

Abstract

We demonstrate photocatalytic hydrogen evolution using COF photosensitizers with molecular proton reduction catalysts for the first time. With azine-linked N2-COF photosensitizer, chloro(pyridine)cobaloxime co-catalyst, and TEOA donor, H2 evolution rate of 782 μmol h–1 g–1 and TON of 54.4 has been obtained in a water/acetonitrile mixture. PXRD, solid-state spectroscopy, EM analysis, and quantum-chemical calculations suggest an outer sphere electron transfer from the COF to the co-catalyst which subsequently follows a monometallic pathway of H2 generation from the CoIII-hydride and/or CoII-hydride species.
Introduction
With fossil fuel reserves dwindling every day, there is an urgent need for clean and sustainable alternative energy sources. Artificial photosynthesis, the conversion of solar energy into energy stored in the bonds of “solar fuels” like hydrogen, could be one of the most viable and nonintermittent solution in this regard.1,2 Development of efficient photocatalytic systems for hydrogen evolution via photoinduced water splitting is thus a very active field of energy research. In this context, covalent organic frameworks (COFs) have recently emerged as a new class of photoactive materials for light-induced hydrogen evolution.3 Similar to related polymeric carbon nitrides, but even more so, COFs are modular, versatile, and adaptive as they are characterized by an easy tunability of (opto)electronic properties, structure, crystallinity, and porosity.4,5 In addition, COFs are solely composed of light elements and thus have enormous prospects as earth-abundant and synthetically versatile platforms for modular, heterogeneous photocatalysis.2−6 The π-electron conjugation in-plane together with the possibility of axial charge transport in the stacking direction by the overlap of π-orbitals can result in high charge carrier mobilities, thus making COFs promising supramolecular architectures for efficient light harvesting and charge transport.7,8 Already, even with the very limited number of reports of H2 evolution with COFs, hydrogen evolution rates as high as 1700 μmol h–1 g–1 have been achieved.9−12 However, in all such studies platinum has been used as the co-catalyst to reduce the overpotential of H2 generation. Despite the excellent activity of metallic platinum, it is rare and expensive and should thus be replaced by earth-abundant, non-precious-metal-based co-catalysts in the long run.13−15 The combination of a COF as the molecularly defined photoabsorber with an earth-abundant molecular co-catalyst could provide a highly tunable, single-site heterogeneous photocatalytic platform which is fully accessible to the toolbox of organic synthesis. It would thus be an important stepping stone toward sustainable and inexpensive photocatalytic systems. However, development of such a system is challenging because of the limited photostability of molecular co-catalysts and generally slow multielectron diffusion-controlled proton reduction processes which need to be coupled efficiently to the light-harvesting and charge-percolation processes on the COF.
We report here, for the first time, light-induced proton reduction catalysis with COFs using cobaloximes as noble-metal-free molecular co-catalysts (Scheme 1). Efficient hydrogen evolution is seen with an azine-linked COF (N2) and a chloro(pyridine)cobaloxime co-catalyst (Co-1) in the presence of triethanolamine (TEOA) as a sacrificial electron donor in a water/acetonitrile mixture under AM 1.5 illumination. The methodology can also be extended to other azine- and hydrazone-based COFs and other cobaloximes as co-catalysts. The results lead way to the development of efficient and robust, noble-metal-free, single-site heterogenized systems for artificial photosynthesis that offer a precise control over the nature, density, and arrangement of the photocatalytically active sites.
Scheme 1. Structures of N2-COF and the Cobaloxime Co-Catalysts Used in This Study.

Schematic representation of photocatalytic H2 evolution with N2-COF and Co-1 is shown on the left.
Results and Discussion
Photocatalysis
The azine-based Nx-COFs were chosen as the photoabsorber, owing to their robustness and efficient hydrogen evolution activity with metallic platinum.9 All our primary investigations have been carried out with N2-COF (Scheme 1) because of a relatively easier synthesis protocol as compared to that of the most active member of the series, N3-COF.
Of the different transition metal based co-catalysts reported for proton transfer catalysis, cobalt complexes with dimethylglyoxime ligands, also known as cobaloximes, are among the most efficient. They feature low overpotentials for H2 generation, easy synthesis, and oxygen tolerance, and can be easily incorporated covalently into natural and artificial photocatalytic systems.14−16 Cobaloximes have been used as earth abundant molecular H2 evolution co-catalysts, e.g., with MOF17 and carbon nitride photosensitizers.18,19 We thus chose the complex chloro(pyridine)cobaloxime(III) (Co-1, Scheme 1) for our studies.
In a typical photocatalytic experiment, 5 mg of N2-COF was dispersed in 10 mL of 4:1 ACN/H2O solvent together with 100 μL of TEOA (0.075 M final concentration) as the sacrificial electron donor and 400 μL of a 2.48 mM solution of Co-1 in acetonitrile (0.1 mM final concentration). When irradiated with 100 mW/cm2 AM 1.5 radiation, the resulting mixture produces hydrogen actively at a rate of 160 μmol g–1 h–1 over a period of 7 h (Figure 1a) with a peak hydrogen as high as 701 μ mol g–1 corresponding to a turnover number (TON) of 3.54 (based on Co-1), after which the activity of the system levels off. An induction period of about 1.5 h is however seen at the onset, which possibly corresponds to the photogeneration of CoII and then finally CoI and CoIII–H and/or CoII–H species from the initial CoIII for H2 evolution to occur (vide infra).13−16,20−22 In control experiments without either the COF or TEOA, no H2 evolution was observed in a period of 3 h. The control experiment without Co-1 produced only 5 μmol g–1 H2 in 3 h. This implies that all the aforementioned three components are necessary for the photocatalytic system to work and that there is a charge transfer in the ensemble. The negative Gibbs free energy of the photoinduced electron transfer reaction (Table 1), from either the conduction band of N2-COF or the reduced radical anion species to either CoIII or to CoII calculated according to the Weller equation,23,24 suggests that electron transfer is thermodynamically feasible.
