliced XBP1 signature correlated with HIF1α and hypoxia-associated signatures, underlying the importance of the IRE1/Xbp1 signaling pathway in TNBC. In addition, depletion of Xbp1 in TNBC cell lines inhibits tumor growth and relapse [162].
Another example in which the UPR can be either a pro-survival or a cell death signal is in the epithelial to mesenchymal transition (EMT). The EMT favors tumorigenesis and drug resistance in mammary gland tumors [163, 164]. In human tumor tissues, EMT gene expression correlates with PERK–eIF2α responsive genes, but not with other branches of the UPR [163].
Endocrine therapies (such as tamoxifen and fulvestrant; Table 1) remain a mainstay for the treatment of ER-positive breast cancer patients. Although there is an initial positive response of a ~ 70% reduction in tumor volume (in treated patients), acquired resistance is ultimately evident in about half of all patients [28]. Interestingly, a rapid “anticipatory” UPR that is independent of endoplasmic reticulum stress is induced upon ligand binding to ER. By contrast, long-term treatments with tamoxifen and fulvestrant cause endoplasmic reticulum stress and induce the UPR, promoting cancer survival and drug resistance in ER-positive breast cancers [165–169].
Anti-estrogen-resistant breast cancer cell lines express elevated levels of BiP and Xbp1 [170–172], suggesting that UPR induction contributes to therapy resistance. One compound, HA15, targets BiP, which is one of the most highly induced targets of the UPR and plays a central role in UPR signaling (see above; Fig. 1). HA15 is effective in a breast cancer cell line and in other tumors, such as melanomas, and kills these cells after inducting an endoplasmic reticulum stress response [173]. These and other studies indicate that modulation of BiP activity and the UPR should be further coopted as therapeutic targets.
As mentioned above, the initiation factor eIF2α is phosphorylated by PERK, which attenuates the synthesis of new proteins and reduces the concentration of unfolded proteins in the endoplasmic reticulum [145, 174]. However, eIF2α can also be phosphorylated by other kinases, such as GCN2 (general control non-depressible 2), PKR (interferon-inducible dsRNA-activated protein kinase), and HRI (heme-regulated inhibitory) [175–178]. During induction of the integrate stress response (ISR)—which is a specialized form of the UPR—GCN2-dependent eIF2α phosphorylation occurs after amino acid starvation, which also induces ATF4 [179]. In this case, ATF4 enhances the expression of autophagy-requiring genes (for example ATG5, 7, and 10) that facilitate cellular recovery after starvation. Consistent with these data, GCN2 silencing decreases cancer cell survival after amino acid starvation and attenuates tumor growth in murine models [180]. Elevated levels of ATF4 also correlate with resistance to chemotherapeutic agents, including DNA damaging agents (ionizing radiation, aphidicolin, hydroxyurea, cytarabine, etoposide, doxorubicin, and mafosfamide), celecoxib (a nonsteroidal anti-inflammatory drug), and bortezomib, in different cancers [181–186]. These data suggest that differential sensitivities to proteasome inhibitors, like bortezomib, in breast cancer and multiple myeloma could be due to selective activation/upregulation of UPR components. Together, the UPR and ISR enable cancer cells to integrate multiple stress stimuli into a common control mechanism, centering on eIF2α phosphorylation and, depending on the duration and magnitude of the stress, acting as either a pro- or anti-tumorigenic signal [30]. Consequently, studies on UPR modulation are ongoing in breast cancer cells as well as in other cancers. For example, a small molecule, BHPI, was used to demonstrate that hyperactivation of the PERK branch of the UPR blocks proliferation of ER-positive breast, ovarian, and endometrial cancer cells due to persistent inhibition of protein synthesis [168, 187, 188].
BiP upregulation is a marker for drug resistance in breast cancer
The endoplasmic reticulum folding capacity is in part limited by the chaperone reservoir. The expression of chaperones like BiP, GRP94, and calreticulin are tightly regulated [45, 47, 189, 190]. BiP was first discovered as a glucose responsive protein (hence the alternative name, GRP78) as well as via its association with immunoglobulin heavy chain [191–193]. In addition, as described above, BiP plays a crucial role during UPR induction [144, 194, 195]. BiP protein levels are normally maintained at moderate levels in adult tissues such as brain, lung, and heart, but this chaperone is strongly induced in breast, melanoma, colon, and adenocarcinoma cancer cells [196–200]. In many cases, BiP expression is associated with poor prognosis and chemotherapy resistance [201–205]. A retrospective study also examined if BiP can be used as a marker of chemotherapy resistance in tumors. As hypothesized, one-third of breast cancer patients have high levels of BiP before treatment, and the risk of recurrence was greater in chemotherapy-treated patients with moderate to high BiP levels [202].
Even though BiP is an endoplasmic reticulum resident, where it regulates the UPR and catalyzes protein folding and ERAD, recent studies demonstrate that this chaperone can also reside at the cell surface, as well as in the cytosol, mitochondria, and nucleus; these data and other results suggest that BiP performs a novel function to regulate cell proliferation, invasion, and apoptosis [206–208]. More generally, a cell surface proteomic analysis of tumor cells confirmed that several cytosolic heat shock proteins and endoplasmic reticulum chaperones, like BiP, reside on the extracellular surface of the plasma membrane, suggesting that chaperone relocation may be an adaptive response to stress induced by perturbations in proteostasis [38, 208]. Moreover, cell surface-resident BiP is more abundant in pancreatic and breast cancers [208].
The extracellular pool of BiP is associated with specific membrane glycoproteins and its amount is enriched after ER stress [209, 210]. As anticipated for a bona fide chaperone-based interaction, the substrate-binding site in BiP is required to interact with these partners [208]. However, the mechanism by which BiP translocates to the extracellular face of the plasma membrane is unclear. Nevertheless, UPR induction favors BiP secretion, possibly due to its overexpression, which in turn may overwhelm the machinery that retains proteins in the secretory pathway [211].
Several studies have demonstrated that BiP also binds BIK, a pro-apoptotic member of the Bcl2 family, impairing apoptotic cell death in ER-positive tumors [212]. The pro-apoptotic Bcl2 family members facilitate the release of cytochrome c from the mitochondrial membrane to the cytosol, which induces the apoptotic cascade [213–215]. In contrast to other pro-apoptotic Bcl2 members that reside in the mitochondria, BIK is an endoplasmic reticulum membrane protein and does not interact directly with pro-apoptotic family members, such as BAX and BAK [214, 216]. BIK plays a critical role in promoting estrogen starvation or anti-estrogen-mediated cell death in human breast cancer cells [217], and BIK knock down impairs estrogen starvation-induced cell death in MCF7 cells [212]. Recent studies suggest that BiP upregulation suppresses BIK activity by inhibiting the apoptotic response after anti-estrogen therapies in ER-positive breast cancer [212, 218]. For this reason, the development of specific BiP inhibitors represents an important goal. In fact, a natural product of the naphthoquinone family, plumbagin, was identified that initiates cell death in ER-positive breast cancer cells by upregulating BIK levels [219, 220]. Plumbagin-mediated BiP inhibition also sensitized breast cancer cells to tamoxifen-mediated cell death. In addition, BiP knock down impaired the plumbagin-mediated increase in Bik, suggesting an inhibitory role of this compound on BiP-mediated downregulation of BIK in breast cancers [220]. Overall, BiP is emerging as a novel target to predict cancer outcomes and therapeutic options [209, 221–223].
The role of autophagy in breast cancer
As outlined in the preceding sections, the autophagy pathway degrades misfolded and aggregating proteins in the cytoplasm, thereby helping to maintain cellular homeostasis after cellular stress. Autophagy also provides a mechanism to replenish cellular energy stores during starvation [38–44, 224]. The autophagy pathway is associated with a variety of processes such as tumor suppression, aging, development, and microorganism elimination [41, 45]. The dependence of cancer cells on autophagy is dictated in part by the nature and the length of the stress [225]. For example, UPR induction together with autophagy upregulation are linked to endocrine tumor therapy resistance [158, 170, 226–228]. More specifically, autophagy inhibition increases the anti-estrogen response in tumors [166, 167, 229–233], and UPR induction promotes autophagy [146, 234, 235]. In addition, UPR induction, and in particular PERK activation, increases eIF2α phosphorylation and the translation of select mRNAs, like ATF4 [236–238]. ATF4 then triggers the expression of genes involved in the ISR as well as those required for autophagy [178, 184, 239–241] (also see above).
Several studies have demonstrated that lapatinib- or trastuzumab-resistant HER2-positive cancer cell lines can be obtained by upregulating the autophagy pathway [242, 243]. Interestingly, ATG12, a protein required for autophagosome elongation, was induced in HER2-positive resistant cells and its downregulation compromised acquired resistance [243, 244]. Up-regulation of ATG5, which is covalently attached to ATG12 during autophagy, also facilitated lapatinib resistance in HER2-positive cells [245]. Therefore, inhibition of these autophagic components might sensitize HER2-positive cancer cells to established treatments.
Autophagy also plays an important role during metastases in neoplastic breast cancer [246, 247]. Chloroquine and chloroquine derivatives (which inhibit autophagolysosome formation), as well as Bcl2 inhibitors (which inhibit early steps in the autophagy pathway), are currently in clinical trials to treat metastatic/invasive breast cancer, ductal carcinomas, and HER2-positive cancers [248–250].
Recently, a correlation between autophagy and the expression of Runx2, a Runt-related transcription factor involved in cell survival during metabolic stress and breast cancer progression, was uncovered [247]. Runx2 promotes the metastatic spread of mammary tumors to the bone, which is a recurrent location for TNBC and luminal breast cancer metastasis [251]. Indeed, aberrant Runx2 expression in metastatic breast cancers altered the activity of PI3K, mTORC1, and AMPK, which function as upstream modulators of autophagy [251–255]. Misregulation of PI3K/Akt/mTOR pathways is often associated with endocrine resistance in ER-positive breast cancers [3]. As might be anticipated, then, Runx2 knock down in a breast cancer cell line reduced metastatic dissemination, suggesting a link between Runx2 and autophagy [247].
A microtubule associated deacetylase, known as HDAC6, has also been suggested to link Runx2 and autophagosome formation/lysosomal fusion [247]. HDAC6 binds polyubiquitylated, misfolded proteins and couples them to the dynein motor complex, facilitating the trafficking of aggregated proteins to autophagosomes [235, 256]. In the absence of stress, HDAC6 is associated with p97, Hsp90, and HSF1 (Fig. 1) [126]. In turn, p97 function is associated with HDAC6-dependent fusion of aggresomes—which are microtubule-associated clusters of ubiquitylated and aggregated proteins—with autophagosomes [257, 258]. Based on its protein “segragase” activity, p97 dissociates HDAC6 from polyubiquitylated proteins and regulates HDAC6 shuttling, governing both proteasome- and autophagy-dependent clearance of misfolded proteins [259–261]. In solid tumors, such as breast cancer, the use of a proteasome inhibitor has minimal effect [262, 263]. However, in other cancers, such as multiple myeloma, inhibition of both proteasome activity and HDAC6 using tubacin synergistically promote cell death through the accumulation of toxic polyubiquitylated protein aggregates [256, 264, 265]. These data suggest again that inhibition of both proteasome and autophagy-mediated degradation could be beneficial in some cancers.
Consistent with this view, treatment with the proteasome inhibitor bortezomib in a breast cancer cell line increased the levels of LC3B, a marker of autophagy, at both the protein and mRNA levels [235]. However, ATF4 knock-down limited LC3B induction after bortezomib treatment, confirming the importance of ATF4 as a mediator of the compensatory response in ER-positive cancer cells. In turn, HDAC6 is also linked to bortezomib resistance in MCF7 cells: Knock down of HDAC6 led to a synergistic effect on MCF7 cell death after bortezomib treatment [164, 266]. These data indicate that HDAC6 may be an important therapeutic target when proteasome activity is impaired. As new and improved inhibitors of the proteasome come on-line, synergistic effects of these compounds with specific HDAC6 inhibitors—and other autophagy inhibitors—should be assessed for their efficacy in breast cancer cells [267, 268].
Open questions and new therapeutic approaches in breast cancer
Based on emerging discoveries from basic research programs and from clinical trials, it is clear that protein quality control and stress response pathways are crucial for cancer cell survival. Therefore, the targeting of these pathways will not only improve patient treatment but also help answer fundamental questions related to cancer biology. To this end, novel compounds that modulate proteostasis pathways and cellular stress responses, such as the UPR, are currently being evaluated in cell and rodent models [14, 27, 29, 30, 80, 269, 270].
Even with these recent developments, numerous questions remain. For example, the sensitivity of different cancers (even within distinct cancer sub-types) to proteasome inhibitors is quite heterogeneous, and in some cases the lack of a correlation between preclinical models and patient outcomes remains mysterious [269]. Thus, it is vital that we define the factors and pathways that give rise to these phenomena. It is also important to identify novel biomarkers that predict drug efficacy, and in particular the efficacy of proteasome inhibitors [29]. One such biomarker might be the extent of autophagy induction, as discussed above, and studies with solid tumors suggest that autophagy is a source of bortezomib resistance [271, 272].
