

Abstract
Group X (GX) and group V (GV) secretory phospholipase A2 (sPLA2) potently release arachidonic acid (AA) from the plasma membrane of intact cells. We previously demonstrated that GX sPLA2 negatively regulates glucose-stimulated insulin secretion (GSIS) by a prostaglandin E2 (PGE2)-dependent mechanism. In this study we investigated whether GV sPLA2 similarly regulates GSIS. GSIS was significantly decreased in islets isolated from GV sPLA2-deficient (GV KO) mice compared to wild-type (WT) mice. Similarly, GSIS was significantly decreased in MIN6 cells, a murine pancreatic beta cell line with siRNA-mediated GV sPLA2 suppression. MIN6 cells overexpressing GV sPLA2 (MIN6-GV) showed a significant increase in GSIS compared to control cells. The amount of AA released into the media by MIN6-GV cells was significantly higher compared to control cells. However, MIN6-GV cells did not exhibit enhanced PGE2 production or decreased cAMP content compared to control MIN6 cells. Surprisingly, GV KO mice exhibited a significant increase in plasma insulin levels following i.p. injection of glucose compared to WT mice. This increase in GSIS in GV KO mice was associated with a significant increase in pancreatic islet size and number of proliferating cells in β-islets compared to WT mice. Thus, deficiency of GV sPLA2 results in diminished GSIS in isolated pancreatic beta-cells. However, the reduced GSIS in islets lacking GV sPLA2 appears to be compensated by increased islet mass in GV KO mice.
Keywords: GV sPLA2, type 2 diabetes, insulin secretion, Arachidonic acid, β-islet mass
Introduction
Type 2 diabetes (T2D) is increasing at an alarming rate worldwide, largely due to an epidemic of obesity and reduced physical activity. T2D results from chronic insulin resistance that ultimately leads to a progressive decline in pancreatic β-cell function. Although insulin resistance precedes the development of hyperglycemia in subjects that eventually develop T2D, the disease manifests itself in insulin-resistant subjects only with the onset of β-cell dysfunction and reduced β-islet mass [1].
Accumulating evidence indicates that arachidonic acid (AA) and its metabolites play key roles in regulating β-cell function. AA constitutes >30% of the glycerolipid fatty acid mass in rodent islets [2], and stimulation of β-cells with glucose is accompanied by release of free AA. Inhibition of AA release impairs glucose-stimulated insulin secretion (GSIS) [3]. The exact mechanisms linking AA and insulin secretion are not clearly understood. While AA is in general considered to be an activator of GSIS, a major metabolite of AA, prostaglandin E2 (PGE2), is a known inhibitor of GSIS [4-9]. It is believed that PGE2 exerts its effect by interacting with its receptor, EP3, thus decreasing adenylyl cyclase activity with a subsequent reduction in cAMP, a known activator of GSIS [10, 11]. Apart from their effects on GSIS, AA and its metabolites have also been implicated in the regulation of β-cell mass. For example, transgenic overexpression of cyclooxygenase-2 (COX-2), a rate-limiting enzyme for prostanoid biosynthesis including PGE2, resulted in significantly decreased pancreatic β-cell number in mice, leading to a significant increase in blood glucose levels. The reduction in β-cell mass in COX-2 transgenic mice was shown to be due to inhibited β-cell proliferation [12].
The phospholipase A2 (PLA2) family comprises a group of intracellular and secreted enzymes that hydrolyze phospholipids to yield free fatty acids and lysophosholipids. This reaction is considered to be the initial, rate-limiting step of AA metabolism leading to the production of bioactive lipids including prostaglandins and leukotrienes. Calcium-independent PLA2 (iPLA2) enzymes have been implicated in glucose-stimulated AA release. Moreover, inhibition of iPLA2β using a pharmacological inhibitor, bromoenol lactone suicide substrate, inhibited AA release and GSIS in vitro [3, 13]. Similarly, knockdown of iPLA2β expression in INS1 cells decreased insulin secretion [14]. Transgenic overexpression of iPLA2β in islet β-cells resulted in enhanced GSIS; consistently, islets from male iPLA2β -null mice exhibited blunted insulin secretion. The role of cytosolic phospholipase A2 (cPLA2) in insulin secretion has also been investigated. Overexpression of cPLA2 enhances exocytosis of insulin from β-cells [15]. However, sustained expression of cPLA2 has been reported to upregulate uncoupling protein-2 (UCP-2) and consequent mitochondrial uncoupling, which drastically reduces the capacity of β-cells to respond to nutrients [16]. Consistently, another study documented that overexpression of cPLA2 has no effect on insulin content or the basal rate of insulin secretion, yet negatively effects GSIS, probably due to the prolonged exposure of β-cells to AA [17].