Figure 1.

(a) H2 evolution using N2-COF and Co-1 (see text for details) as well as N2-COF and metallic platinum (5 μL of 8 wt % H2PtCl6 solution in water) in the presence of TEOA, when irradiated with 100 mW cm–2 AM 1.5 light. Control experiments in absence of either of the three components, with all other conditions being the same, show no H2 evolution in 3 h. (b) H2 evolution using optimized parameters, 5 mg of N2-COF dispersed in 10 mL of 4:1 ACN/H2O solvent together with 100 μL of TEOA, 400 μL of a 2.48 mM solution of Co-1 in ACN, and 4.69 mM dmgH2 at a final pH of 8. The reaction mixture is illuminated with 100 mW cm–2 AM 1.5 light.
Table 1. Gibbs Free Energy of formation of CoII and CoI by Oxidative and Reductive Electron Transfer Pathwaysa.
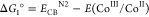
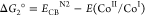
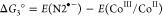
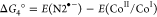
After photocatalysis, the COF sample was then fully characterized to check for any decomposition. The framework structure and crystallinity is fully retained after photocatalysis, as seen in the PXRD pattern of the post photocatalysis N2-COF sample (Figure S1). FTIR and ssNMR spectra (Figures 3 and S2) again remain unchanged, demonstrating that molecular connectivity and hence the structure of the COF remains intact after photocatalysis. SEM images evidence that the rod-like morphology of N2-COF is unchanged (Figure S3), and TEM images confirm retention of the hexagonally ordered crystalline domains after photocatalysis (Figure S4). Also, no trace of cobalt oxide or metallic cobalt was seen on the surface of the COF.
Figure 3.

(a) 13C CPMAS NMR spectra of N2-COF under different conditions. No change in chemical shift in the COF signals is seen. Please see Figure S12 for peak assignments. (b) ATR-IR spectra of N2-COF under different conditions. Again, no shift in the frequencies of the bands is seen.
We then tried to find the optimum working conditions for the hybrid photocatalytic system. Solvent variation was found to have a profound influence on H2 production.26 Different solvents have different coordination abilities for binding to cobalt; they have different polarities and dielectric constants which differently stabilize the reduction intermediates. Also, the solvent dependence of the CoII/CoI redox potential, and/or the reduction of the CoIII and/or CoII-hydride intermediate greatly affects the driving force for the H2 generation reaction. While in DMF/H2O 4:1, H2 evolution is seen at a rate of 22.6 μmol g–1 h–1; most efficient H2 evolution is seen with ACN/H2O 4:1 (160 μmol g–1 h–1) (Figure S5). H2 evolves at a rate of only 4.75 μmol g–1 h–1 in a THF/H2O 4:1 system. The ratio of ACN to H2O in the solvent was found to have an influence on the H2 evolution efficiency as well and the rate of hydrogen production increases when the ratio is increased from 2:3 to 3:2 and to finally 4:1 where it reaches a maximum (Figure S6). The induction period also seems to be somewhat shortened when using a higher ACN content.
As seen commonly for many H2 production systems, the pH of the reaction mixture was also found to have a profound influence on H2 evolution efficiency.20 The amount of H2 generated from the photochemical reaction is maximum at around pH 8. Significantly less H2 evolution is seen at lower pH values because TEOA is either protonated or else due to inhibition of proton loss from TEOA+.20 Likewise, very little H2 evolution is seen at pH 12 (Figure S7) because of the reduced thermodynamic driving force and because of protonation of the cobalt catalyst becoming greatly unfavorable.
Next, we varied the sacrificial donor. Triethylamine (TEA) as the electron donor led to significantly reduced hydrogen generation (17 μmol g–1 h–1) as compared to TEOA (160 μmol g–1 h–1; Figure S8). Interestingly, a TEOA concentration as low as 0.075 M led to the most efficient H2 production in our system. When [TEOA] was increased to 0.375 M, H2 evolution was reduced (110 μmol g–1 h–1), most likely as a result of an increase in pH.
Cobaloxime complexes are unstable because of the labile dimethylglyoxime ligands which undergo exchange with free dimethylglyoxime in solution.14,15,27 We thus added 8 equiv of dmgH2 to the photocatalytic reaction mixture when absolutely no further H2 evolution was seen with the initially added Co-1. H2 evolution duly renewed and continued for an additional 9 h at the rate 170 μmol g–1 h–1 in comparison to H2 evolution for only 6 h with a slightly lower rate of 150 μmol g–1 h–1 before dmgH2 addition (Figure S9). The improvement in the efficiency of H2 production with dimethylglyoxime led us to explore its use as the sacrificial electron donor, replacing TEOA, for long-term hydrogen evolution. With 0.05 M dimethylglyoxime (this is the limit of solubility of dmgH2 in 4:1 ACN/H2O solvent), H2 however evolves at an extremely poor rate of 0.63 μmol g–1 h–1 for 24 h after an initial induction period of about 3 h (Figure S10).