A second question relates to the dual role of UPR induction as either a pro-survival or a pro-death signal in cancer [146, 184, 241]. The recently discovered anticipatory UPR, which can be induced by ER signaling in breast cancer cells, is an intriguing candidate pathway to treat ER-positive breast cancer patients. At the same time, however, patients treated with endocrine therapies might not benefit from this potential treatment.
Nevertheless, induction of the PERK branch of the UPR is associated with increased autophagy [239–241]. These data suggest that a combination of proteasome and PERK inhibitors, which place an increased burden on the proteasome, may sensitize tumors to canonical treatments. This would also allow for lower drug dosages, which may help avoid potential side effects and decrease the likelihood of drug-resistant point mutations in HER and ER [30]. However, it is unknown if blocking UPR-induced autophagy or inhibiting upstream UPR effectors will prove more effective. Regardless, combinatorial treatments remain the best option to avoid cancer cell resistance and cancer relapse [273], especially since most therapeutic treatments for HER2-positive breast cancers (radiotherapy, chemotherapy and HER2 inhibitors) activate autophagy [249, 250].
A third question is why markers to predict chemotherapy or radiotherapy effectiveness have not been fully verified [220]. However, as outlined in this review, cell surface BiP is a prime candidate to predict chemotherapy effectiveness. On the contrary, increased expression of BiP in neuroblastoma patients correlates with longer survival [274]. These contradictory findings in human clinical samples highlight the necessity for further studies on the role of BiP in different tumorigenic contexts. It is also important to consider whether induction of intra- versus extracellular BiP hint at different outcomes. Another problem in identifying biomarkers is that metastasis formation can occur at early stages or during late stage tumor development [275, 276]. Metastases arising from late stage primary tumors are more heterogeneous, which favors chemotherapeutic resistance.
Finally, it is worth noting that drug discovery efforts and clinical trials to modulate the activity of distinct components of the “proteostatis network”, including components of the proteasome-ubiquitin machinery, Hsp90, p97, and the autophagy pathway are in progress [86, 267, 268, 270, 277–286]. When combined with standard and emerging therapies for each breast cancer cell sub-class, we envision that synergistic effects to improve clinical outcomes for patients will then become evident [8, 224, 287, 288].
Acknowledgements
The authors give a special thanks to Dr. Amit Sabnis for his comments on the manuscript.
Related work in the Brodsky lab is supported by National Institutes of Health grants GM75061 and DK79307. S.S. acknowledges the receipt of a long-term EMBO post-doctoral fellowship (ALTF 823-2016).
Authors’ contribution
Both the authors have read and agreed on the content of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kohler BA, Sherman RL, Howlader N, Jemal A, Ryerson AB, Henry KA, et al. Annual Report to the Nation on the Status of Cancer, 1975-2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. J Natl Cancer Inst. 2015;107(6):djv048. doi: 10.1093/jnci/djv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howell A, Howell SJ, Evans DG. New approaches to the endocrine prevention and treatment of breast cancer. Cancer Chemother Pharmacol. 2003;52(Suppl 1):S39–44. doi: 10.1007/s00280-003-0645-5. [DOI] [PubMed] [Google Scholar]
- 5.Cyrus K, Wehenkel M, Choi EY, Swanson H, Kim KB. Two-headed PROTAC: an effective new tool for targeted protein degradation. Chembiochem. 2010;11(11):1531–4. doi: 10.1002/cbic.201000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1. Receptor interactions. J Med Chem. 2003;46(6):883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 7.Jordan VC. The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer. Endocr Relat Cancer. 2015;22(1):R1–31. doi: 10.1530/ERC-14-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bondeson DP, Crews CM. Targeted protein degradation by small molecules. Annu Rev Pharmacol Toxicol. 2017;57:107–23. doi: 10.1146/annurev-pharmtox-010715-103507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991;51(15):3867–73. [PubMed] [Google Scholar]
- 10.Dauvois S, White R, Parker MG. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J Cell Sci. 1993;106(Pt 4):1377–88. doi: 10.1242/jcs.106.4.1377. [DOI] [PubMed] [Google Scholar]
- 11.Wittmann BM, Sherk A, McDonnell DP. Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res. 2007;67(19):9549–60. doi: 10.1158/0008-5472.CAN-07-1590. [DOI] [PubMed] [Google Scholar]
- 12.Jankowitz RC, Oesterreich S, Lee AV, Davidson NE. New strategies in metastatic hormone receptor-positive breast cancer: searching for biomarkers to tailor endocrine and other targeted therapies. Clin Cancer Res. 2017;23(5):1126–31. doi: 10.1158/1078-0432.CCR-16-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Early Breast Cancer Trialists' Collaborative G. Davies C, Godwin J, Gray R, Clarke M, Cutter D, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378(9793):771–84. doi: 10.1016/S0140-6736(11)60993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer--An overview and update. Mol Cell Endocrinol. 2015;418(Pt 3):220–34. doi: 10.1016/j.mce.2015.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Odiete O, Hill MF, Sawyer DB. Neuregulin in cardiovascular development and disease. Circ Res. 2012;111(10):1376–85. doi: 10.1161/CIRCRESAHA.112.267286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertelsen V, Stang E. The mysterious ways of ErbB2/HER2 trafficking. Membranes (Basel) 2014;4(3):424–46. doi: 10.3390/membranes4030424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilboa L, Ben-Levy R, Yarden Y, Henis YI. Roles for a cytoplasmic tyrosine and tyrosine kinase activity in the interactions of Neu receptors with coated pits. J Biol Chem. 1995;270(13):7061–7. doi: 10.1074/jbc.270.13.7061. [DOI] [PubMed] [Google Scholar]
- 18.Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19(53):6102–14. doi: 10.1038/sj.onc.1203973. [DOI] [PubMed] [Google Scholar]
- 19.Citri A, Alroy I, Lavi S, Rubin C, Xu W, Grammatikakis N, et al. Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J. 2002;21(10):2407–17. doi: 10.1093/emboj/21.10.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Citri A, Kochupurakkal BS, Yarden Y. The achilles heel of ErbB-2/HER2: regulation by the Hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle. 2004;3(1):51–60. [PubMed] [Google Scholar]
- 21.Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11(2):495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 22.Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008;68(14):5878–87. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- 23.Salami J, Crews CM. Waste disposal--an attractive strategy for cancer therapy. Science. 2017;355(6330):1163–7. doi: 10.1126/science.aam7340. [DOI] [PubMed] [Google Scholar]
- 24.Sun B, Zhang S, Zhang D, Li Y, Zhao X, Luo Y, et al. Identification of metastasis-related proteins and their clinical relevance to triple-negative human breast cancer. Clin Cancer Res. 2008;14(21):7050–9. doi: 10.1158/1078-0432.CCR-08-0520. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Chen M, Zhou J, Zhang X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy. Int J Oncol. 2014;45(1):18–30. doi: 10.3892/ijo.2014.2399. [DOI] [PubMed] [Google Scholar]
- 26.Shajahan AN, Riggins RB, Clarke R. The role of X-box binding protein-1 in tumorigenicity. Drug News Perspect. 2009;22(5):241–6. doi: 10.1358/dnp.2009.22.5.1378631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajapaksa G, Nikolos F, Bado I, Clarke R, Gustafsson JA, Thomas C. ERbeta decreases breast cancer cell survival by regulating the IRE1/XBP-1 pathway. Oncogene. 2015;34(31):4130–41. doi: 10.1038/onc.2014.343. [DOI] [PubMed] [Google Scholar]
- 28.Clarke R, Cook KL. Unfolding the role of stress response signaling in endocrine resistant breast cancers. Front Oncol. 2015;5:140. doi: 10.3389/fonc.2015.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McConkey DJ. The integrated stress response and proteotoxicity in cancer therapy. Biochem Biophys Res Commun. 2017;482(3):450–3. doi: 10.1016/j.bbrc.2016.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ojha R, Amaravadi RK. Targeting the unfolded protein response in cancer. Pharmacol Res. 2017;120:258–66. doi: 10.1016/j.phrs.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9(12):944–57. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai YC, Weissman AM. Ubiquitylation in ERAD: reversing to go forward? PLoS Biol. 2011;9(3):e1001038. doi: 10.1371/journal.pbio.1001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol. 2013;5(9):a013185. [DOI] [PMC free article] [PubMed]
- 35.Lam YA, Xu W, DeMartino GN, Cohen RE. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature. 1997;385(6618):737–40. doi: 10.1038/385737a0. [DOI] [PubMed] [Google Scholar]
- 36.Preston GM, Brodsky JL. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem J. 2017;474(4):445–69. doi: 10.1042/BCJ20160582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haq S, Ramakrishna S. Deubiquitylation of deubiquitylases. Open Biol. 2017;7(6):170016. doi: 10.1098/rsob.170016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306(5698):990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klionsky DJ. Autophagy. Curr Biol. 2005;15(8):R282–3. doi: 10.1016/j.cub.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 40.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 41.Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr. 2007;27:19–40. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- 42.Mizushima N. The exponential growth of autophagy-related research: from the humble yeast to the Nobel Prize. FEBS Lett. 2017;591(5):681–9. doi: 10.1002/1873-3468.12594. [DOI] [PubMed] [Google Scholar]
- 43.Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263(1):55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 44.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 45.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4(3):181–91. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 46.Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426(6968):891–4. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 47.Anelli T, Sitia R. Protein quality control in the early secretory pathway. EMBO J. 2008;27(2):315–27. doi: 10.1038/sj.emboj.7601974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92(2):537–76. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cavo M, Tacchetti P, Patriarca F, Petrucci MT, Pantani L, Galli M, et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet. 2010;376(9758):2075–85. doi: 10.1016/S0140-6736(10)61424-9. [DOI] [PubMed] [Google Scholar]
- 50.Dimopoulos MA, Goldschmidt H, Niesvizky R, Joshua D, Chng WJ, Oriol A, et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(10):1327–37. doi: 10.1016/S1470-2045(17)30578-8. [DOI] [PubMed] [Google Scholar]
- 51.Hideshima T, Chauhan D, Ishitsuka K, Yasui H, Raje N, Kumar S, et al. Molecular characterization of PS-341 (bortezomib) resistance: implications for overcoming resistance using lysophosphatidic acid acyltransferase (LPAAT)-beta inhibitors. Oncogene. 2005;24(19):3121–9. doi: 10.1038/sj.onc.1208522. [DOI] [PubMed] [Google Scholar]
- 52.Stessman HA, Baughn LB, Sarver A, Xia T, Deshpande R, Mansoor A, et al. Profiling bortezomib resistance identifies secondary therapies in a mouse myeloma model. Mol Cancer Ther. 2013;12(6):1140–50. doi: 10.1158/1535-7163.MCT-12-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stessman HA, Mansoor A, Zhan F, Linden MA, Van Ness B, Baughn LB. Bortezomib resistance can be reversed by induced expression of plasma cell maturation markers in a mouse in vitro model of multiple myeloma. PLoS One. 2013;8(10):e77608. doi: 10.1371/journal.pone.0077608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016;26(4):484–98. doi: 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raedler LA. Kyprolis (carfilzomib) received new indications as combination therapy for use in relapsed and/or refractory multiple myeloma. Am Health Drug Benefits. 2016;9(Spec Feature):93–6. [PMC free article] [PubMed] [Google Scholar]
- 56.Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell. 2014;25(5):563–73. doi: 10.1016/j.ccr.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 57.Kim H, Bhattacharya A, Qi L. Endoplasmic reticulum quality control in cancer: friend or foe. Semin Cancer Biol. 2015;33:25–33. doi: 10.1016/j.semcancer.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams BR, Amon A. Aneuploidy: cancer's fatal flaw? Cancer Res. 2009;69(13):5289–91. doi: 10.1158/0008-5472.CAN-09-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8(1):7–12. doi: 10.1016/j.ccr.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 60.Deshais MA, Fisher AB, Hausman NL, Kahng S. Further investigation of a rapid restraint analysis. J Appl Behav Anal. 2015;48(4):845–59. doi: 10.1002/jaba.251. [DOI] [PubMed] [Google Scholar]