Among the secretory PLA2 (sPLA2) family members, group IB sPLA2 is reportedly expressed in pancreatic islets of rodents and localized within insulin granules. Stimulation with glucose results in the co-secretion of insulin and group IB sPLA2 [18]. However, the role of the enzyme in GSIS or pancreatic β-cell function is not known. Recently, we reported that another member of the sPLA2 family, group X sPLA2 (GX sPLA2) is expressed in insulin-producing cells of mouse pancreatic islets and negatively regulates GSIS by enhancing the production of PGE2. Moreover, C57BL/6 mice deficient in GX sPLA2 had increased plasma insulin levels following glucose challenge compared to wild-type mice [19].
Thus, based on available evidence, it appears that during glucose-stimulation, the liberation of AA positively modulates insulin secretion, whereas the generation of AA metabolites such as PGE2 negatively modulates insulin secretion. Therefore, the regulatory effect of individual PLA2's likely depends on the potency and kinetics of AA/PGE2 production. In this study, we investigated the role of yet another member of the sPLA2 family, group V sPLA2 (GV sPLA2) in pancreatic β-cell function. In vitro studies using artificial phospholipid substrates indicate that GV and GX sPLA2s are the most potent in hydrolyzing phosphatidylcholine [20], the major phospholipid on mammalian cell membranes. However, it is not clear whether these two closely related sPLA2 family members perform redundant physiological functions in vivo.
Materials and Methods
Biochemical Reagents and Assays
Assays for Insulin (Crystal Chem Inc), PGE2 metabolites (Cayman) and cAMP (ENZO Life Sciences) were performed according to the manufacturers' instructions. Phospholipase activity in conditioned media was measured using a colorimetric assay as we previously described [21] with 1-palmitoyl-2-oleoylphosphatidylglycerol (POPG; Matreya LLC) as a substrate. Briefly, mixed micelles were prepared by warming 7 mg of POPG to 37°C in a 0.2 mL mixture of 4.0% (wt/vol) Nonidet-40 and 2.0% sodium deoxycholate, and then adding 1.8 mL warm assay buffer (0.12 mol/L Tris-HCl pH 8, 12 mmol/L CaCl2, 0.1 mmol/L EDTA). For enzyme assays, 10 μL of conditioned media was added to 40 μL of substrate solution. After incubating for 20 minutes at 37°C, the amount of free fatty acids (FFA) released was quantified using a NEFA-C kit (Wako Chemicals); phospholipase activity was calculated as the nmol of FFA released in 20 minutes per mg cell protein.
Animals
GV sPLA2-deficient (GV KO) mice, backcrossed >10 times with C57BL/6 mice, were originally provided by Dr. Jonathan Arm [22]. Mice were maintained on a 10-h light/14-h dark cycle and received standard mouse chow and water ad libitum. Male mice were used throughout the study. All procedures were in accordance with the guidelines of the University of Kentucky Institutional Animal Care and Use Committees.
Islet isolation
Mouse islets were isolated via intraductal collagenase (Sigma) digestion and ficoll gradient centrifugation as described earlier [19]. A detailed method will be provided on request. Following isolation, similar-sized islets were selected and hand-picked and maintained in RPMI containing 5 mM glucose, 10% (v/v) FBS and penicillin and streptomycin.
Immunohistochemistry
Pancreata from WT and GV KO mice were embedded in paraffin and 5 μm-thick sections were mounted on glass slides; processing of the tissues on glass slides was done as described earlier [19]. The sections were immunostained using rabbit anti-mouse GV sPLA2 (gift from Dr. M. Gelb, University of Washington) and goat anti-mouse insulin (Santa Cruz Biotechnology), at a dilution of 1:100 for anti-GV sPLA2 and 1:500 for anti-insulin. For fluorescent images, Alexa Fluor-conjugated secondary antibodies were used (Invitrogen). For Ki67 staining, the sections were immunostained using rabbit anti-Ki67 (Abcam,1:150).