Crystallinity and porosity of the COF also seem to have an effect on the efficiency of H2 evolution. Poorly crystalline samples (with typically lower porosity) led to poorer H2 generation. This is most likely because of a smaller extension of the π-system in the less crystalline sample and/or stacking faults which could impede lateral and/or vertical charge carrier transport in the COF photosensitizer and likely also the interfacial charge transfer from the COF to the cobaloxime. We would also expect a less porous COF sample to impede accessibility to Co-1 and thus limit effective transfer of charges.
With all the above variables optimized, a H2 evolution rate of 782 μmol g–1 h–1 is achieved corresponding to a TON of 54.4 at 20 h (Figure 1b) and an initial TOF of 3.96 h–1. The amount of H2 evolved thus makes this system competitive with carbon nitride based benchmark photocatalytic systems such as Pt-modified amorphous melon (720 μmol g–1 h–1),28 g–C3N4 (840 μmol g–1 h–1),29 or crystalline poly(triazine imide) (864 μmol g–1 h–1).28 The TONs obtained are comparable to that obtained for a homogeneous photocatalytic system comprising of a Pt-terpyridyl acetylide chromophore and Co-1 co-catalyst in MeOH/H2O 3:2 (TON of 56).26 Even higher TONs may be attained in our system by adding dmgH2 periodically because, as shown above, the COF photosensitizer is quite stable under photocatalytic conditions. In fact, our previous report shows it to be stable for more than 120 h under photocatalytic conditions.9 The apparent quantum efficiency (AQE) in the present system was estimated to be 0.027% under AM 1.5 illumination. Under 400 nm irradiation, the AQE is estimated to be as high as 0.16%. To put this into perspective, the AQE of the photocatalytic reaction of the Ni bis(diphosphine) catalyst, NiP, in combination with the heptazine carbon nitride polymer melon in water is (0.04 ± 0.01) % using 460 nm irradiation.30
In order to further optimize the hydrogen evolution efficiency of N2-COF with cobaloximes, we tried to circumvent the instability of the dimethylglyoxime ligands. Indeed, a higher H2 evolution rate (414 μmol g–1 h–1) and a higher TON of 9.79 are obtained with the more stable BF2-annulated complex Co-2 as compared to that with Co-1 (160 μmol g–1 h–1, TON 3.54) under the same conditions (Figure 2a and Table S1).24 However, cobaloxime Co-3, despite the stable tetradentate diimine-dioxime ligand, produces very little hydrogen (20 μmol g–1 in 6 h).25 The low activity could arise from the difficulty of Co-3 to undergo protonation at the oxime moieties since they are linked covalently to the boron atom. This makes adjustment of the redox potentials to the acido-basic conditions of the reaction mixture difficult and thus probably disfavors proton reduction in this system.31 As compared to Co-2, which is also a BF2-annulated complex, H2 evolution with Co-3 is further hindered because of the single diimine dioxime ligand, whose other diimine end cannot be protonated. Interestingly, the H2 evolution efficiency of N2-COF with Co-1 (160 μmol g–1 h–1) is higher as compared to that in the presence of colloidal platinum (52 μmol g–1 h–1) (Figure 1a, the mol % of platinum being the same as Co-1). As seen from our previous report, photocatalytic hydrogen evolution with N2-COF in the presence of platinum takes place with much higher efficiency in water (438 μmol g–1 h–1).9 The lower H2 evolution efficiency of N2-COF in this report is thus probably a reflection of the choice of solvent (4:1 ACN/H2O, instead of pure water). TEM images of the post photocatalysis (with Pt in 4:1 ACN/H2O) N2-COF sample shows a distribution of ∼2 nm nanoparticles on the surface of the COF (Figure S11). Such distributions were however seen only in some areas. In comparison, well-distributed, though larger, nanoparticle clusters of 10–15 nm size were seen when the reaction was done in water where a significantly higher H2 evolution was observed. Thus, while smaller nanoparticles indeed form in 4:1 ACN/H2O and should make H2 evolution more efficient because of a higher availability of surface Pt atoms, the overall poorer distribution and/or poorer photodeposition of Pt nanoparticles in this solvent probably reverses the trend in H2 evolution reaction. The energetics of the charge transfer processes involved, in 4:1 ACN/H2O vs H2O, might also vary and could also contribute to the lower H2 evolution efficiency in the former solvent. A comparison between the activities of N2-COF with Co-1 and with platinum in 4:1 ACN/H2O is thus difficult.32Co-1 is insoluble in pure water, whereas Co-2 is soluble. However, no hydrogen evolution is seen with N2-COF in the presence of Co-2 in water.
Figure 2.

(a) H2 evolution with N2-COF and different co-catalysts. The co-catalyst concentration is 0.1 mM in all measurements. All other conditions are the same including a pH of 10. (b) H2 evolution with different COFs at pH 8. 5 mg COF sample has been used in all the measurements. All other conditions are the same. Rates are 233, 390, 163, and 100 μmol g–1 h–1 for COF-42, N2, N3 and N1 COFs, respectively. TON for the reaction with N2-COF is 10.89 at 6.5 h.