- 61.Powers MV, Clarke PA, Workman P. Death by chaperone: HSP90, HSP70 or both? Cell Cycle. 2009;8(4):518–26. doi: 10.4161/cc.8.4.7583. [DOI] [PubMed] [Google Scholar]
- 62.Lanneau D, Wettstein G, Bonniaud P, Garrido C. Heat shock proteins: cell protection through protein triage. ScientificWorldJournal. 2010;10:1543–52. doi: 10.1100/tsw.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Booth L, Roberts JL, Ecroyd H, Tritsch SR, Bavari S, Reid SP, et al. AR-12 inhibits multiple chaperones concomitant with stimulating autophagosome formation collectively preventing virus replication. J Cell Physiol. 2016;231(10):2286–302. doi: 10.1002/jcp.25431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anelli T, Sannino S, Sitia R. Proteostasis and "redoxtasis" in the secretory pathway: Tales of tails from ERp44 and immunoglobulins. Free Radic Biol Med. 2015;83:323–30. doi: 10.1016/j.freeradbiomed.2015.02.020. [DOI] [PubMed] [Google Scholar]
- 65.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 66.Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334(6059):1086–90. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raasi S, Wolf DH. Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin Cell Dev Biol. 2007;18(6):780–91. doi: 10.1016/j.semcdb.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 68.Kostova Z, Wolf DH. For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003;22(10):2309–17. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McCaffrey K, Braakman I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016;60(2):227–35. doi: 10.1042/EBC20160003. [DOI] [PubMed] [Google Scholar]
- 70.Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414(6864):652–6. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 71.Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem. 2004;279(44):45676–84. doi: 10.1074/jbc.M409034200. [DOI] [PubMed] [Google Scholar]
- 72.Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A. 2005;102(40):14132–8. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, et al. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2011;14(1):93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187(6):875–88. doi: 10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ju JS, Weihl CC. p97/VCP at the intersection of the autophagy and the ubiquitin proteasome system. Autophagy. 2010;6(2):283–5. doi: 10.4161/auto.6.2.11063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heo JM, Livnat-Levanon N, Taylor EB, Jones KT, Dephoure N, Ring J, et al. A stress-responsive system for mitochondrial protein degradation. Mol Cell. 2010;40(3):465–80. doi: 10.1016/j.molcel.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Y, Ballar P, Zhong Y, Zhang X, Liu C, Zhang YJ, et al. SVIP induces localization of p97/VCP to the plasma and lysosomal membranes and regulates autophagy. PLoS One. 2011;6(8):e24478. doi: 10.1371/journal.pone.0024478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chapman E, Fry AN, Kang M. The complexities of p97 function in health and disease. Mol Biosyst. 2011;7(3):700–10. doi: 10.1039/c0mb00176g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Verma R, Oania RS, Kolawa NJ, Deshaies RJ. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. Elife. 2013;2:e00308. doi: 10.7554/eLife.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vekaria PH, Home T, Weir S, Schoenen FJ, Rao R. Targeting p97 to disrupt protein homeostasis in cancer. Front Oncol. 2016;6:181. doi: 10.3389/fonc.2016.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singh N, Joshi R, Komurov K. HER2-mTOR signaling-driven breast cancer cells require ER-associated degradation to survive. Sci Signal. 2015;8(378):ra52. doi: 10.1126/scisignal.aaa6922. [DOI] [PubMed] [Google Scholar]
- 82.Fiebiger E, Hirsch C, Vyas JM, Gordon E, Ploegh HL, Tortorella D. Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol Biol Cell. 2004;15(4):1635–46. doi: 10.1091/mbc.E03-07-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Q, Shinkre BA, Lee JG, Weniger MA, Liu Y, Chen W, et al. The ERAD inhibitor Eeyarestatin I is a bifunctional compound with a membrane-binding domain and a p97/VCP inhibitory group. PLoS One. 2010;5(11):e15479. doi: 10.1371/journal.pone.0015479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cross BC, McKibbin C, Callan AC, Roboti P, Piacenti M, Rabu C, et al. Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J Cell Sci. 2009;122(Pt 23):4393–400. doi: 10.1242/jcs.054494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang Q, Li L, Ye Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem. 2008;283(12):7445–54. doi: 10.1074/jbc.M708347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, et al. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell. 2015;28(5):653–65. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer. 2008;8(11):835–50. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- 88.Fry WH, Simion C, Sweeney C, Carraway KL., 3rd Quantity control of the ErbB3 receptor tyrosine kinase at the endoplasmic reticulum. Mol Cell Biol. 2011;31(14):3009–18. doi: 10.1128/MCB.05105-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qiu XB, Goldberg AL. Nrdp1/FLRF is a ubiquitin ligase promoting ubiquitination and degradation of the epidermal growth factor receptor family member, ErbB3. Proc Natl Acad Sci U S A. 2002;99(23):14843–8. doi: 10.1073/pnas.232580999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cao Z, Wu X, Yen L, Sweeney C, Carraway KL., 3rd Neuregulin-induced ErbB3 downregulation is mediated by a protein stability cascade involving the E3 ubiquitin ligase Nrdp1. Mol Cell Biol. 2007;27(6):2180–8. doi: 10.1128/MCB.01245-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ingalla EQ, Miller JK, Wald JH, Workman HC, Kaur RP, Yen L, et al. Post-transcriptional mechanisms contribute to the suppression of the ErbB3 negative regulator protein Nrdp1 in mammary tumors. J Biol Chem. 2010;285(37):28691–7. doi: 10.1074/jbc.M110.127977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100(15):8933–8. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yen L, Cao Z, Wu X, Ingalla ER, Baron C, Young LJ, et al. Loss of Nrdp1 enhances ErbB2/ErbB3-dependent breast tumor cell growth. Cancer Res. 2006;66(23):11279–86. doi: 10.1158/0008-5472.CAN-06-2319. [DOI] [PubMed] [Google Scholar]
- 94.Stern DF. ERBB3/HER3 and ERBB2/HER2 duet in mammary development and breast cancer. J Mammary Gland Biol Neoplasia. 2008;13(2):215–23. doi: 10.1007/s10911-008-9083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carraway KL., 3rd E3 ubiquitin ligases in ErbB receptor quantity control. Semin Cell Dev Biol. 2010;21(9):936–43. doi: 10.1016/j.semcdb.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hatakeyama J, Wald JH, Rafidi H, Cuevas A, Sweeney C, Carraway KL., 3rd The ER structural protein Rtn4A stabilizes and enhances signaling through the receptor tyrosine kinase ErbB3. Sci Signal. 2016;9(434):ra65. doi: 10.1126/scisignal.aaf1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174:138–44. doi: 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- 98.Okuhira K, Demizu Y, Hattori T, Ohoka N, Shibata N, Kurihara M, et al. Molecular design, synthesis, and evaluation of SNIPER(ER) that induces proteasomal degradation of ERalpha. Methods Mol Biol. 2016;1366:549–60. doi: 10.1007/978-1-4939-3127-9_42. [DOI] [PubMed] [Google Scholar]
- 99.Valley CC, Metivier R, Solodin NM, Fowler AM, Mashek MT, Hill L, et al. Differential regulation of estrogen-inducible proteolysis and transcription by the estrogen receptor alpha N terminus. Mol Cell Biol. 2005;25(13):5417–28. doi: 10.1128/MCB.25.13.5417-5428.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xie T, Lim SM, Westover KD, Dodge ME, Ercan D, Ficarro SB, et al. Pharmacological targeting of the pseudokinase Her3. Nat Chem Biol. 2014;10(12):1006–12. doi: 10.1038/nchembio.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Donnelly N, Storchova Z. Aneuploidy and proteotoxic stress in cancer. Mol Cell Oncol. 2015;2(2):e976491. doi: 10.4161/23723556.2014.976491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Calderwood SK, Gong J. Heat shock proteins promote cancer: it's a protection racket. Trends Biochem Sci. 2016;41(4):311–23. doi: 10.1016/j.tibs.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Workman P, Clarke PA, Al-Lazikani B. Blocking the survival of the nastiest by HSP90 inhibition. Oncotarget. 2016;7(4):3658–61. doi: 10.18632/oncotarget.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Powers MV, Valenti M, Miranda S, Maloney A, Eccles SA, Thomas G, et al. Mode of cell death induced by the HSP90 inhibitor 17-AAG (tanespimycin) is dependent on the expression of pro-apoptotic BAX. Oncotarget. 2013;4(11):1963–75. doi: 10.18632/oncotarget.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013;9(11):677–84. doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18(6):345–60. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 108.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277(42):39858–66. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 109.Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, Neckers L. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol. 2005;12(2):120–6. doi: 10.1038/nsmb885. [DOI] [PubMed] [Google Scholar]
- 110.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5(10):761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 111.Pick E, Kluger Y, Giltnane JM, Moeder C, Camp RL, Rimm DL, et al. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007;67(7):2932–7. doi: 10.1158/0008-5472.CAN-06-4511. [DOI] [PubMed] [Google Scholar]
- 112.Brennan PJ, Kumagai T, Berezov A, Murali R, Greene MI. HER2/neu: mechanisms of dimerization/oligomerization. Oncogene. 2000;19(53):6093–101. doi: 10.1038/sj.onc.1203967. [DOI] [PubMed] [Google Scholar]
- 113.Colomer R, Montero S, Lluch A, Ojeda B, Barnadas A, Casado A, et al. Circulating HER2 extracellular domain and resistance to chemotherapy in advanced breast cancer. Clin Cancer Res. 2000;6(6):2356–62. [PubMed] [Google Scholar]
- 114.Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem. 1996;271(37):22796–801. doi: 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- 115.Marx C, Held JM, Gibson BW, Benz CC. ErbB2 trafficking and degradation associated with K48 and K63 polyubiquitination. Cancer Res. 2010;70(9):3709–17. doi: 10.1158/0008-5472.CAN-09-3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A. 2002;99(20):12847–52. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim S, Zhang S, Choi KH, Reister R, Do C, Baykiz AF, et al. An E3 ubiquitin ligase, Really Interesting New Gene (RING) Finger 41, is a candidate gene for anxiety-like behavior and beta-carboline-induced seizures. Biol Psychiatry. 2009;65(5):425–31. doi: 10.1016/j.biopsych.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene. 2009;28(6):803–14. doi: 10.1038/onc.2008.432. [DOI] [PubMed] [Google Scholar]
- 119.Voellmy R, Boellmann F. Chaperone regulation of the heat shock protein response. Adv Exp Med Biol. 2007;594:89–99. doi: 10.1007/978-0-387-39975-1_9. [DOI] [PubMed] [Google Scholar]
- 120.McCollum AK, Teneyck CJ, Sauer BM, Toft DO, Erlichman C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 2006;66(22):10967–75. doi: 10.1158/0008-5472.CAN-06-1629. [DOI] [PubMed] [Google Scholar]
- 121.Guo F, Rocha K, Bali P, Pranpat M, Fiskus W, Boyapalle S, et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005;65(22):10536–44. doi: 10.1158/0008-5472.CAN-05-1799. [DOI] [PubMed] [Google Scholar]
- 122.Sidera K, Patsavoudi E. HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat Anticancer Drug Discov. 2014;9(1):1–20. [PubMed] [Google Scholar]
- 123.Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S. Heat shock proteins and cancer. Trends Pharmacol Sci. 2017;38(3):226–56. doi: 10.1016/j.tips.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 124.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94(4):471–80. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 125.Ali A, Bharadwaj S, O'Carroll R, Ovsenek N. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol. 1998;18(9):4949–60. doi: 10.1128/mcb.18.9.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Home T, Jensen RA, Rao R. Heat shock factor 1 in protein homeostasis and oncogenic signal integration. Cancer Res. 2015;75(6):907–12. doi: 10.1158/0008-5472.CAN-14-2905. [DOI] [PubMed] [Google Scholar]
- 127.Bagatell R, Paine-Murrieta GD, Taylor CW, Pulcini EJ, Akinaga S, Benjamin IJ, et al. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res. 2000;6(8):3312–8. [PubMed] [Google Scholar]
- 128.Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150(3):549–62. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Santarosa M, Favaro D, Quaia M, Galligioni E. Expression of heat shock protein 72 in renal cell carcinoma: possible role and prognostic implications in cancer patients. Eur J Cancer. 1997;33(6):873–7. doi: 10.1016/s0959-8049(97)00002-6. [DOI] [PubMed] [Google Scholar]