β-cell mass, average islet size, and β-cell proliferation
β-cell mass and average islet size were determined as described earlier [23]. Entire pancreata were removed from 4 GV KO and 4 WT mice (16 weeks old), adhering fat tissues as well as other nonpancreatic tissues were removed, and the organ was then weighed and fixed as described above and cut into 5 μm-thick sections. Every 30th section (a total of 7-8 sections per pancreas) was immunostained for insulin, and then imaged under X10 magnification using NIS elements software (Nikon Instruments, Inc.). β-cell mass was calculated by first obtaining the fraction of the total cross-sectional area of the pancreatic tissue immunopositive for insulin and then multiplying the pancreatic weight by this fraction. Average islet size was calculated by dividing the total islet area by the total number of islets analyzed.
The number of nuclei positive for Ki67 within insulin-positive cells was counted to determine β-cell proliferation. Approximately 40 islets from 2 sections per mouse were analyzed in the proliferation assay (n= 3 mice per group).
In vivo GSIS, glucose tolerance and insulin tolerance tests
In vivo GSIS was performed as described earlier [19]. Mice were fasted for 16 h, and then plasma samples were collected from the retro-orbital sinus before and 15 min after i.p. glucose injection (3g/kg). For glucose tolerance tests, mice were fasted for 6 h. Blood glucose concentrations were quantified using a glucometer (Contour; Bayer Laboratories) immediately before and 15, 30, 60, 90, and 120 min following intraperitoneal (i.p) administration of glucose (1.5 g/kg body wt). Insulin tolerance was assessed following a 4-h fast by quantifying blood glucose concentrations at 0, 30, 60, 90 and 120 min after administration of human insulin (0.5 U/kg body wt i.p; Novolin, Novo Nordisk).
Cell culture and transfections
MIN6 cells were cultured in complete media (DMEM supplemented with 15% heat-inactivated fetal bovine serum, 2 mmol/l L-glutamine, 45 mmol/l β-mercaptoethanol, 100 units/ml pencillin and 100 μg/ml streptomycin). C-terminal Flag-tagged cDNA for GV sPLA2 was constructed by PCR using forward (F) and reverse (R) primers containing Hind III and EcoRI restriction sites respectively: 5′- TACCCAAGCTTATGAAGGGTCTCCTCACA-3′(F) and 5′- GCGGAATTCTTACTTGTCATCGTCGTCCTTGTAGTCGCAGAGGAAGTTGGG-3′ (R) and mouse GV sPLA2 cDNA as a template. The PCR product was inserted into the mammalian expression vector pcDNA 3.0 (Invitrogen, Carlsbad, CA) to yield a coding sequence that expressed GV sPLA2 with a C-terminal FLAG epitope tag. DNA sequencing was performed to confirm the integrity of the expression construct. The plasmids were transfected into MIN6 cells using Nucleofector Kit following the manufacturer's instructions and the program T-016 (Nucleofector Kit V; Lonza).
Arachidonic acid (AA) release assay
MIN6 cells transiently transfected with either the “empty” pcDNA 3.0 plasmid (MIN6-C) or the pcDNA3.0 GV sPLA2 expression construct (MIN6-GV) were assayed 24 h after transfection. The transfected cells (∼5×105cells/well pf 12-well plate) were incubated in 1 ml of complete medium (DMEM medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin) containing 0.1 μCi [3H]arachidonate (200 Ci/mmol; American Radiochemicals Inc, Saint Louis, MO) for 12-16 h. The cells were then washed three times with complete medium followed by 6 h incubation in the same medium. The amount of tritium associated with cells and released into the medium was determined by scintillation counting.