We also measured the activity of other COFs which are known to produce H2 photocatalytically with metallic platinum, namely, the azine-linked COFs N1 and N3, and the hydrazone linked COF-42 (Figure 2b, Schemes S1 and S2 and Table S2). With COFs N1 and N3, nonoptimized TONs of 2.03 and 5.65 could be obtained at pH 8 with Co-1 co-catalyst, respectively, while a TON of 5.79 was obtained with COF-42 under similar conditions. The reaction methodology can thus be extended to different types of COFs producing H2 under photocatalytic conditions.
Interestingly, the H2 evolution rate of N3-COF (163 μmol g–1 h–1) is lower than that of N2-COF (390 μmol g–1 h–1) with Co-1 at pH 8 in 4:1 ACN/H2O. This is contrary to our previously reported results with Pt co-catalyst in water where N3-COF was seen to be 4 times as active as N2-COF (1703 vs 438 μmol g–1 h–1, respectively).9 However, the H2 evolution rate of N3-COF (175 μmol g–1 h–1) with metallic Pt in 4:1 ACN/H2O is still about 3.5 times higher than that of N2-COF (52 μmol g–1 h–1) with Pt under the same conditions (Table S3). Therefore, the charge transfer processes between the COF and Co-1 seem to dictate the lower reaction rate of N3-COF with Co-1 as compared to N2-COF.
Outer versus Inner Sphere Electron Transfer
Cobaloximes, as discussed before, are known to be quite labile complexes, more so under photocatalytic conditions. The dimethylgloxime ligands as well as the axial pyridine ligands exchange readily and this limits the long-term usability of such catalysts.27 This ligand exchange could have far reaching implications in the present photocatalytic system in terms of what the actual proton reduction catalyst is or what way the electron is actually transferred from the COF photosensitizer to the cobalt center. The lability of the dimethylglyoxime ligands might lead to the formation of an entirely different H2 evolution catalyst, with the primary coordination sphere of cobalt being occupied by N atoms of the azine linkers (the N atoms of the pyrimidine nodes might be too sterically hindered to interact). However, this seems unlikely looking at the importance of the dimethylglyoxime ligands in keeping the catalyst active for proton reduction.13−15,26,27 A quite possible alternative could be axial coordination of a N atom of the azine linker to the cobalt center after the labile pyridine is lost. This would mean that the COF backbone forms a part of the coordination sphere of the co-catalyst and electrons are transferred from the COF photosensitizer to the catalyst in an inner sphere mechanism.33
In order to probe any interaction between N2-COF and Co-1, we recorded 13C cross-polarization magic angle spinning (CPMAS) NMR spectra of N2-COF post photocatalysis and found it absolutely identical to pristine N2-COF including the signal for the azine carbon at 162 ppm (Figures 3a and S12), thus suggesting no chemical interaction between the COF and Co-1. Neither peaks corresponding to Co-1 could be seen, nor were effects due to the presence of any paramagnetic cobalt species such as line broadening or loss of signal intensity observed. No interactions could again be seen in an illuminated and dried mixture of N2-COF and 8 or 35 wt % Co-1 in ACN. This time, while peaks corresponding to Co-1 are seen owing to higher amounts of Co-1 in the sample, the chemical shifts again remain unchanged. No interactions were observed in the 1H MAS NMR spectra as well (Figure S2). ATR-IR spectra of the COF sample before and after photocatalysis are again identical, including the ν(C=N)stretch appearing at 1620 cm–1, as is the IR spectrum of an illuminated and dried mixture of N2-COF and 8 wt % Co-1 in acetonitrile. In the latter sample, the new features arising can easily be assigned to Co-1 and the spectrum is simply additive (Figure 3b). Energy-dispersive X-ray (EDX) spectroscopic analysis in TEM shows no trace of cobalt in the post-photocatalysis sample (Figure S14). However, in the illuminated and dried mixture of N2-COF and 8 wt % Co-1, cobalt and chlorine can easily be detected (Figure S15). Also, the filtered, washed, and thus recovered N2-COF sample after photocatalysis does not produce any H2 in the presence of TEOA without Co-1, all other conditions being exactly the same as before. These results combined prove beyond doubt that (i) Co-1 rather than the photochemically decomposed metallic cobalt is the catalytically active species and (ii) that it does not chemically interact with N2-COF. Also, physisorption, if any, is weak enough for Co-1 to be washed away very easily with standard solvents.
Quantum-chemical calculations with Co-1 and model compounds further confirm this argument. Four different cobaloxime-COF composites were modeled in order to mimic possible binding sites of the cobalt co-catalyst to the framework (Figure 4 and Quantum-Chemical Calculations section). Two different cobaloximes with pyridine and ACN as the axial N donor ligands (Figures S16 and S17) were also modeled in order to compare cobalt–axial nitrogen bond lengths of these optimized compounds against the corresponding distances in cobaloxime-COF models, in order to estimate their binding strength. For cobaloxime-COF models, the shortest cobalt–nitrogen distance obtained is 2.79 Å for the surface-diazene cobaloxime-COF model (Table S5), which is still significantly larger than the longest cobalt–axial nitrogen bond distance of 1.96 Å observed among the modeled cobaloximes (Table S4). A distance-based approximation thus suggests that cobalt tends to form more stable complexes with its axial N donor ligands in the parent complexes, pyridine or ACN, than with a N center on the COF framework. Interaction energies were also calculated on PBE0-D3/def2-TZVP level of theory34−39 using the FermiONs++ program package40,41 and, as anticipated from the analysis of cobalt–nitrogen distances, all four COF-cobaloxime models, especially pore-diazene and pore-diazene-90°, were seen to be distinctly unfavored in comparison to the parent complexes with either pyridine, ACN or H2O as the axial ligands (Table S6). Combined experimental and quantum-chemical investigations thus refute the possibility of an inner sphere electron transfer from the COF to the co-catalyst via covalent interactions and suggest possibly an outer sphere collisional electron transfer mechanism.