- 130.Uozaki H, Ishida T, Kakiuchi C, Horiuchi H, Gotoh T, Iijima T, et al. Expression of heat shock proteins in osteosarcoma and its relationship to prognosis. Pathol Res Pract. 2000;196(10):665–73. doi: 10.1016/S0344-0338(00)80118-1. [DOI] [PubMed] [Google Scholar]
- 131.Nanbu K, Konishi I, Mandai M, Kuroda H, Hamid AA, Komatsu T, et al. Prognostic significance of heat shock proteins HSP70 and HSP90 in endometrial carcinomas. Cancer Detect Prev. 1998;22(6):549–55. doi: 10.1046/j.1525-1500.1998.00069.x. [DOI] [PubMed] [Google Scholar]
- 132.Vargas-Roig LM, Gago FE, Tello O, Aznar JC, Ciocca DR. Heat shock protein expression and drug resistance in breast cancer patients treated with induction chemotherapy. Int J Cancer. 1998;79(5):468–75. doi: 10.1002/(sici)1097-0215(19981023)79:5<468::aid-ijc4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 133.Brondani Da Rocha A, Regner A, Grivicich I, Pretto Schunemann D, Diel C, Kovaleski G, et al. Radioresistance is associated to increased Hsp70 content in human glioblastoma cell lines. Int J Oncol. 2004;25(3):777–85. [PubMed] [Google Scholar]
- 134.Seo SJ, Kim HT, Cho G, Rho HM, Jung G. Sp1 and C/EBP-related factor regulate the transcription of human Cu/Zn SOD gene. Gene. 1996;178(1-2):177–85. doi: 10.1016/0378-1119(96)00383-6. [DOI] [PubMed] [Google Scholar]
- 135.Rodina A, Vilenchik M, Moulick K, Aguirre J, Kim J, Chiang A, et al. Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat Chem Biol. 2007;3(8):498–507. doi: 10.1038/nchembio.2007.10. [DOI] [PubMed] [Google Scholar]
- 136.Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200(4):425–35. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nylandsted J, Jaattela M, Hoffmann EK, Pedersen SF. Heat shock protein 70 inhibits shrinkage-induced programmed cell death via mechanisms independent of effects on cell volume-regulatory membrane transport proteins. Pflugers Arch. 2004;449(2):175–85. doi: 10.1007/s00424-004-1332-z. [DOI] [PubMed] [Google Scholar]
- 138.Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463(7280):549–53. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- 139.Sabnis AJ, Guerriero CJ, Olivas V, Sayana A, Shue J, Flanagan J, et al. Combined chemical-genetic approach identifies cytosolic HSP70 dependence in rhabdomyosarcoma. Proc Natl Acad Sci U S A. 2016;113(32):9015–20. doi: 10.1073/pnas.1603883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 141.Fourie AM, Sambrook JF, Gething MJ. Common and divergent peptide binding specificities of hsp70 molecular chaperones. J Biol Chem. 1994;269(48):30470–8. [PubMed] [Google Scholar]
- 142.Skowronek MH, Hendershot LM, Haas IG. The variable domain of nonassembled Ig light chains determines both their half-life and binding to the chaperone BiP. Proc Natl Acad Sci U S A. 1998;95(4):1574–8. doi: 10.1073/pnas.95.4.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rothman JE, Schekman R. Molecular mechanism of protein folding in the cell. Cell. 2011;146(6):851–4. doi: 10.1016/j.cell.2011.08.041. [DOI] [PubMed] [Google Scholar]
- 144.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–32. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 145.Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332(6025):91–4. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- 146.B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41(16):7683–99. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12(7):982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22(53):8608–18. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- 149.Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. 2004;136(3):343–50. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- 150.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13(3):365–76. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 151.Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013;3(4):1279–92. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Urano F, Bertolotti A, Ron D. IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci. 2000;113(Pt 21):3697–702. doi: 10.1242/jcs.113.21.3697. [DOI] [PubMed] [Google Scholar]
- 153.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23(21):7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hazari YM, Bashir A, Haq EU, Fazili KM. Emerging tale of UPR and cancer: an essentiality for malignancy. Tumour Biol. 2016;37(11):14381–90. doi: 10.1007/s13277-016-5343-0. [DOI] [PubMed] [Google Scholar]
- 155.Shu CW, Huang CM. HSP70s: from tumor transformation to cancer therapy. Clin Med Oncol. 2008;2:335–45. doi: 10.4137/cmo.s475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bonora M, Wieckowsk MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, et al. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene. 2015;34(12):1608. doi: 10.1038/onc.2014.462. [DOI] [PubMed] [Google Scholar]
- 157.Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4(11):e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Clarke R, Cook KL, Hu R, Facey CO, Tavassoly I, Schwartz JL, et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72(6):1321–31. doi: 10.1158/0008-5472.CAN-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rhodes DR, Chinnaiyan AM. Bioinformatics strategies for translating genome-wide expression analyses into clinically useful cancer markers. Ann N Y Acad Sci. 2004;1020:32–40. doi: 10.1196/annals.1310.005. [DOI] [PubMed] [Google Scholar]
- 160.Kharabi Masouleh B, Geng H, Hurtz C, Chan LN, Logan AC, Chang MS, et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2014;111(21):E2219–28. doi: 10.1073/pnas.1400958111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Montagner M, Enzo E, Forcato M, Zanconato F, Parenti A, Rampazzo E, et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature. 2012;487(7407):380–4. doi: 10.1038/nature11207. [DOI] [PubMed] [Google Scholar]
- 162.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508(7494):103–7. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014;4(6):702–15. doi: 10.1158/2159-8290.CD-13-0945. [DOI] [PubMed] [Google Scholar]
- 164.Feng YX, Sokol ES, Gupta PB. The endoplasmic reticulum may be an Achilles' heel of cancer cells that have undergone an epithelial-to-mesenchymal transition. Mol Cell Oncol. 2014;1(2):e961822. doi: 10.4161/23723548.2014.961822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ishii Y, Papa L, Bahadur U, Yue Z, Aguirre-Ghiso J, Shioda T, et al. Bortezomib enhances the efficacy of fulvestrant by amplifying the aggregation of the estrogen receptor, which leads to a proapoptotic unfolded protein response. Clin Cancer Res. 2011;17(8):2292–300. doi: 10.1158/1078-0432.CCR-10-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cook KL, Shajahan AN, Warri A, Jin L, Hilakivi-Clarke LA, Clarke R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012;72(13):3337–49. doi: 10.1158/0008-5472.CAN-12-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cook KL, Warri A, Soto-Pantoja DR, Clarke PA, Cruz MI, Zwart A, et al. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin Cancer Res. 2014;20(12):3222–32. doi: 10.1158/1078-0432.CCR-13-3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Andruska N, Zheng X, Yang X, Helferich WG, Shapiro DJ. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer. Oncogene. 2015;34(29):3760–9. doi: 10.1038/onc.2014.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yu L, Andruska N, Zheng X, Shapiro DJ. Anticipatory activation of the unfolded protein response by epidermal growth factor is required for immediate early gene expression and cell proliferation. Mol Cell Endocrinol. 2016;422:31–41. doi: 10.1016/j.mce.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gomez BP, Riggins RB, Shajahan AN, Klimach U, Wang A, Crawford AC, et al. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J. 2007;21(14):4013–27. doi: 10.1096/fj.06-7990com. [DOI] [PubMed] [Google Scholar]
- 171.Davies MP, Barraclough DL, Stewart C, Joyce KA, Eccles RM, Barraclough R, et al. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int J Cancer. 2008;123(1):85–8. doi: 10.1002/ijc.23479. [DOI] [PubMed] [Google Scholar]
- 172.Zhang X, Cook KL, Warri A, Cruz IM, Rosim M, Riskin J, et al. Lifetime genistein intake increases the response of mammary tumors to tamoxifen in rats. Clin Cancer Res. 2017;23(3):814–24. doi: 10.1158/1078-0432.CCR-16-1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cerezo M, Lehraiki A, Millet A, Rouaud F, Plaisant M, Jaune E, et al. Compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell. 2016;30(1):183. doi: 10.1016/j.ccell.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 174.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066–77. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109(7):2693–9. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chen JJ, Throop MS, Gehrke L, Kuo I, Pal JK, Brodsky M, et al. Cloning of the cDNA of the heme-regulated eukaryotic initiation factor 2 alpha (eIF-2 alpha) kinase of rabbit reticulocytes: homology to yeast GCN2 protein kinase and human double-stranded-RNA-dependent eIF-2 alpha kinase. Proc Natl Acad Sci U S A. 1991;88(17):7729–33. doi: 10.1073/pnas.88.17.7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011;22(22):4390–405. doi: 10.1091/mbc.E11-06-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rzymski T, Milani M, Singleton DC, Harris AL. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle. 2009;8(23):3838–47. doi: 10.4161/cc.8.23.10086. [DOI] [PubMed] [Google Scholar]
- 179.Sood R, Porter AC, Olsen DA, Cavener DR, Wek RC. A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2alpha. Genetics. 2000;154(2):787–801. doi: 10.1093/genetics/154.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell. 2005;16(12):5493–501. doi: 10.1091/mbc.E05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Levenson VV, Davidovich IA, Roninson IB. Pleiotropic resistance to DNA-interactive drugs is associated with increased expression of genes involved in DNA replication, repair, and stress response. Cancer Res. 2000;60(18):5027–30. [PubMed] [Google Scholar]
- 182.Tsutsumi S, Namba T, Tanaka KI, Arai Y, Ishihara T, Aburaya M, et al. Celecoxib upregulates endoplasmic reticulum chaperones that inhibit celecoxib-induced apoptosis in human gastric cells. Oncogene. 2006;25(7):1018–29. doi: 10.1038/sj.onc.1209139. [DOI] [PubMed] [Google Scholar]
- 183.Fung H, Liu P, Demple B. ATF4-dependent oxidative induction of the DNA repair enzyme Ape1 counteracts arsenite cytotoxicity and suppresses arsenite-mediated mutagenesis. Mol Cell Biol. 2007;27(24):8834–47. doi: 10.1128/MCB.00974-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, et al. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29(31):4424–35. doi: 10.1038/onc.2010.191. [DOI] [PubMed] [Google Scholar]
- 185.Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29(27):3881–95. doi: 10.1038/onc.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kim JE, Lee JI, Jin DH, Lee WJ, Park GB, Kim S, et al. Sequential treatment of HPV E6 and E7-expressing TC-1 cells with bortezomib and celecoxib promotes apoptosis through p-p38 MAPK-mediated downregulation of cyclin D1 and CDK2. Oncol Rep. 2014;31(5):2429–37. doi: 10.3892/or.2014.3082. [DOI] [PubMed] [Google Scholar]
- 187.Andruska ND, Zheng X, Yang X, Mao C, Cherian MM, Mahapatra L, et al. Estrogen receptor alpha inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc Natl Acad Sci U S A. 2015;112(15):4737–42. doi: 10.1073/pnas.1403685112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shapiro DJ, Livezey M, Yu L, Zheng X, Andruska N. Anticipatory UPR activation: a protective pathway and target in cancer. Trends Endocrinol Metab. 2016;27(10):731–41. doi: 10.1016/j.tem.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 190.Benham AM. Protein secretion and the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2012;4(8):a012872. doi: 10.1101/cshperspect.a012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bole DG, Hendershot LM, Kearney JF. Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J Cell Biol. 1986;102(5):1558–66. doi: 10.1083/jcb.102.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26(8):504–10. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 193.van Anken E, Pena F, Hafkemeijer N, Christis C, Romijn EP, Grauschopf U, et al. Efficient IgM assembly and secretion require the plasma cell induced endoplasmic reticulum protein pERp1. Proc Natl Acad Sci U S A. 2009;106(40):17019–24. doi: 10.1073/pnas.0903036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun. 2000;279(2):445–50. doi: 10.1006/bbrc.2000.3987. [DOI] [PubMed] [Google Scholar]
- 195.van Anken E, Braakman I. Endoplasmic reticulum stress and the making of a professional secretory cell. Crit Rev Biochem Mol Biol. 2005;40(5):269–83. doi: 10.1080/10409230500315352. [DOI] [PubMed] [Google Scholar]
- 196.Dent P, Yacoub A, Grant S, Curiel DT, Fisher PB. MDA-7/IL-24 regulates proliferation, invasion and tumor cell radiosensitivity: a new cancer therapy? J Cell Biochem. 2005;95(4):712–9. doi: 10.1002/jcb.20502. [DOI] [PubMed] [Google Scholar]