Gene silencing with Small Interfering RNA (siRNA)
A set of pre-designed synthetic oligonucleotides directed to mouse GV sPLA2 (Thermo Scientific, On-TARGETplus SMARTpool siRNA; L-042189-01-0010; mouse PLA2G5 18784) was used. The oligonucleotides were transfected into MIN6 cells using Nucleofector Kit (Nucleofector Kit V; Lonza) and the program T-016. Scrambled siRNA (Thermo Scientific; Non-targeting siRNA#1; D-001810-20) was used as control. The siRNAs were transfected at a final concentration of 2 μg of siRNA pool or non-targeting siRNA per 2 × 106 MIN6 cells. Cells were collected 24 h after transfection for RNA preparation to confirm gene silencing by real-time RT-PCR and used for GSIS assay 48 h after transfection.
Real time RT-PCR
Total RNA was prepared from MIN6 cells using the RNeasy Mini kit (Promega). Quantification was performed in duplicate using the standard curve method and normalized to 18S RNA. The primer sequences used for GV sPLA2 mRNA detection were 5′-AGG GGG CTT GCT AGA ACT CA -3′ (F) and 5′-CAA TCA GTG CCA TCC TTG G -3′ (R).
In vitro insulin secretion assays
In vitro GSIS assays were carried out as described earlier [19]. Briefly, after isolation and handpicking, islets from WT and GV KO mice were cultured overnight in RPMI media. Islets were then selected and transferred to culture inserts (Greiner bio-one) in 12-well plates (25 islets per well), washed, and equilibrated for 1 h in Buffer 1 (DMEM supplemented with 38 mM sodium bicarbonate, 4 mM L-glutamine, 1 mM sodium pyruvate, 4.65 mM HEPES and 1 g/l BSA) containing 5 mM glucose, and then incubated successively for 40 min in Buffer 1 containing 5 mM glucose (low glucose) followed by 40 min in Buffer 1 containing 20 mM glucose (high glucose). At the end of the incubations, insulin content in the low and high glucose-containing buffers was assayed and normalized to total cellular insulin content, which was determined after lysing islets in acid-ethanol (75% ethanol, 0.2 mol/l HCl). To ensure that islets from the two strains of mice were challenged sufficiently to detect differences in response to glucose, we performed additional in vitro GSIS assays in which islets were equilibrated for 1 h in Buffer 1 containing low glucose (1 mM) and then incubated successively for 40 min in Buffer 1 containing physiological glucose (5 mM) followed by 40 min in Buffer 1 containing glucose (20 mM). The impact of GV sPLA2 on GSIS was not different under the two pre-equilibration conditions (data not shown). For GSIS in MIN6-C and MIN6-GV cells, cells (70-80% confluent) in 24-well plates were washed once with Kreb's Ringer buffer containing 0.2% BSA (KRB-BSA) and 5 mM glucose and then equilibrated in the same buffer for 1 h. The media was replaced with fresh KRB-BSA supplemented with either 5 mM (low) glucose or 20 mM (high) glucose for 40 min. Insulin concentrations in conditioned media were normalized to total cell protein.
Statistics
Data are expressed as mean ± SEM. Results were analyzed by t test or by 1-way ANOVA followed by a Bonferroni post-test. P<0.05 was considered statistically significant. All statistical analyses were carried out using GraphPad Prism 4.
Results
GV sPLA2 is expressed in mouse pancreatic islets
Recently, we reported that group X secretory phospholipase A2 (GX sPLA2) is expressed in pancreatic β-islet cells and negatively regulates insulin secretion through a mechanism mediated by cyclooxygenase-2-dependent prostaglandin E2 production [19]. In this study, we investigated whether a closely related member of the sPLA2 family, group V sPLA2, is expressed and functional in mouse β-islet cells. Double immunofluorescent staining of pancreatic sections from C57BL/6 mice showed positive immunoreactivity for GV sPLA2 (Fig. 1; green staining) that was most intense in regions immunopositive for insulin (red staining), indicating that GV sPLA2 is expressed in insulin-producing β-cells in mouse pancreas (Fig. 1; yellow on merged image). As expected, GV sPLA2 immunoreactivity was not detected in pancreatic sections from GV KO mice (Fig. 1).
Fig 1.