Figure 4.

Constrained optimized geometry of (a) pore-diazene, (b) pore-diazene-90°, (c) surface-diazene, and (d) surface-triazine cobaloxime-COF models, obtained on the PBE0-D3/def2-SVP level of theory using the Turbomole program package.34−39 The surface-diazene and triazine models are for possible interactions on the surface of the COF microstructure. Other details of the calculations can be found in the Supporting Information. The dashed pink lines show the shortest Co–N distance obtained and are 4.197, 4.082, 2.792, and 3.00 Å, respectively, in panels a–d.
Mechanism
A general mechanism of proton reduction by cobalt complexes involves stepwise reduction of the CoIII complex to the resting state of the complex, CoII, then to CoI which is then protonated to form a CoIII hydride intermediate.14−16,20,22,27 A direct proton coupled electron transfer step from CoII to H–CoIII has also been proposed.21 Likewise, in the present COF-cobaloxime photocatalytic system, CoII and the CoI intermediates can actually be identified in the photolysis solutions owing to their unique spectroscopic signatures.
Prior to irradiation of the reaction mixture containing COF-42 as the photosensitizer and Co-1 as the co-catalyst, cobalt is only present in the +3 oxidation state and has no significant absorption in the visible region. After irradiation for 2 h at pH 8, an absorption band centered at 440 nm corresponding to CoII can be seen, and the reaction mixture is visibly dark yellow (Figure 5a).20,24,26,31,42,43 Measurements were impeded by the use of N2-COF, since the COF particles took an extraordinarily long time to settle down for us to be able to record an absorption spectrum of the supernate. This problem could be avoided with COF-42. We also recorded an X-band EPR spectrum of this photocatalytic reaction mixture before and after illumination and could observe formation of the one electron reduced paramagnetic CoII species with Lorentzian line broadening corresponding to geff = 2.006 (Figure 5b) as has been reported previously.17,18,44−46 Before illumination there seems to be a weak signal at geff = 2.058 possibly corresponding to paramagnetic impurities in the starting complex Co-1.17,18 At low pH the formation rate of CoI is itself very low. At high pH, H2 production is supposed to be greatly decreased and photoaccumulation of the CoI state should be possible.20 Nevertheless, our efforts to spectroscopically monitor the CoI state at pH 12 proved unsuccessful. However, a 4 h illumination of the reaction mixture at pH 10 with 5 equiv of added P(n-Bu)3 led to an intense blue color corresponding to an absorption band at 500–700 nm (Figure 5a). The blue color which disappears immediately upon air exposure can have three possible origins. It can be attributed to the phosphine coordinated CoI species,31,47 namely, [CoI(dmgH)2(P(n-Bu)3)]− or bridge protonated [CoI(dmgH)(dmgH2)(P(n-Bu)3)]. It could also be attributed to a solvent stabilized charge-transfer state of [CoIIIH(dmgH)2(P(n-Bu)3)], i.e., the H–CoIII species,47,48 as all of these have a similar absorption spectrum. However, an initially formed photoreduced CoI species uncoordinated to P(n-Bu)3 or the CoII-hydride species can safely be ruled out.20,21,24,26,31,42,43 P(n-Bu)3 is actually reported to increase the efficiency in some hydrogen evolving photocatalytic systems by stabilizing the aforesaid intermediate CoI state.31,49However, the fact that no hydrogen evolution is seen in our system with added P(n-Bu)3 makes us believe that it is the CoIII hydride [CoH(dmgH)2(P(n-Bu)3)], known to produce H2 only on thermolysis at 150 °C,50 which is actually formed.
Figure 5.

(a) Red trace: UV–vis spectra of the degassed photocatalytic reaction dispersion containing 2.5 mg of COF-42, 50 μL of TEOA and 200 μL of Co-1 (2.48 mM in ACN) in 5 mL 4:1 ACN/H2O mixture at pH 8 illuminated with 100 mW cm–2 AM 1.5 light. The reaction mixture was allowed to stand for 1 h after illumination before a spectrum was recorded. Blue trace: similar reaction conditions as before except at pH 10 of the reaction mixture and 5 equiv of externally added P(n-Bu)3. The noise in the spectra is from the still suspended COF particles. (b) X-band EPR spectrum at 4K of the photocatalytic reaction dispersion containing COF-42 before and after illumination. The microwave frequencies are 9.47614 GHz in both cases. The reaction conditions are identical to those in Figure 5a. (c) H2 evolution at 3 h after illumination under different [Co-1]. In all measurements, 5 mg of N2-COF and 100 μL of TEOA in 10 mL of 4:1 ACN/H2O has been used. The reaction pH is 8.