- 197.Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67(8):3496–9. doi: 10.1158/0008-5472.CAN-07-0325. [DOI] [PubMed] [Google Scholar]
- 198.Ni M, Zhou H, Wey S, Baumeister P, Lee AS. Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS One. 2009;4(8):e6868. doi: 10.1371/journal.pone.0006868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kaira K, Toyoda M, Shimizu A, Imai H, Sakakura K, Nikkuni O, et al. Decreasing expression of glucose-regulated protein GRP78/BiP as a significant prognostic predictor in patients with advanced laryngeal squamous cell carcinoma. Head Neck. 2016;38(10):1539–44. doi: 10.1002/hed.24471. [DOI] [PubMed] [Google Scholar]
- 200.Kaira K, Toyoda M, Shimizu A, Mori K, Shino M, Sakakura K, et al. Expression of ER stress markers (GRP78/BiP and PERK) in patients with tongue cancer. Neoplasma. 2016;63(4):588–94. doi: 10.4149/neo_2016_412. [DOI] [PubMed] [Google Scholar]
- 201.Davidson DJ, Haskell C, Majest S, Kherzai A, Egan DA, Walter KA, et al. Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res. 2005;65(11):4663–72. doi: 10.1158/0008-5472.CAN-04-3426. [DOI] [PubMed] [Google Scholar]
- 202.Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006;66(16):7849–53. doi: 10.1158/0008-5472.CAN-06-1660. [DOI] [PubMed] [Google Scholar]
- 203.Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66(3):1702–11. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ermakova SP, Kang BS, Choi BY, Choi HS, Schuster TF, Ma WY, et al. (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006;66(18):9260–9. doi: 10.1158/0008-5472.CAN-06-1586. [DOI] [PubMed] [Google Scholar]
- 205.Schwarze S, Rangnekar VM. Targeting plasma membrane GRP78 for cancer growth inhibition. Cancer Biol Ther. 2010;9(2):153–5. doi: 10.4161/cbt.9.2.10760. [DOI] [PubMed] [Google Scholar]
- 206.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32(7):805–18. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434(2):181–8. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tsai YL, Zhang Y, Tseng CC, Stanciauskas R, Pinaud F, Lee AS. Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J Biol Chem. 2015;290(13):8049–64. doi: 10.1074/jbc.M114.618736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell. 2004;6(3):275–84. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 210.Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009;11(9):2299–306. doi: 10.1089/ARS.2009.2568. [DOI] [PubMed] [Google Scholar]
- 211.Barlowe CK, Miller EA. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics. 2013;193(2):383–410. doi: 10.1534/genetics.112.142810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67(8):3734–40. doi: 10.1158/0008-5472.CAN-06-4594. [DOI] [PubMed] [Google Scholar]
- 213.Kluck RM, Esposti MD, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, et al. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol. 1999;147(4):809–22. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8(3):705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 215.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Germain M, Mathai JP, Shore GC. BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J Biol Chem. 2002;277(20):18053–60. doi: 10.1074/jbc.M201235200. [DOI] [PubMed] [Google Scholar]
- 217.Hur J, Chesnes J, Coser KR, Lee RS, Geck P, Isselbacher KJ, et al. The Bik BH3-only protein is induced in estrogen-starved and antiestrogen-exposed breast cancer cells and provokes apoptosis. Proc Natl Acad Sci U S A. 2004;101(8):2351–6. doi: 10.1073/pnas.0307337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhou H, Zhang Y, Fu Y, Chan L, Lee AS. Novel mechanism of anti-apoptotic function of 78-kDa glucose-regulated protein (GRP78): endocrine resistance factor in breast cancer, through release of B-cell lymphoma 2 (BCL-2) from BCL-2-interacting killer (BIK) J Biol Chem. 2011;286(29):25687–96. doi: 10.1074/jbc.M110.212944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ahmad A, Banerjee S, Wang Z, Kong D, Sarkar FH. Plumbagin-induced apoptosis of human breast cancer cells is mediated by inactivation of NF-kappaB and Bcl-2. J Cell Biochem. 2008;105(6):1461–71. doi: 10.1002/jcb.21966. [DOI] [PubMed] [Google Scholar]
- 220.Kawiak A, Domachowska A, Jaworska A, Lojkowska E. Plumbagin sensitizes breast cancer cells to tamoxifen-induced cell death through GRP78 inhibition and Bik upregulation. Sci Rep. 2017;7:43781. doi: 10.1038/srep43781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Miao YR, Eckhardt BL, Cao Y, Pasqualini R, Argani P, Arap W, et al. Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin Cancer Res. 2013;19(8):2107–16. doi: 10.1158/1078-0432.CCR-12-2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Liu R, Li X, Gao W, Zhou Y, Wey S, Mitra SK, et al. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clin Cancer Res. 2013;19(24):6802–11. doi: 10.1158/1078-0432.CCR-13-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zhang Y, Zhao Q, Jiang Y, Yuan Z, Yang L. ATP-tumor chemosensitivity assay directed chemotherapy in patients with cervical cancer. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2013;38(12):1223–7. doi: 10.3969/j.issn.1672-7347.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 224.Zambrano J, Yeh ES. Autophagy and apoptotic crosstalk: mechanism of therapeutic resistance in HER2-positive breast cancer. Breast Cancer (Auckl) 2016;10:13–23. doi: 10.4137/BCBCR.S32791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Maycotte P, Thorburn A. Targeting autophagy in breast cancer. World J Clin Oncol. 2014;5(3):224–40. doi: 10.5306/wjco.v5.i3.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Clarke R, Shajahan AN, Wang Y, Tyson JJ, Riggins RB, Weiner LM, et al. Endoplasmic reticulum stress, the unfolded protein response, and gene network modeling in antiestrogen resistant breast cancer. Horm Mol Biol Clin Investig. 2011;5(1):35–44. doi: 10.1515/hmbci.2010.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gu Z, Lee RY, Skaar TC, Bouker KB, Welch JN, Lu J, et al. Association of interferon regulatory factor-1, nucleophosmin, nuclear factor-kappaB, and cyclic AMP response element binding with acquired resistance to Faslodex (ICI 182,780) Cancer Res. 2002;62(12):3428–37. [PubMed] [Google Scholar]
- 228.Parmar JH, Cook KL, Shajahan-Haq AN, Clarke PA, Tavassoly I, Clarke R, et al. Modelling the effect of GRP78 on anti-oestrogen sensitivity and resistance in breast cancer. Interface Focus. 2013;3(4):20130012. doi: 10.1098/rsfs.2013.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Clarke R, Shajahan AN, Riggins RB, Cho Y, Crawford A, Xuan J, et al. Gene network signaling in hormone responsiveness modifies apoptosis and autophagy in breast cancer cells. J Steroid Biochem Mol Biol. 2009;114(1-2):8–20. doi: 10.1016/j.jsbmb.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Crawford AC, Riggins RB, Shajahan AN, Zwart A, Clarke R. Co-inhibition of BCL-W and BCL2 restores antiestrogen sensitivity through BECN1 and promotes an autophagy-associated necrosis. PLoS One. 2010;5(1):e8604. doi: 10.1371/journal.pone.0008604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol Cancer Ther. 2008;7(9):2977–87. doi: 10.1158/1535-7163.MCT-08-0447. [DOI] [PubMed] [Google Scholar]
- 232.Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy. 2009;5(3):400–3. doi: 10.4161/auto.5.3.7784. [DOI] [PubMed] [Google Scholar]
- 233.Schwartz-Roberts JL, Shajahan AN, Cook KL, Warri A, Abu-Asab M, Clarke R. GX15-070 (obatoclax) induces apoptosis and inhibits cathepsin D- and L-mediated autophagosomal lysis in antiestrogen-resistant breast cancer cells. Mol Cancer Ther. 2013;12(4):448–59. doi: 10.1158/1535-7163.MCT-12-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Milani M, Harris AL. Targeting tumour hypoxia in breast cancer. Eur J Cancer. 2008;44(18):2766–73. doi: 10.1016/j.ejca.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 236.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 237.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318(5):1351–65. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 238.Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. 2007;9(12):2357–71. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- 239.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11(3):619–33. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 240.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167(1):27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120(1):127–41. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Chen S, Chen CM, Yu KD, Yang WT, Shao ZM. A prognostic model to predict outcome of patients failing to achieve pathological complete response after anthracycline-containing neoadjuvant chemotherapy for breast cancer. J Surg Oncol. 2012;105(6):577–85. doi: 10.1002/jso.22140. [DOI] [PubMed] [Google Scholar]
- 243.Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Corominas-Faja B, Cuyas E, Lopez-Bonet E, et al. The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci Rep. 2013;3:2469. doi: 10.1038/srep02469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Corominas-Faja B, Urruticoechea A, Martin-Castillo B, et al. Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget. 2012;3(12):1600–14. doi: 10.18632/oncotarget.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Yoon JH, Her S, Kim M, Jang IS, Park J. The expression of damage-regulated autophagy modulator 2 (DRAM2) contributes to autophagy induction. Mol Biol Rep. 2012;39(2):1087–93. doi: 10.1007/s11033-011-0835-x. [DOI] [PubMed] [Google Scholar]
- 246.White J. Defining target volumes in breast cancer radiation therapy for the future: back to basics. Int J Radiat Oncol Biol Phys. 2015;93(2):277–80. doi: 10.1016/j.ijrobp.2015.06.027. [DOI] [PubMed] [Google Scholar]
- 247.Tandon M, Othman AH, Ashok V, Stein GS, Pratap J. The role of Runx2 in facilitating autophagy in metastatic breast cancer cells. J Cell Physiol. 2017;233(1):559–71. doi: 10.1002/jcp.25916. [DOI] [PubMed] [Google Scholar]
- 248.Martin AP, Mitchell C, Rahmani M, Nephew KP, Grant S, Dent P. Inhibition of MCL-1 enhances lapatinib toxicity and overcomes lapatinib resistance via BAK-dependent autophagy. Cancer Biol Ther. 2009;8(21):2084–96. doi: 10.4161/cbt.8.21.9895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zarzynska JM. The importance of autophagy regulation in breast cancer development and treatment. Biomed Res Int. 2014;2014:710345. doi: 10.1155/2014/710345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Jain K, Paranandi KS, Sridharan S, Basu A. Autophagy in breast cancer and its implications for therapy. Am J Cancer Res. 2013;3(3):251–65. [PMC free article] [PubMed] [Google Scholar]
- 251.Tandon M, Chen Z, Othman AH, Pratap J. Role of Runx2 in IGF-1Rbeta/Akt- and AMPK/Erk-dependent growth, survival and sensitivity towards metformin in breast cancer bone metastasis. Oncogene. 2016;35(36):4730–40. doi: 10.1038/onc.2015.518. [DOI] [PubMed] [Google Scholar]
- 252.Ferrari N, McDonald L, Morris JS, Cameron ER, Blyth K. RUNX2 in mammary gland development and breast cancer. J Cell Physiol. 2013;228(6):1137–42. doi: 10.1002/jcp.24285. [DOI] [PubMed] [Google Scholar]
- 253.Onodera Y, Miki Y, Suzuki T, Takagi K, Akahira J, Sakyu T, et al. Runx2 in human breast carcinoma: its potential roles in cancer progression. Cancer Sci. 2010;101(12):2670–5. doi: 10.1111/j.1349-7006.2010.01742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Tandon M, Chen Z, Pratap J. Runx2 activates PI3K/Akt signaling via mTORC2 regulation in invasive breast cancer cells. Breast Cancer Res. 2014;16(1):R16. doi: 10.1186/bcr3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Yang Z, Zhang B, Liu B, Xie Y, Cao X. Combined Runx2 and Snail overexpression is associated with a poor prognosis in breast cancer. Tumour Biol. 2015;36(6):4565–73. doi: 10.1007/s13277-015-3101-3. [DOI] [PubMed] [Google Scholar]
- 256.Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu C, Sakamoto KM. Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008;68(8):2557–60. doi: 10.1158/0008-5472.CAN-07-5989. [DOI] [PubMed] [Google Scholar]
- 257.Yao TP. The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer. 2010;1(7):779–86. doi: 10.1177/1947601910383277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lee JY, Yao TP. Quality control autophagy: a joint effort of ubiquitin, protein deacetylase and actin cytoskeleton. Autophagy. 2010;6(4):555–7. doi: 10.4161/auto.6.4.11812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Garcia-Mata R, Gao YS, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic. 2002;3(6):388–96. doi: 10.1034/j.1600-0854.2002.30602.x. [DOI] [PubMed] [Google Scholar]
- 260.Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21(17):2172–81. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Dargemont C, Ossareh-Nazari B. Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim Biophys Acta. 2012;1823(1):138–44. doi: 10.1016/j.bbamcr.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 262.Papandreou CN, Logothetis CJ. Bortezomib as a potential treatment for prostate cancer. Cancer Res. 2004;64(15):5036–43. doi: 10.1158/0008-5472.CAN-03-2707. [DOI] [PubMed] [Google Scholar]