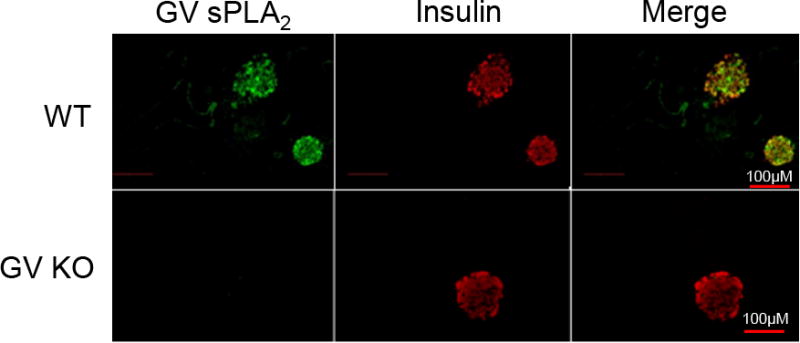
GV sPLA2 is expressed in mouse pancreatic islet cells. Pancreatic sections from WT and GV KO mice were co-stained by indirect immunofluorescence for GV sPLA2 (green) and insulin (red) and visualized (20 × magnification) by fluorescence microscopy. A merged image shows expression of GV sPLA2 in insulin-producing cells of WT pancreas (yellow).
GV sPLA2 augments glucose-stimulated insulin secretion (GSIS) in mouse pancreatic β-cell line
GV sPLA2 is known to potentiate the release of arachidonic acid (AA) leading to eicosanoid generation, including PGE2, in several cell types [22, 24]. PGE2 is a negative regulator of glucose-stimulated insulin secretion (GSIS), whereas non-esterified AA is an activator of GSIS [6, 8, 25-28]. Therefore, to assess whether GV sPLA2 regulates GSIS, we investigated the impact of forced overexpression in MIN6 cells, a mouse pancreatic β-cell line that expresses endogenous GV sPLA2 (data not shown). Transient transfection of GV sPLA2 cDNA resulted in a 1.8-fold increase in phospholipase activity secreted into the culture media of MIN6 cells (MIN6-GV) compared to media from cells transfected with control plasmid (MIN6-C; Fig. 2a). Insulin secretion was similar in MIN6-C and MIN6-GV cells incubated in low glucose (5 mM), indicating that overexpression of GV sPLA2 does not impact insulin secretion under basal conditions (Fig. 2b). In contrast, insulin secretion in cells incubated in high glucose (20 mM) was significantly increased in MIN6-GV (2.8±0.19 μg/mg cell protein) compared to MIN6-C (1.95±0.06 μg/mg cell protein; p=0.0015). Moreover, when expressed as fold-increase over basal, the magnitude of GSIS was significantly greater in cells overexpressing GV sPLA2 (6.4±0.43) compared to control cells (4.8±0.16; p=0.0052; data not shown). The effect of GV sPLA2 overexpression to enhance GSIS occurred in the absence of alterations in total cellular insulin content (Fig. 2c).
Fig 2.
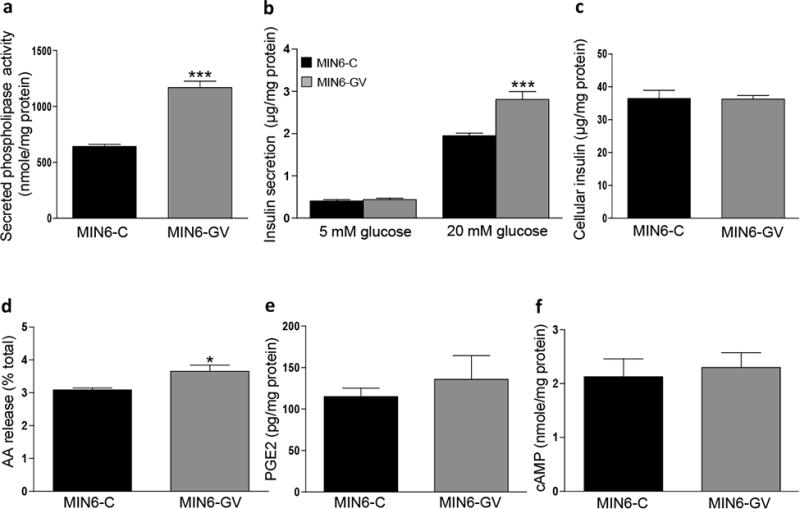
GV sPLA2 activates glucose-stimulated insulin secretion (GSIS) and enhances AA release in MIN6 cells. MIN6 cells were transfected with a control expression plasmid (MIN6-C) or an expression plasmid encoding GV sPLA2 (MIN6-GV) as described under “Materials and Methods.” a, Phospholipase activity in 48 h conditioned media from MIN6-C or MIN6-GV cell cultures was determined and normalized to cell protein. b, GSIS was performed in MIN6-C and MIN6-GV cells 48 h after transfection as described in “Materials and Methods.” Insulin levels in the media were determined and