The cobaltIII and/or cobaltII hydride formed in the reaction mixture can produce hydrogen by either a homolytic/bimetallic pathway involving two cobalt centers or a kinetically distinguishable heterolytic/monometallic pathway involving a single cobalt center.14−16,26,51,52 In order to distinguish between these two pathways for the present photocatalytic system, we studied the amount of hydrogen evolved for different concentrations of Co-1, while keeping all other conditions the same. From Figure 5c it can be seen that H2 evolution after 3 h of photolysis exhibits a linear dependence on [Co-1], thus supporting a single cobalt mechanism for hydrogen generation (Scheme S4).14,26,52
The other reversible cycle, i.e., the photochemical COF cycle, can proceed along either oxidative or reductive quenching of the COF upon photoexcitation. Our previously published theoretical studies on the Nx-COFs show that the formation of a radical cation intermediate during the photocatalytic cycle is less likely for these COFs for energetic reasons.9 In fact, a radical anionic state has been identified in an ongoing experimental study. This speculation however does not undermine the importance of a correct identification of the reaction pathway adopted in our COF-cobaloxime photocatalytic system. Detailed transient absorption measurements are underway in this regard and will be reported elsewhere.
We also tried to explore the charge transfer pathways in our photocatalytic system by photoluminescence measurements. Unfortunately, N2-COF is scarcely emissive; thus, it was not possible to collect reproducible emission spectra or quantum yields of the photolysis dispersions to check whether Co-1 or TEOA quench emission. Measurements were further impeded by simultaneous absorption of Co-1. Photoluminescence lifetimes recorded using time-correlated single-photon counting method (TCSPC), however, show almost no change in the decay of N2-COF in the presence of either TEOA, Co-1 or both (Figure S23 and Table S8), which probably suggests a different time scale of the electron transfer process from TEOA and to Co-1 under these conditions.53
Conclusions and Outlook
Photocatalytic hydrogen evolution with COF photosensitizers using molecular, earth-abundant co-catalysts has been demonstrated with large H2 evolution rates and good TONs, as exemplified with the Nx-COF series and COF-42 with Co-1 and other cobaloximes. No external proton source is required for H2 evolution. Metallic cobalt, which could possibly form by photodecomposition of Co-1, could be ruled out as the hydrogen evolving co-catalyst; Co-1 in solution thus acts as the proton reduction catalyst transferring reducing equivalents from the photosensitizer to the protons. Experimental results and quantum-chemical calculations suggest an outer sphere electron transfer from N2-COF to co-catalyst Co-1 and a monometallic, i.e., a single cobalt pathway was identified in the present system for H2 generation from the intermediate CoIII- and/or CoII-hydride. Long-term stability needs to be further addressed with other more stable and efficient H2 evolving co-catalysts or by engineering the COF so as to prevent ligand dissociation.17 A possible improvement of the H2 evolution efficiency by optimizing the electron transfer process between the COF and the co-catalyst by covalently linking the molecular co-catalyst to the COF backbone also needs to be explored and is currently underway.
To conclude, it is important to understand the implications of the results presented in this article. The quest for earth abundant molecular replacements of co-catalyst platinum for photocatalytic H2 evolution using COF photosensitizers is a big challenge because: (i) COFs that produce H2 photocatalytically are themselves rare. A number of factors come into play such as crystallinity, porosity, rigidity, and stability on the one hand and light harvesting, charge separation/recombination, and charge transport on the other, which have to be retained throughout the course of the photocatalytic reaction. (ii) Electron transfer from the COF to the co-catalyst has to be thermodynamically and kinetically favorable. There needs to be an efficient coupling of single-photon electron events with the multielectron redox reactions necessary for H2 evolution. (iii) Molecular co-catalysts, unlike metallic platinum, possess limited photostability and could have slow multielectron diffusion controlled rates.
The observation of photocatalytic H2 evolution from COFs with molecular cobaloxime based co-catalysts is thus the first step in overcoming these challenges. The results presented herein lead way to the development of efficient “COF-molecular co-catalyst” based photocatalytic systems entirely free of noble metals which, with the robustness and tunability of the COF backbone, enables a precise control over the nature, the arrangement and the density of photocatalytically active sites for optimal competence.54 The results show that water splitting or CO2 reduction catalysts could be combined with COF-based light-harvesting systems in a “leaf”-like architecture for stable generation of solar fuels in future. While covalently linked COF-co-catalyst architectures could be envisaged for better performance, our results also show that design and optimization of the COF photosensitizer and the co-catalyst as independent components is another worthwhile avenue.
Acknowledgments
B.V.L. acknowledges financial support by an ERC Starting Grant (project COF Leaf, grant number 639233), the Max Planck Society, the cluster of excellence Nanosystems Initiative Munich (NIM), and the Center for Nanoscience (CeNS). We thank Viola Duppel for recording the SEM and TEM images, Igor Moudrakovski for the measurement of the solid-state NMR, Marie-Luise Schreiber for recording the ATR-IR spectra, and Prof. Reinhard Kremer for recording the EPR spectra.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07489.
Structures of COFs used, synthesis and methods, variation of H2 evolution rates under different conditions, post photocatalysis characterization, ΔG values for electron transfer between COF and co-catalysts, details of quantum-chemical calculations, cobaloxime catalytic cycle and TCSPC results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tachibana A. Y.; Vayssieres L.; Durrant J. R. Nat. Photonics 2012, 6, 511–518. 10.1038/nphoton.2012.175. [DOI] [Google Scholar]; and references therein.