- 263.Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007;67(13):6383–91. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 264.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101(52):18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100(8):4389–94. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Shenkman M, Groisman B, Ron E, Avezov E, Hendershot LM, Lederkremer GZ. A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J Biol Chem. 2013;288(4):2167–78. doi: 10.1074/jbc.M112.438275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Wang S, Huang J, Lyu H, Cai B, Yang X, Li F, et al. Therapeutic targeting of erbB3 with MM-121/SAR256212 enhances antitumor activity of paclitaxel against erbB2-overexpressing breast cancer. Breast Cancer Res. 2013;15(5):R101. doi: 10.1186/bcr3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Papadopoulos KP, Burris HA, 3rd, Gordon M, Lee P, Sausville EA, Rosen PJ, et al. A phase I/II study of carfilzomib 2-10-min infusion in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;72(4):861–8. doi: 10.1007/s00280-013-2267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014;12:94. doi: 10.1186/s12915-014-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zhou HJ, Wang J, Yao B, Wong S, Djakovic S, Kumar B, et al. Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB-5083) J Med Chem. 2015;58(24):9480–97. doi: 10.1021/acs.jmedchem.5b01346. [DOI] [PubMed] [Google Scholar]
- 271.Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy. 2007;3(6):643–5. doi: 10.4161/auto.5050. [DOI] [PubMed] [Google Scholar]
- 272.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155(6):1216–9. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Glickman MS, Sawyers CL. Converting cancer therapies into cures: lessons from infectious diseases. Cell. 2012;148(6):1089–98. doi: 10.1016/j.cell.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Gifford JB, Huang W, Zeleniak AE, Hindoyan A, Wu H, Donahue TR, et al. Expression of GRP78, master regulator of the unfolded protein response, increases chemoresistance in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2016;15(5):1043–52. doi: 10.1158/1535-7163.MCT-15-0774. [DOI] [PubMed] [Google Scholar]
- 275.Wuicik L, Cavalli LR, Cornelio DA, Schmid Braz AT, Barbosa ML, Lima RS, et al. Chromosome alterations associated with positive and negative lymph node involvement in breast cancer. Cancer Genet Cytogenet. 2007;173(2):114–21. doi: 10.1016/j.cancergencyto.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 276.Okita R, Ohsumi S, Takashima S, Saeki T, Aogi K, Nishimura R. Synchronous liver metastases of intracystic papillary carcinoma with invasion of the breast. Breast Cancer. 2005;12(4):327–30. doi: 10.2325/jbcs.12.327. [DOI] [PubMed] [Google Scholar]
- 277.Ellard SL, Clemons M, Gelmon KA, Norris B, Kennecke H, Chia S, et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J Clin Oncol. 2009;27(27):4536–41. doi: 10.1200/JCO.2008.21.3033. [DOI] [PubMed] [Google Scholar]
- 278.Jerusalem G, Fasolo A, Dieras V, Cardoso F, Bergh J, Vittori L, et al. Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat. 2011;125(2):447–55. doi: 10.1007/s10549-010-1260-x. [DOI] [PubMed] [Google Scholar]
- 279.Morrow PK, Wulf GM, Ensor J, Booser DJ, Moore JA, Flores PR, et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol. 2011;29(23):3126–32. doi: 10.1200/JCO.2010.32.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Chandarlapaty S, Scaltriti M, Angelini P, Ye Q, Guzman M, Hudis CA, et al. Inhibitors of HSP90 block p95-HER2 signaling in trastuzumab-resistant tumors and suppress their growth. Oncogene. 2010;29(3):325–34. doi: 10.1038/onc.2009.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149(5):1098–111. doi: 10.1016/j.cell.2012.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379(9816):633–40. doi: 10.1016/S0140-6736(11)61847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–9. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366(2):109–19. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Baselga J, Segalla JG, Roche H, Del Giglio A, Pinczowski H, Ciruelos EM, et al. Sorafenib in combination with capecitabine: an oral regimen for patients with HER2-negative locally advanced or metastatic breast cancer. J Clin Oncol. 2012;30(13):1484–91. doi: 10.1200/JCO.2011.36.7771. [DOI] [PubMed] [Google Scholar]
- 286.Auner HW, Moody AM, Ward TH, Kraus M, Milan E, May P, et al. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS One. 2013;8(9):e74415. doi: 10.1371/journal.pone.0074415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Lopez-Tarruella S, Jerez Y, Marquez-Rodas I, Martin M. Neratinib (HKI-272) in the treatment of breast cancer. Future Oncol. 2012;8(6):671–81. doi: 10.2217/fon.12.66. [DOI] [PubMed] [Google Scholar]
- 288.Leignadier J, Dalenc F, Poirot M, Silvente-Poirot S. Improving the efficacy of hormone therapy in breast cancer: The role of cholesterol metabolism in SERM-mediated autophagy, cell differentiation and death. Biochem Pharmacol. 2017;144:18–28. doi: 10.1016/j.bcp.2017.06.120. [DOI] [PubMed] [Google Scholar]
- 289.Nawaz Z, Stancel GM, Hyder SM. The pure antiestrogen ICI 182,780 inhibits progestin-induced transcription. Cancer Res. 1999;59(2):372–6. [PubMed] [Google Scholar]
- 290.Howell A. Fulvestrant ('Faslodex'): current and future role in breast cancer management. Crit Rev Oncol Hematol. 2006;57(3):265–73. doi: 10.1016/j.critrevonc.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 291.Long X, Nephew KP. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha. J Biol Chem. 2006;281(14):9607–15. doi: 10.1074/jbc.M510809200. [DOI] [PubMed] [Google Scholar]
- 292.Perey L, Paridaens R, Hawle H, Zaman K, Nole F, Wildiers H, et al. Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK 21/00) Ann Oncol. 2007;18(1):64–9. doi: 10.1093/annonc/mdl341. [DOI] [PubMed] [Google Scholar]
- 293.Manni A, Trujillo J, Marshall JS, Pearson OH. Antiestrogen-induced remissions in stage IV breast cancer. Cancer Treat Rep. 1976;60(10):1445–50. [PubMed] [Google Scholar]
- 294.Veronesi U, Maisonneuve P, Rotmensz N, Bonanni B, Boyle P, Viale G, et al. Tamoxifen for the prevention of breast cancer: late results of the Italian Randomized Tamoxifen Prevention Trial among women with hysterectomy. J Natl Cancer Inst. 2007;99(9):727–37. doi: 10.1093/jnci/djk154. [DOI] [PubMed] [Google Scholar]
- 295.Poirot M, Chailleux C, Mesange F, Bayard F, Faye JC. Characterization of the membranous antiestrogen binding protein: II. Purification to homogeneity. J Recept Res. 1994;14(1):37–46. doi: 10.3109/10799899409066995. [DOI] [PubMed] [Google Scholar]
- 296.Grese TA, Cho S, Finley DR, Godfrey AG, Jones CD, Lugar CW, 3rd, et al. Structure-activity relationships of selective estrogen receptor modulators: modifications to the 2-arylbenzothiophene core of raloxifene. J Med Chem. 1997;40(2):146–67. doi: 10.1021/jm9606352. [DOI] [PubMed] [Google Scholar]
- 297.Cummings SR, Eckert S, Krueger KA, Grady D, Powles TJ, Cauley JA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple outcomes of Raloxifene evaluation. JAMA. 1999;281(23):2189–97. doi: 10.1001/jama.281.23.2189. [DOI] [PubMed] [Google Scholar]
- 298.Liu H, Lee ES, Gajdos C, Pearce ST, Chen B, Osipo C, et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95(21):1586–97. doi: 10.1093/jnci/djg080. [DOI] [PubMed] [Google Scholar]
- 299.Kawagoe J, Ohmichi M, Takahashi T, Ohshima C, Mabuchi S, Takahashi K, et al. Raloxifene inhibits estrogen-induced up-regulation of telomerase activity in a human breast cancer cell line. J Biol Chem. 2003;278(44):43363–72. doi: 10.1074/jbc.M304363200. [DOI] [PubMed] [Google Scholar]
- 300.Morishima S, Shibata MA, Ohmichi M, Otsuki Y. Raloxifene, a selective estrogen receptor modulator, induces mitochondria-mediated apoptosis in human endometrial carcinoma cells. Med Mol Morphol. 2008;41(3):132–8. doi: 10.1007/s00795-008-0403-1. [DOI] [PubMed] [Google Scholar]
- 301.Brennan M, Lim B. The actual role of receptors as cancer markers, biochemical and clinical aspects: receptors in breast cancer. Adv Exp Med Biol. 2015;867:327–37. doi: 10.1007/978-94-017-7215-0_20. [DOI] [PubMed] [Google Scholar]
- 302.Vogel CL. Update on the current use of hormonals as therapy in advanced breast cancer. Anticancer Drugs. 2003;14(4):265–73. doi: 10.1097/00001813-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 303.Xiong R, Zhao J, Gutgesell LM, Wang Y, Lee S, Karumudi B, et al. Novel selective estrogen receptor downregulators (SERDs) developed against treatment-resistant breast cancer. J Med Chem. 2017;60(4):1325–42. doi: 10.1021/acs.jmedchem.6b01355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem. 2015;58(12):4888–904. doi: 10.1021/acs.jmedchem.5b00054. [DOI] [PubMed] [Google Scholar]
- 305.De Savi C, Bradbury RH, Rabow AA, Norman RA, de Almeida C, Andrews DM, et al. Optimization of a novel binding motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetra hydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic acid (AZD9496), a potent and orally bioavailable selective estrogen receptor downregulator and antagonist. J Med Chem. 2015;58(20):8128–40. doi: 10.1021/acs.jmedchem.5b00984. [DOI] [PubMed] [Google Scholar]
- 306.Weir HM, Bradbury RH, Lawson M, Rabow AA, Buttar D, Callis RJ, et al. AZD9496: An oral estrogen receptor inhibitor that blocks the growth of ER-positive and ESR1-mutant breast tumors in preclinical models. Cancer Res. 2016;76(11):3307–18. doi: 10.1158/0008-5472.CAN-15-2357. [DOI] [PubMed] [Google Scholar]
- 307.Garner F, Shomali M, Paquin D, Lyttle CR, Hattersley G. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs. 2015;26(9):948–56. doi: 10.1097/CAD.0000000000000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wardell SE, Nelson ER, Chao CA, Alley HM, McDonnell DP. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr Relat Cancer. 2015;22(5):713–24. doi: 10.1530/ERC-15-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Bihani T, Patel HK, Arlt H, Tao N, Jiang H, Brown JL, et al. Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor activity in multiple ER+ breast cancer patient-derived xenograft models. Clin Cancer Res. 2017;23(16):4793–804. doi: 10.1158/1078-0432.CCR-16-2561. [DOI] [PubMed] [Google Scholar]
- 310.Trunet PF, Bhatnagar AS, Chaudri HA, Hornberger U. Letrozole (CGS 20267), a new oral aromatase inhibitor for the treatment of advanced breast cancer in postmenopausal patients. Acta Oncol. 1996;35(Suppl 5):15–8. doi: 10.3109/02841869609083962. [DOI] [PubMed] [Google Scholar]
- 311.Dixon JM, Love CD, Bellamy CO, Cameron DA, Leonard RC, Smith H, et al. Letrozole as primary medical therapy for locally advanced and large operable breast cancer. Breast Cancer Res Treat. 2001;66(3):191–9. doi: 10.1023/a:1010669403283. [DOI] [PubMed] [Google Scholar]
- 312.Buzdar AU, Robertson JF, Eiermann W, Nabholtz JM. An overview of the pharmacology and pharmacokinetics of the newer generation aromatase inhibitors anastrozole, letrozole, and exemestane. Cancer. 2002;95(9):2006–16. doi: 10.1002/cncr.10908. [DOI] [PubMed] [Google Scholar]
- 313.Blackwell K, Burris H, Gomez P, Lynn Henry N, Isakoff S, Campana F, et al. Phase I/II dose-escalation study of PI3K inhibitors pilaralisib or voxtalisib in combination with letrozole in patients with hormone-receptor-positive and HER2-negative metastatic breast cancer refractory to a non-steroidal aromatase inhibitor. Breast Cancer Res Treat. 2015;154(2):287–97. doi: 10.1007/s10549-015-3615-9. [DOI] [PubMed] [Google Scholar]
- 314.Geisler J, King N, Dowsett M, Ottestad L, Lundgren S, Walton P, et al. Influence of anastrozole (Arimidex), a selective, non-steroidal aromatase inhibitor, on in vivo aromatisation and plasma oestrogen levels in postmenopausal women with breast cancer. Br J Cancer. 1996;74(8):1286–91. doi: 10.1038/bjc.1996.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Dowsett M. Theoretical considerations for the ideal aromatase inhibitor. Breast Cancer Res Treat. 1998;49(Suppl 1):S39–44. doi: 10.1023/a:1006088405721. [DOI] [PubMed] [Google Scholar]
- 316.Bonneterre J, Buzdar A, Nabholtz JM, Robertson JF, Thurlimann B, von Euler M, et al. Anastrozole is superior to tamoxifen as first-line therapy in hormone receptor positive advanced breast carcinoma. Cancer. 2001;92(9):2247–58. doi: 10.1002/1097-0142(20011101)92:9<2247::aid-cncr1570>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 317.Choueiri TK, Alemany CA, Abou-Jawde RM, Budd GT. Role of aromatase inhibitors in the treatment of breast cancer. Clin Ther. 2004;26(8):1199–214. doi: 10.1016/s0149-2918(04)80017-1. [DOI] [PubMed] [Google Scholar]
- 318.Evans TR, Di Salle E, Ornati G, Lassus M, Benedetti MS, Pianezzola E, et al. Phase I and endocrine study of exemestane (FCE 24304), a new aromatase inhibitor, in postmenopausal women. Cancer Res. 1992;52(21):5933–9. [PubMed] [Google Scholar]
- 319.Geisler J, King N, Anker G, Ornati G, Di Salle E, Lonning PE, et al. In vivo inhibition of aromatization by exemestane, a novel irreversible aromatase inhibitor, in postmenopausal breast cancer patients. Clin Cancer Res. 1998;4(9):2089–93. [PubMed] [Google Scholar]