normalized to total cell protein. c, Total cellular insulin content of MIN6-C and MIN6-GV cells was assessed as described in “Materials and Methods.” d, [3H]-AA release by MIN6-C and MIN6-GV cells was quantified and expressed as the percent of total cellular [3H]-AA as described in “Materials and Methods.” e, PGE2 levels in culture media from MIN6-C and MIN6-GV cells 48 h after transfection, normalized to total cell protein. f, cellular cAMP content in MIN6-C and MIN6-GV cells was determined 48 h after transfection and normalized to total cell protein. Data are from 4 independent transfections per construct and are presented as mean ± S.E; *p<0.05. ***, p < 0.001.
GV sPLA2 enhances AA release but not PGE2 production in β-cells
Given the recognized importance of AA and its metabolite, PGE2, in regulating GSIS [6, 8, 27, 28], we next investigated the extent to which GV sPLA2 overexpression enhances AA and PGE2 production in β-cells. MIN6-GV cells demonstrated a modest but significant increase in AA release compared to MIN6-C cells, consistent with GV sPLA2's known ability to hydrolyze glycerophospholipids on mammalian cell membranes (Fig. 2d) [29]. However, increased AA release was not associated with a significant increase in PGE2 secretion by MIN6-GV cells (Fig. 2e). Moreover, cellular cAMP was not significantly different in MIN6-GV compared to MIN6-C cells (Fig. 2f), as would be expected if enhanced GV sPLA2 activity resulted in increased PGE2 generation. Thus, in contrast to what we observed with GX sPLA2 [19], it appears that AA released by MIN6 cells through the action of GV sPLA2 is of insufficient magnitude, or is not effectively coupled to the prostaglandin synthetic pathway, to provide a detectable increase in PGE2 production.
Deficiency of endogenous GV sPLA2 attenuates GSIS in β-cells
To understand the role of endogenous GV sPLA2 in β -cell function, we next investigated the impact of siRNA-mediated silencing on GSIS in MIN6 cells. GV sPLA2 expression was suppressed ∼90% in MIN6 cells 24 h after transient transfection with siRNA targeting GV sPLA2 (GV-siRNA; Fig. 3a). While the magnitude of GSIS in GV-siRNA cells (7.03±0.18 μg/mg cell protein) was significantly reduced compared to Scr-siRNA cells (8.8±0.35 μg/mg cell protein; p=0.004), basal insulin secretion was also significantly lower in GV-siRNA cells compared to Scr-siRNA cells (Fig. 3b). As a consequence, the fold-increase in insulin secretion over basal conditions was not significantly different for the two cell lines (6.5±0.16 for GV-siRNA versus 6.1±0.24 for Scr-siRNA).
Fig 3.