- Berardi S.; Drouet S.; Francàs L.; Gimbert-Suriňach C.; Guttentag M.; Richmond C.; Stoll T.; Llobet A. Chem. Soc. Rev. 2014, 43, 7501–7519. 10.1039/C3CS60405E. [DOI] [PubMed] [Google Scholar]
- Vyas V. S.; Lau V. W.; Lotsch B. V. Chem. Mater. 2016, 28, 5191–5204. 10.1021/acs.chemmater.6b01894. [DOI] [Google Scholar]
- Ding S.-Y.; Wang W. Chem. Soc. Rev. 2013, 42, 548–568. 10.1039/C2CS35072F. [DOI] [PubMed] [Google Scholar]
- Bisbey R. P.; Dichtel W. R. ACS Cent. Sci. 2017, 3, 533–543. 10.1021/acscentsci.7b00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N.; Wang P.; Jiang D. Nat. Rev. Mater. 2016, 1, 16068. 10.1038/natrevmats.2016.68. [DOI] [Google Scholar]
- Ding X.; Guo J.; Feng X.; Honsho Y.; Guo J.; Seki S.; Maitarad P.; Saeki A.; Nagase S.; Jiang D. Angew. Chem., Int. Ed. 2011, 50, 1289–1293. 10.1002/anie.201005919. [DOI] [PubMed] [Google Scholar]
- Butchosa C.; McDonald T. O.; Cooper A. I.; Adams D. J.; Zwijnenburg M. A. J. Phys. Chem. C 2014, 118, 4314–4324. 10.1021/jp411854f. [DOI] [Google Scholar]
- Vyas V. S.; Haase F.; Stegbauer L.; Savasci G.; Podjaski F.; Ochsenfeld C.; Lotsch B. V. Nat. Commun. 2015, 6, 8508. 10.1038/ncomms9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegbauer L.; Schwinghammer K.; Lotsch B. V. Chem. Sci. 2014, 5, 2789–2793. 10.1039/C4SC00016A. [DOI] [Google Scholar]
- Haase F.; Banerjee T.; Savasci G.; Ochsenfeld C.; Lotsch B. V. Faraday Discuss. 2017, 201, 247–264. 10.1039/C7FD00051K. [DOI] [PubMed] [Google Scholar]
- Thote J.; Aiyappa H. B.; Deshpande A.; Díaz Díaz D. D.; Kurungot S.; Banerjee R. Chem. - Eur. J. 2014, 20, 15961–15965. 10.1002/chem.201403800. [DOI] [PubMed] [Google Scholar]
- Du A. P.; Eisenberg R. Energy Environ. Sci. 2012, 5, 6012–6021. 10.1039/c2ee03250c. [DOI] [Google Scholar]
- Artero V.; Chavarot-Kerlidou M.; Fontecave M. Angew. Chem., Int. Ed. 2011, 50, 7238–7266. 10.1002/anie.201007987. [DOI] [PubMed] [Google Scholar]
- Eckenhoff W. T.; McNamara W. R.; Du P.; Eisenberg R. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 958–973. 10.1016/j.bbabio.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Dempsey J. l.; Brunschwig B. S.; Winkler J. R.; Gray H. B. Acc. Chem. Res. 2009, 42, 1995–2004. 10.1021/ar900253e. [DOI] [PubMed] [Google Scholar]
- Nasalevich M. A.; Becker R.; Ramos-Fernandez E. V.; Castellanos S.; Veber S. L.; Fedin M. V.; Kapteijn F.; Reek J. N. H.; van der Vlugt J. I.; Gascon J. Energy Environ. Sci. 2015, 8, 364–375. 10.1039/C4EE02853H. [DOI] [Google Scholar]
- Gao L.-F.; Zhu Z.-Y.; Feng W.-S.; Wang Q.; Zhang H.-L. J. Phys. Chem. C 2016, 120, 28456–28462. 10.1021/acs.jpcc.6b09767. [DOI] [Google Scholar]
- Cao S.-W.; Liu X.-F.; Yuan Y.-P.; Zhang Z.-Y.; Fang J.; Loo S. C. J.; Barber J.; Sum T. C.; Xue C. Phys. Chem. Chem. Phys. 2013, 15, 18363–18366. 10.1039/c3cp53350f. [DOI] [PubMed] [Google Scholar]
- Du P.; Knowles K.; Eisenberg R. J. Am. Chem. Soc. 2008, 130, 12576–12577. 10.1021/ja804650g. [DOI] [PubMed] [Google Scholar]
- Muckerman J. T.; Fujita E. Chem. Commun. 2011, 47, 12456–12458. 10.1039/c1cc15330g. [DOI] [PubMed] [Google Scholar]
- Solis B. H.; Hammes-Schiffer S. Inorg. Chem. 2011, 50, 11252–11262. 10.1021/ic201842v. [DOI] [PubMed] [Google Scholar]
- Kavarnos G. J.; Turro N. J. Chem. Rev. 1986, 86, 401–449. 10.1021/cr00072a005. [DOI] [Google Scholar]
- Zhang P.; Wang M.; Dong J.; Li X.; Wang F.; Wu L.; Sun L. J. Phys. Chem. C 2010, 114, 15868–15874. 10.1021/jp106512a. [DOI] [Google Scholar]
- Jacques P. – A.; Artero V.; Pécaut J.; Fontecave M. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 20627–20632. 10.1073/pnas.0907775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P.; Schneider J.; Luo G.; Brennessel W. W.; Eisenberg R. Inorg. Chem. 2009, 48, 4952–4962. 10.1021/ic900389z. [DOI] [PubMed] [Google Scholar]
- McCormick T. M.; Han Z.; Weinberg D. J.; Brennessel W. W.; Holland P. L.; Eisenberg R. Inorg. Chem. 2011, 50, 10660–10666. 10.1021/ic2010166. [DOI] [PubMed] [Google Scholar]
- Schwinghammer K.; Tuffy B.; Mesch M. B.; Wirnhier E.; Martineau C.; Taulelle F.; Schnick W.; Senker J.; Lotsch B. V. Angew. Chem., Int. Ed. 2013, 52, 2435–2439. 10.1002/anie.201206817. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen X.; Takanabe K.; Maeda K.; Domen K.; Epping J. D.; Fu X.; Antonietti M.; Wang X. Angew. Chem., Int. Ed. 2010, 49, 441–444. 10.1002/anie.200903886. [DOI] [PubMed] [Google Scholar]
- Caputo C. A.; Gross M. A.; Lau V. W.; Cavazza C.; Lotsch B. V.; Reisner E. Angew. Chem., Int. Ed. 2014, 53, 11538–11542. 10.1002/anie.201406811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Jacques P. – A.; Chavarot-Kerlidou M.; Wang M.; Sun L.; Fontecave M.; Artero V. Inorg. Chem. 2012, 51, 2115–2120. 10.1021/ic2019132. [DOI] [PubMed] [Google Scholar]
- It needs to be mentioned that a comparison between the activities with a co-catalyst in solution versus photodeposited metallic platinum is not reasonable in any case, even in the absence of all the factors discussed above. This is because only a fraction of atoms on the surface of the nanoparticles are photocatalytically active; the rest, in the bulk of the nanoparticles, are inactive.