- 320.Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31(17):2128–35. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Robarge JD, Desta Z, Nguyen AT, Li L, Hertz D, Rae JM, et al. Effects of exemestane and letrozole therapy on plasma concentrations of estrogens in a randomized trial of postmenopausal women with breast cancer. Breast Cancer Res Treat. 2017;161(3):453–61. doi: 10.1007/s10549-016-4077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21(2):227–39. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Leung E, Rewcastle GW, Joseph WR, Rosengren RJ, Larsen L, Baguley BC. Identification of cyclohexanone derivatives that act as catalytic inhibitors of topoisomerase I: effects on tamoxifen-resistant MCF-7 cancer cells. Invest New Drugs. 2012;30(6):2103–12. doi: 10.1007/s10637-011-9768-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Ji Y, Di W, Yang Q, Lu Z, Cai W, Wu J. Inhibition of autophagy increases proliferation inhibition and apoptosis induced by the PI3K/mTOR inhibitor NVP-BEZ235 in breast cancer cells. Clin Lab. 2015;61(8):1043–51. doi: 10.7754/clin.lab.2015.150144. [DOI] [PubMed] [Google Scholar]
- 325.Hamunyela RH, Serafin AM, Akudugu JM. Strong synergism between small molecule inhibitors of HER2, PI3K, mTOR and Bcl-2 in human breast cancer cells. Toxicol In Vitro. 2017;38:117–23. doi: 10.1016/j.tiv.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 326.Baselga J, Cortes J, Im SA, Clark E, Ross G, Kiermaier A, et al. Biomarker analyses in CLEOPATRA: a phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J Clin Oncol. 2014;32(33):3753–61. doi: 10.1200/JCO.2013.54.5384. [DOI] [PubMed] [Google Scholar]
- 327.Hoeflich KP, Guan J, Edgar KA, O'Brien C, Savage H, Wilson TR, et al. The PI3K inhibitor taselisib overcomes letrozole resistance in a breast cancer model expressing aromatase. Genes Cancer. 2016;7(3-4):73–85. doi: 10.18632/genesandcancer.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Juric D, Krop I, Ramanathan RK, Wilson TR, Ware JA, Sanabria Bohorquez SM, et al. Phase I dose-escalation study of taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Discov. 2017;7(7):704–15. doi: 10.1158/2159-8290.CD-16-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Vaillant F, Merino D, Lee L, Breslin K, Pal B, Ritchie ME, et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell. 2013;24(1):120–9. doi: 10.1016/j.ccr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 330.Deeks ED. Venetoclax: first global approval. Drugs. 2016;76(9):979–87. doi: 10.1007/s40265-016-0596-x. [DOI] [PubMed] [Google Scholar]
- 331.Fleming GF, Ma CX, Huo D, Sattar H, Tretiakova M, Lin L, et al. Phase II trial of temsirolimus in patients with metastatic breast cancer. Breast Cancer Res Treat. 2012;136(2):355–63. doi: 10.1007/s10549-011-1910-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104(12):1828–35. doi: 10.1038/bjc.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Ward CS, Eriksson P, Izquierdo-Garcia JL, Brandes AH, Ronen SM. HDAC inhibition induces increased choline uptake and elevated phosphocholine levels in MCF7 breast cancer cells. PLoS One. 2013;8(4):e62610. doi: 10.1371/journal.pone.0062610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Deming DA, Ninan J, Bailey HH, Kolesar JM, Eickhoff J, Reid JM, et al. A Phase I study of intermittently dosed vorinostat in combination with bortezomib in patients with advanced solid tumors. Invest New Drugs. 2014;32(2):323–9. doi: 10.1007/s10637-013-0035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Wardley AM, Pivot X, Morales-Vasquez F, Zetina LM, de Fatima Dias Gaui M, Reyes DO, et al. Randomized phase II trial of first-line trastuzumab plus docetaxel and capecitabine compared with trastuzumab plus docetaxel in HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28(6):976–83. doi: 10.1200/JCO.2008.21.6531. [DOI] [PubMed] [Google Scholar]
- 336.Yardley DA, Peacock NW, Dickson NR, White MB, Vazquez ER, Foust JT, et al. A phase II trial of neoadjuvant gemcitabine, epirubicin, and docetaxel as primary treatment of patients with locally advanced or inflammatory breast cancer. Clin Breast Cancer. 2010;10(3):217–23. doi: 10.3816/CBC.2010.n.029. [DOI] [PubMed] [Google Scholar]
- 337.Rao R, Nalluri S, Kolhe R, Yang Y, Fiskus W, Chen J, et al. Treatment with panobinostat induces glucose-regulated protein 78 acetylation and endoplasmic reticulum stress in breast cancer cells. Mol Cancer Ther. 2010;9(4):942–52. doi: 10.1158/1535-7163.MCT-09-0988. [DOI] [PubMed] [Google Scholar]
- 338.Fortunati N, Marano F, Bandino A, Frairia R, Catalano MG, Boccuzzi G. The pan-histone deacetylase inhibitor LBH589 (panobinostat) alters the invasive breast cancer cell phenotype. Int J Oncol. 2014;44(3):700–8. doi: 10.3892/ijo.2013.2218. [DOI] [PubMed] [Google Scholar]
- 339.Tan WW, Allred JB, Moreno-Aspitia A, Northfelt DW, Ingle JN, Goetz MP, et al. Phase I study of Panobinostat (LBH589) and Letrozole in postmenopausal metastatic breast cancer patients. Clin Breast Cancer. 2016;16(2):82–6. doi: 10.1016/j.clbc.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Le Bihan S, Marsaud V, Mercier-Bodard C, Baulieu EE, Mader S, White JH, et al. Calcium/calmodulin kinase inhibitors and immunosuppressant macrolides rapamycin and FK506 inhibit progestin- and glucocorticosteroid receptor-mediated transcription in human breast cancer T47D cells. Mol Endocrinol. 1998;12(7):986–1001. doi: 10.1210/mend.12.7.0128. [DOI] [PubMed] [Google Scholar]
- 341.Pang H, Faber LE. Estrogen and rapamycin effects on cell cycle progression in T47D breast cancer cells. Breast Cancer Res Treat. 2001;70(1):21–6. doi: 10.1023/a:1012570204923. [DOI] [PubMed] [Google Scholar]
- 342.Zhou X, Tan M, Stone Hawthorne V, Klos KS, Lan KH, Yang Y, et al. Activation of the Akt/mammalian target of rapamycin/4E-BP1 pathway by ErbB2 overexpression predicts tumor progression in breast cancers. Clin Cancer Res. 2004;10(20):6779–88. doi: 10.1158/1078-0432.CCR-04-0112. [DOI] [PubMed] [Google Scholar]
- 343.Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13(10):433–42. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 344.Wang G, Yin T. Rapamycin enhances the antiproliferative effect of transforming growth factor-beta on MCF-7 human breast cancer cells. Exp Ther Med. 2017;14(1):748–52. doi: 10.3892/etm.2017.4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Okuhira K, Demizu Y, Hattori T, Ohoka N, Shibata N, Nishimaki-Mogami T, et al. Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci. 2013;104(11):1492–8. doi: 10.1111/cas.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Mao C, Livezey M, Kim JE, Shapiro DJ. Antiestrogen resistant cell lines expressing estrogen receptor alpha mutations upregulate the unfolded protein response and are killed by BHPI. Sci Rep. 2016;6:34753. doi: 10.1038/srep34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Braicu C, Gherman CD, Irimie A, Berindan-Neagoe I. Epigallocatechin-3-Gallate (EGCG) inhibits cell proliferation and migratory behaviour of triple negative breast cancer cells. J Nanosci Nanotechnol. 2013;13(1):632–7. doi: 10.1166/jnn.2013.6882. [DOI] [PubMed] [Google Scholar]
- 348.Luo HQ, Xu M, Zhong WT, Cui ZY, Liu FM, Zhou KY, et al. EGCG decreases the expression of HIF-1alpha and VEGF and cell growth in MCF-7 breast cancer cells. J BUON. 2014;19(2):435–9. [PubMed] [Google Scholar]
- 349.Ranzato E, Magnelli V, Martinotti S, Waheed Z, Cain SM, Snutch TP, et al. Epigallocatechin-3-gallate elicits Ca2+ spike in MCF-7 breast cancer cells: essential role of Cav3.2 channels. Cell Calcium. 2014;56(4):285–95. doi: 10.1016/j.ceca.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 350.Moradzadeh M, Hosseini A, Erfanian S, Rezaei H. Epigallocatechin-3-gallate promotes apoptosis in human breast cancer T47D cells through down-regulation of PI3K/AKT and Telomerase. Pharmacol Rep. 2017;69(5):924–8. doi: 10.1016/j.pharep.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 351.Zeng L, Yan J, Luo L, Ma M, Zhu H. Preparation and characterization of (-)-Epigallocatechin-3-gallate (EGCG)-loaded nanoparticles and their inhibitory effects on Human breast cancer MCF-7 cells. Sci Rep. 2017;7:45521. doi: 10.1038/srep45521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Hong OY, Noh EM, Jang HY, Lee YR, Lee BK, Jung SH, et al. Epigallocatechin gallate inhibits the growth of MDA-MB-231 breast cancer cells via inactivation of the beta-catenin signaling pathway. Oncol Lett. 2017;14(1):441–6. doi: 10.3892/ol.2017.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Mondal A, Bennett LL. Resveratrol enhances the efficacy of sorafenib mediated apoptosis in human breast cancer MCF7 cells through ROS, cell cycle inhibition, caspase 3 and PARP cleavage. Biomed Pharmacother. 2016;84:1906–14. doi: 10.1016/j.biopha.2016.10.096. [DOI] [PubMed] [Google Scholar]
- 354.Andreani C, Bartolacci C, Wijnant K, Crinelli R, Bianchi M, Magnani M, et al. Resveratrol fuels HER2 and ERalpha-positive breast cancer behaving as proteasome inhibitor. Aging (Albany NY) 2017;9(2):508–23. doi: 10.18632/aging.101175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Vogel C, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. First-line, single-agent Herceptin(R) (trastuzumab) in metastatic breast cancer. a preliminary report. Eur J Cancer. 2001;37(Suppl 1):25–9. [PubMed] [Google Scholar]
- 356.Pegram M, Slamon D. Biological rationale for HER2/neu (c-erbB2) as a target for monoclonal antibody therapy. Semin Oncol. 2000;27(5 Suppl 9):13–9. [PubMed] [Google Scholar]
- 357.Baselga J. Clinical trials of Herceptin(R) (trastuzumab) Eur J Cancer. 2001;37(Suppl 1):18–24. [PubMed] [Google Scholar]
- 358.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(3):719–26. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 359.Burstein HJ, Keshaviah A, Baron AD, Hart RD, Lambert-Falls R, Marcom PK, et al. Trastuzumab plus vinorelbine or taxane chemotherapy for HER2-overexpressing metastatic breast cancer: the trastuzumab and vinorelbine or taxane study. Cancer. 2007;110(5):965–72. doi: 10.1002/cncr.22885. [DOI] [PubMed] [Google Scholar]
- 360.Saini KS, Azim HA, Jr, Metzger-Filho O, Loi S, Sotiriou C, de Azambuja E, et al. Beyond trastuzumab: new treatment options for HER2-positive breast cancer. Breast. 2011;20(Suppl 3):S20–7. doi: 10.1016/S0960-9776(11)70289-2. [DOI] [PubMed] [Google Scholar]
- 361.Barok M, Tanner M, Koninki K, Isola J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 2011;13(2):R46. doi: 10.1186/bcr2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17(20):6437–47. doi: 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- 363.Krop IE, Modi S, LoRusso PM, Pegram M, Guardino E, Althaus B, et al. Phase 1b/2a study of trastuzumab emtansine (T-DM1), paclitaxel, and pertuzumab in HER2-positive metastatic breast cancer. Breast Cancer Res. 2016;18(1):34. doi: 10.1186/s13058-016-0691-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Michel LL, Bermejo JL, Gondos A, Marme F, Schneeweiss A. T-DM1 as a new treatment option for patients with metastatic HER2-positive breast cancer in clinical practice. Anticancer Res. 2015;35(9):5085–90. [PubMed] [Google Scholar]
- 365.Kiewe P, Hasmuller S, Kahlert S, Heinrigs M, Rack B, Marme A, et al. Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clin Cancer Res. 2006;12(10):3085–91. doi: 10.1158/1078-0432.CCR-05-2436. [DOI] [PubMed] [Google Scholar]
- 366.Tinoco G, Warsch S, Gluck S, Avancha K, Montero AJ. Treating breast cancer in the 21st century: emerging biological therapies. J Cancer. 2013;4(2):117–32. doi: 10.7150/jca.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(1):25–32. doi: 10.1016/S1470-2045(11)70336-9. [DOI] [PubMed] [Google Scholar]
- 368.Geretti E, van Meeteren LA, Shimizu A, Dudley AC, Claesson-Welsh L, Klagsbrun M. A mutated soluble neuropilin-2 B domain antagonizes vascular endothelial growth factor bioactivity and inhibits tumor progression. Mol Cancer Res. 2010;8(8):1063–73. doi: 10.1158/1541-7786.MCR-10-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Kirouac DC, Du JY, Lahdenranta J, Overland R, Yarar D, Paragas V, et al. Computational modeling of ERBB2-amplified breast cancer identifies combined ErbB2/3 blockade as superior to the combination of MEK and AKT inhibitors. Sci Signal. 2013;6(288):ra68. doi: 10.1126/scisignal.2004008. [DOI] [PubMed] [Google Scholar]
- 370.McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther. 2012;11(3):582–93. doi: 10.1158/1535-7163.MCT-11-0820. [DOI] [PubMed] [Google Scholar]
- 371.Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66(3):1630–9. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 372.Johnston S, Pippen J, Jr, Pivot X, Lichinitser M, Sadeghi S, Dieras V, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009;27(33):5538–46. doi: 10.1200/JCO.2009.23.3734. [DOI] [PubMed] [Google Scholar]
- 373.Yap TA, Vidal L, Adam J, Stephens P, Spicer J, Shaw H, et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol. 2010;28(25):3965–72. doi: 10.1200/JCO.2009.26.7278. [DOI] [PubMed] [Google Scholar]