- Balzani V., Ed. Electron Transfer in Chemistry; WILEY-VCH Verlag GmbH: Weinheim, 2008. [Google Scholar]
- Adamo C.; Barone V. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Ernzerhof M.; Scuseria G. E. J. Chem. Phys. 1999, 110, 5029–5036. 10.1063/1.478401. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg S. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. J. Chem. Phys. 1992, 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Ahlrichs R.; Bär M.; Häser M.; Horn H.; Kölmel C. Chem. Phys. Lett. 1989, 162, 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. J. Chem. Phys. 1994, 100, 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Kussmann J.; Ochsenfeld C. J. Chem. Phys. 2013, 138, 134114. 10.1063/1.4796441. [DOI] [PubMed] [Google Scholar]
- Kussmann J.; Ochsenfeld C. J. Chem. Theory Comput. 2015, 11, 918–922. 10.1021/ct501189u. [DOI] [PubMed] [Google Scholar]
- Hu X.; Brunschwig B. S.; Peters J. C. J. Am. Chem. Soc. 2007, 129, 8988–8998. 10.1021/ja067876b. [DOI] [PubMed] [Google Scholar]
- Lazarides T.; McCormick T.; Du P.; Luo G.; Lindley B.; Eisenberg R. J. Am. Chem. Soc. 2009, 131, 9192–9194. 10.1021/ja903044n. [DOI] [PubMed] [Google Scholar]
- Baumgarten M.; Lubitz W.; Winscom C. J. Chem. Phys. Lett. 1987, 133, 102–108. 10.1016/0009-2614(87)87029-X. [DOI] [Google Scholar]
- Cropek D. M.; Metz A.; Müller A. M.; Gray H. B.; Horne T.; Horton D. C.; Poluektov O.; Tiede D. M.; Weber R. T.; Jarrett W. L.; Phillips J. D.; Holder A. A. Dalton Trans. 2012, 41, 13060–13073. 10.1039/c2dt30309d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubitz W.; Winscom C. J.; Diegruber H.; Möseler R. Z. Naturforsch., A: Phys. Sci. 1987, 42, 970–986. 10.1515/zna-1987-0910. [DOI] [Google Scholar]
- Bhattacharjee A.; Chavarot-Kerlidou M.; Andreiadis E. S.; Fontecave M.; Field M. J.; Artero V. Inorg. Chem. 2012, 51, 7087–7093. 10.1021/ic2024204. [DOI] [PubMed] [Google Scholar]
- Szajna-Fuller E.; Bakac A. Eur. J. Inorg. Chem. 2010, 2010, 2488–2494. 10.1002/ejic.200901176. [DOI] [Google Scholar]
- Hawecker J.; Lehn J. M.; Ziessel R. New J. Chem. 1983, 7, 271–277. [Google Scholar]
- Schrauzer G. N.; Holland R. J. J. Am. Chem. Soc. 1971, 93, 1505–1506. 10.1021/ja00735a040. [DOI] [PubMed] [Google Scholar]
- Wen F.; Yang J.; Zong X.; Ma B.; Wang D.; Li C. J. Catal. 2011, 281, 318–324. 10.1016/j.jcat.2011.05.015. [DOI] [Google Scholar]
- Fihri A.; Artero V.; Pereira A.; Fontecave M. Dalton Trans. 2008, 5567–5569. 10.1039/b812605b. [DOI] [PubMed] [Google Scholar]
- Lau V. W.; Yu V. W.; Ehrat F.; Botari T.; Moudrakovski I.; Simon T.; Duppel V.; Medina E.; Stolarczyk J. K.; Feldmann J.; Blum V.; Lotsch B. V. Adv. Energy Mater. 2017, 7, 1602251. 10.1002/aenm.201602251. [DOI] [Google Scholar]
- Diercks C. S.; Yaghi O. M. Science 2017, 355, eaal1585. 10.1126/science.aal1585. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.