- 374.Lin NU, Winer EP, Wheatley D, Carey LA, Houston S, Mendelson D, et al. A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res Treat. 2012;133(3):1057–65. doi: 10.1007/s10549-012-2003-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Harbeck N, Huang CS, Hurvitz S, Yeh DC, Shao Z, Im SA, et al. Afatinib plus vinorelbine versus trastuzumab plus vinorelbine in patients with HER2-overexpressing metastatic breast cancer who had progressed on one previous trastuzumab treatment (LUX-Breast 1): an open-label, randomised, phase 3 trial. Lancet Oncol. 2016;17(3):357–66. doi: 10.1016/S1470-2045(15)00540-9. [DOI] [PubMed] [Google Scholar]
- 376.Erlichman C, Boerner SA, Hallgren CG, Spieker R, Wang XY, James CD, et al. The HER tyrosine kinase inhibitor CI1033 enhances cytotoxicity of 7-ethyl-10-hydroxycamptothecin and topotecan by inhibiting breast cancer resistance protein-mediated drug efflux. Cancer Res. 2001;61(2):739–48. [PubMed] [Google Scholar]
- 377.Brunner-Kubath C, Shabbir W, Saferding V, Wagner R, Singer CF, Valent P, et al. The PI3 kinase/mTOR blocker NVP-BEZ235 overrides resistance against irreversible ErbB inhibitors in breast cancer cells. Breast Cancer Res Treat. 2011;129(2):387–400. doi: 10.1007/s10549-010-1232-1. [DOI] [PubMed] [Google Scholar]
- 378.Wong KK, Fracasso PM, Bukowski RM, Lynch TJ, Munster PN, Shapiro GI, et al. A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res. 2009;15(7):2552–8. doi: 10.1158/1078-0432.CCR-08-1978. [DOI] [PubMed] [Google Scholar]
- 379.Awada A, Colomer R, Inoue K, Bondarenko I, Badwe RA, Demetriou G, et al. Neratinib plus paclitaxel vs trastuzumab plus paclitaxel in previously untreated metastatic ERBB2-positive breast cancer: the NEfERT-T randomized clinical trial. JAMA Oncol. 2016;2(12):1557–64. doi: 10.1001/jamaoncol.2016.0237. [DOI] [PubMed] [Google Scholar]
- 380.Zuo WJ, Jiang YZ, Wang YJ, Xu XE, Hu X, Liu GY, et al. Dual characteristics of novel HER2 kinase domain mutations in response to HER2-targeted therapies in human breast cancer. Clin Cancer Res. 2016;22(19):4859–69. doi: 10.1158/1078-0432.CCR-15-3036. [DOI] [PubMed] [Google Scholar]
- 381.Echavarria I, Lopez-Tarruella S, Marquez-Rodas I, Jerez Y, Martin M. Neratinib for the treatment of HER2-positive early stage breast cancer. Expert Rev Anticancer Ther. 2017;17(8):669–79. doi: 10.1080/14737140.2017.1338954. [DOI] [PubMed] [Google Scholar]
- 382.Baselga J. Combined anti-EGF receptor and anti-HER2 receptor therapy in breast cancer: a promising strategy ready for clinical testing. Ann Oncol. 2002;13(1):8–9. doi: 10.1093/annonc/mdf092. [DOI] [PubMed] [Google Scholar]
- 383.Lim SM, Xie T, Westover KD, Ficarro SB, Tae HS, Gurbani D, et al. Development of small molecules targeting the pseudokinase Her3. Bioorg Med Chem Lett. 2015;25(16):3382–9. doi: 10.1016/j.bmcl.2015.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Kim TE, Murren JR. Erlotinib OSI/Roche/Genentech. Curr Opin Investig Drugs. 2002;3(9):1385–95. [PubMed] [Google Scholar]
- 385.Liu CY, Huang TT, Huang CT, Hu MH, Wang DS, Wang WL, et al. EGFR-independent Elk1/CIP2A signalling mediates apoptotic effect of an erlotinib derivative TD52 in triple-negative breast cancer cells. Eur J Cancer. 2017;72:112–23. doi: 10.1016/j.ejca.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 386.Fu X, Creighton CJ, Biswal NC, Kumar V, Shea M, Herrera S, et al. Overcoming endocrine resistance due to reduced PTEN levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase B, or mitogen-activated protein kinase kinase. Breast Cancer Res. 2014;16(5):430. doi: 10.1186/s13058-014-0430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Moreno-Aspitia A. Clinical overview of sorafenib in breast cancer. Future Oncol. 2010;6(5):655–63. doi: 10.2217/fon.10.41. [DOI] [PubMed] [Google Scholar]
- 388.Bazzola L, Foroni C, Andreis D, Zanoni V, R Cappelletti M, Allevi G, et al. Combination of letrozole, metronomic cyclophosphamide and sorafenib is well-tolerated and shows activity in patients with primary breast cancer. Br J Cancer. 2015;112(1):52–60. doi: 10.1038/bjc.2014.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Ferrario C, Strepponi I, Esfahani K, Charamis H, Langleben A, Scarpi E, et al. Phase I/II trial of sorafenib in combination with vinorelbine as first-line chemotherapy for metastatic breast cancer. PLoS One. 2016;11(12):e0167906. doi: 10.1371/journal.pone.0167906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Huang J, Wang S, Lyu H, Cai B, Yang X, Wang J, et al. The anti-erbB3 antibody MM-121/SAR256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Mol Cancer. 2013;12(1):134. doi: 10.1186/1476-4598-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010;70(6):2485–94. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Curley MD, Sabnis GJ, Wille L, Adiwijaya BS, Garcia G, Moyo V, et al. Seribantumab, an anti-ERBB3 antibody, delays the onset of resistance and restores sensitivity to letrozole in an estrogen receptor-positive breast cancer model. Mol Cancer Ther. 2015;14(11):2642–52. doi: 10.1158/1535-7163.MCT-15-0169. [DOI] [PubMed] [Google Scholar]
- 393.Espelin CW, Leonard SC, Geretti E, Wickham TJ, Hendriks BS. Dual HER2 Targeting with trastuzumab and liposomal-encapsulated doxorubicin (MM-302) demonstrates synergistic antitumor activity in breast and gastric cancer. Cancer Res. 2016;76(6):1517–27. doi: 10.1158/0008-5472.CAN-15-1518. [DOI] [PubMed] [Google Scholar]
- 394.Martin M, Lopez-Tarruella S. Emerging therapeutic options for HER2-positive breast cancer. Am Soc Clin Oncol Educ Book. 2016;35:e64–70. doi: 10.1200/EDBK_159167. [DOI] [PubMed] [Google Scholar]
- 395.Moulder SL, Borges VF, Baetz T, McSpadden T, Fernetich G, Murthy RK, et al. Phase I study of ONT-380, a HER2 inhibitor, in patients with HER2 + -advanced solid tumors, with an expansion cohort in HER2+ metastatic breast cancer (MBC) Clin Cancer Res. 2017;23(14):3529–36. doi: 10.1158/1078-0432.CCR-16-1496. [DOI] [PubMed] [Google Scholar]
- 396.Doi T, Takiuchi H, Ohtsu A, Fuse N, Goto M, Yoshida M, et al. Phase I first-in-human study of TAK-285, a novel investigational dual HER2/EGFR inhibitor, in cancer patients. Br J Cancer. 2012;106(4):666–72. doi: 10.1038/bjc.2011.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Nakayama A, Takagi S, Yusa T, Yaguchi M, Hayashi A, Tamura T, et al. Antitumor activity of TAK-285, an investigational, non-Pgp substrate HER2/EGFR kinase inhibitor, in cultured tumor cells, mouse and rat xenograft tumors, and in an HER2-positive brain metastasis model. J Cancer. 2013;4(7):557–65. doi: 10.7150/jca.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Takagi S, Banno H, Hayashi A, Tamura T, Ishikawa T, Ohta Y. HER2 and HER3 cooperatively regulate cancer cell growth and determine sensitivity to the novel investigational EGFR/HER2 kinase inhibitor TAK-285. Oncoscience. 2014;1(3):196–204. doi: 10.18632/oncoscience.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Meng X, Li Y, Tang H, Mao W, Yang H, Wang X, et al. Drug response to HER2 gatekeeper T798M mutation in HER2-positive breast cancer. Amino Acids. 2016;48(2):487–97. doi: 10.1007/s00726-015-2102-2. [DOI] [PubMed] [Google Scholar]
- 400.Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28(16):2698–704. doi: 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- 401.Wang L, Yu X, Wang C, Pan S, Liang B, Zhang Y, et al. The anti-ErbB2 antibody H2-18 and the pan-PI3K inhibitor GDC-0941 effectively inhibit trastuzumab-resistant ErbB2-overexpressing breast cancer. Oncotarget. 2017;8(32):52877–88. doi: 10.18632/oncotarget.17907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Tolaney S, Burris H, Gartner E, Mayer IA, Saura C, Maurer M, et al. Phase I/II study of pilaralisib (SAR245408) in combination with trastuzumab or trastuzumab plus paclitaxel in trastuzumab-refractory HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2015;149(1):151–61. doi: 10.1007/s10549-014-3248-4. [DOI] [PubMed] [Google Scholar]
- 403.Abramson VG, Supko JG, Ballinger T, Cleary JM, Hilton JF, Tolaney SM, et al. Phase Ib study of safety and pharmacokinetics of the PI3K inhibitor SAR245408 with the HER3-neutralizing human antibody SAR256212 in patients with solid tumors. Clin Cancer Res. 2017;23(14):3520–8. doi: 10.1158/1078-0432.CCR-16-1764. [DOI] [PubMed] [Google Scholar]
- 404.Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol. 2007;25(34):5410–7. doi: 10.1200/JCO.2007.11.7960. [DOI] [PubMed] [Google Scholar]
- 405.Gartner EM, Silverman P, Simon M, Flaherty L, Abrams J, Ivy P, et al. A phase II study of 17-allylamino-17-demethoxygeldanamycin in metastatic or locally advanced, unresectable breast cancer. Breast Cancer Res Treat. 2012;131(3):933–7. doi: 10.1007/s10549-011-1866-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N, et al. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011;17(15):5132–9. doi: 10.1158/1078-0432.CCR-11-0072. [DOI] [PubMed] [Google Scholar]
- 407.Hanson BE, Vesole DH. Retaspimycin hydrochloride (IPI-504): a novel heat shock protein inhibitor as an anticancer agent. Expert Opin Investig Drugs. 2009;18(9):1375–83. doi: 10.1517/13543780903158934. [DOI] [PubMed] [Google Scholar]
- 408.Modi S, Saura C, Henderson C, Lin NU, Mahtani R, Goddard J, et al. A multicenter trial evaluating retaspimycin HCL (IPI-504) plus trastuzumab in patients with advanced or metastatic HER2-positive breast cancer. Breast Cancer Res Treat. 2013;139(1):107–13. doi: 10.1007/s10549-013-2510-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823(3):742–55. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Xiang L, Gilkes DM, Chaturvedi P, Luo W, Hu H, Takano N, et al. Ganetespib blocks HIF-1 activity and inhibits tumor growth, vascularization, stem cell maintenance, invasion, and metastasis in orthotopic mouse models of triple-negative breast cancer. J Mol Med (Berl) 2014;92(2):151–64. doi: 10.1007/s00109-013-1102-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Friedland JC, Smith DL, Sang J, Acquaviva J, He S, Zhang C, et al. Targeted inhibition of Hsp90 by ganetespib is effective across a broad spectrum of breast cancer subtypes. Invest New Drugs. 2014;32(1):14–24. doi: 10.1007/s10637-013-9971-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Jiang J, Lu Y, Li Z, Li L, Niu D, Xu W, et al. Ganetespib overcomes resistance to PARP inhibitors in breast cancer by targeting core proteins in the DNA repair machinery. Invest New Drugs. 2017;35(3):251–9. doi: 10.1007/s10637-016-0424-x. [DOI] [PubMed] [Google Scholar]
- 413.Gril B, Palmieri D, Qian Y, Anwar T, Liewehr DJ, Steinberg SM, et al. Pazopanib inhibits the activation of PDGFRbeta-expressing astrocytes in the brain metastatic microenvironment of breast cancer cells. Am J Pathol. 2013;182(6):2368–79. doi: 10.1016/j.ajpath.2013.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Cristofanilli M, Johnston SR, Manikhas A, Gomez HL, Gladkov O, Shao Z, et al. A randomized phase II study of lapatinib + pazopanib versus lapatinib in patients with HER2+ inflammatory breast cancer. Breast Cancer Res Treat. 2013;137(2):471–82. doi: 10.1007/s10549-012-2369-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Zagouri F, Sergentanis TN, Chrysikos D, Papadimitriou CA, Dimopoulos MA, Psaltopoulou T. Hsp90 inhibitors in breast cancer: a systematic review. Breast. 2013;22(5):569–78. doi: 10.1016/j.breast.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 416.Arnold LD, Calderwood DJ, Dixon RW, Johnston DN, Kamens JS, Munschauer R, et al. Pyrrolo[2,3-d]pyrimidines containing an extended 5-substituent as potent and selective inhibitors of lck I. Bioorg Med Chem Lett. 2000;10(19):2167–70. doi: 10.1016/s0960-894x(00)00441-8. [DOI] [PubMed] [Google Scholar]
- 417.Burchat AF, Calderwood DJ, Hirst GC, Holman NJ, Johnston DN, Munschauer R, et al. Pyrrolo[2,3-d]pyrimidines containing an extended 5-substituent as potent and selective inhibitors of lck II. Bioorg Med Chem Lett. 2000;10(19):2171–4. doi: 10.1016/s0960-894x(00)00442-x. [DOI] [PubMed] [Google Scholar]
- 418.Mukai H, Saeki T, Aogi K, Naito Y, Matsubara N, Shigekawa T, et al. Patritumab plus trastuzumab and paclitaxel in human epidermal growth factor receptor 2-overexpressing metastatic breast cancer. Cancer Sci. 2016;107(10):1465–70. doi: 10.1111/cas.13017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell. 2011;20(4):472–86. doi: 10.1016/j.ccr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 420.Tao JJ, Castel P, Radosevic-Robin N, Elkabets M, Auricchio N, Aceto N, et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal. 2014;7(318):ra29. doi: 10.1126/scisignal.2005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Jiang H, Rugo HS. Human epidermal growth factor receptor 2 positive (HER2+) metastatic breast cancer: how the latest results are improving therapeutic options. Ther Adv Med Oncol. 2015;7(6):321–39. doi: 10.1177/1758834015599389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Ruttala HB, Ko YT. Liposome encapsulated albumin-paclitaxel nanoparticle for enhanced antitumor efficacy. Pharm Res. 2015;32(3):1002–16. doi: 10.1007/s11095-014-1512-2. [DOI] [PubMed] [Google Scholar]