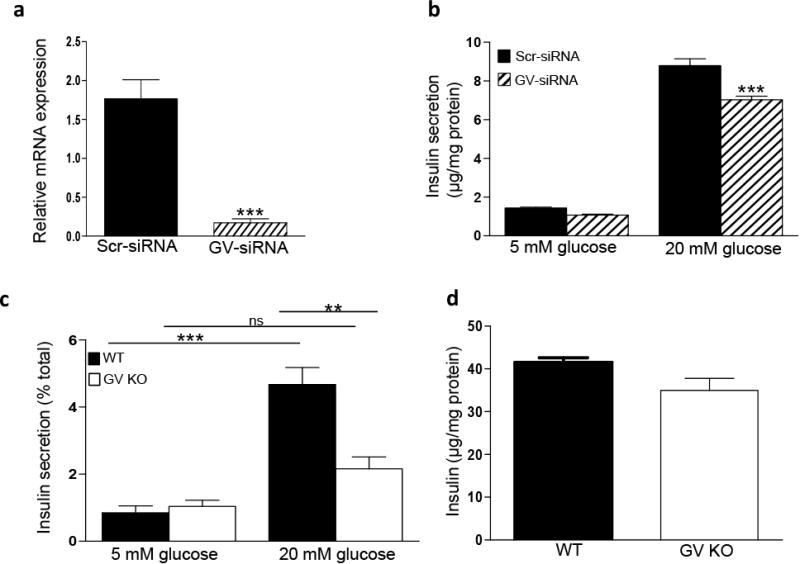
GV sPLA2 deficiency suppresses GSIS in MIN6 cells and primary mouse pancreatic islet cells. MIN6 cells were transfected with a control siRNA (Scr-siRNA) or siRNA directed to GV sPLA2 (GV-siRNA), as described under “Materials and Methods.” a, RNAs were prepared from Scr-siRNA and GV-siRNA cells 24 h after transfection for quantification of GV sPLA2 mRNA abundance. b, GSIS was performed in Scr-siRNA cells and GV-siRNA cells as described in “Materials and Methods.” Insulin secreted into the media was assayed and normalized to total cell protein. c, GSIS assay was performed using islets (25 similar-sized islets per mouse) isolated from 4-month-old WT and GV KO mice (n=4). Islets were incubated successively for 40 min in buffer containing 5 mM glucose (low glucose) and for 40 min in buffer containing 20 mM glucose (high glucose). Insulin in the assay media was determined and normalized to total cellular insulin in the corresponding islets. d, Total islet insulin content in 25 similar-sized islets from WT and GV KO mice were estimated and normalized to total protein. Data are presented as mean ± S.E; ns-not significant; **p<0.01;***p<0.001.
The lack of a pronounced effect of siRNA-mediated GV suppression on GSIS in MIN6 cells may be the result of residual GV sPLA2 activity in the silenced cells. It is also possible that GV sPLA2 plays a relatively small role in regulating GSIS in this immortalized cell line. Thus, to more definitively assess the impact of endogenous GV sPLA2 on β -cell function, we performed experiments with islets isolated from WT and GV KO mice. To rule out any potential differences in GSIS due to variations in islet size [30], similar-sized islets from WT and GV KO mice were used for the assay. We also confirmed that GX sPLA2 expression was not significantly different in pancreatic islets isolated from GV KO mice compared to WT mice (data not shown). There was no significant difference in basal insulin secretion (5 mM glucose) between islets from WT and GV KO mice (Fig. 3c). As expected, insulin secretion by WT islets increased significantly in response to high glucose (20 mM). Conversely, GSIS was almost totally abolished in islets lacking GV sPLA2 (Fig. 3c). Deficiency of GV sPLA2 in primary islets did not significantly change total insulin content (Fig. 3d), consistent with findings in MIN6 cells (Fig. 2c). Taken together, our results demonstrate that GV sPLA2 enhances GSIS in pancreatic β-cells, in marked contrast to our previous findings with GX sPLA2 [19].
GV KO mice demonstrate enhanced GSIS in vivo
Our in vitro findings that GV sPLA2 augments GSIS in cultured MIN6 cells and isolated islets prompted us to investigate the in vivo effect of GV sPLA2 deficiency on glucose homeostasis in mice. 6-month-old GV KO mice showed a trend for decreased fasting glucose levels compared to age-matched WT mice (Fig. 4a), which was associated with a non-significant increase in fasting insulin levels in GV KO mice compared to WT (0 min, Fig. 4b). As expected, plasma insulin levels were significantly increased in both WT and GV KO mice 15 minutes after i.p glucose injection (3 mg/kg body weight). Surprisingly, in contrast to in vitro and ex vivo results, plasma insulin levels 15 min following glucose injection were significantly higher in GV KO mice compared to WT mice (Fig. 4b). Glucose disposal and insulin tolerance were not significantly different between the two strains of mice (Fig. 4c, d), nor was there a significant difference in body weights (data not shown).
Fig 4.
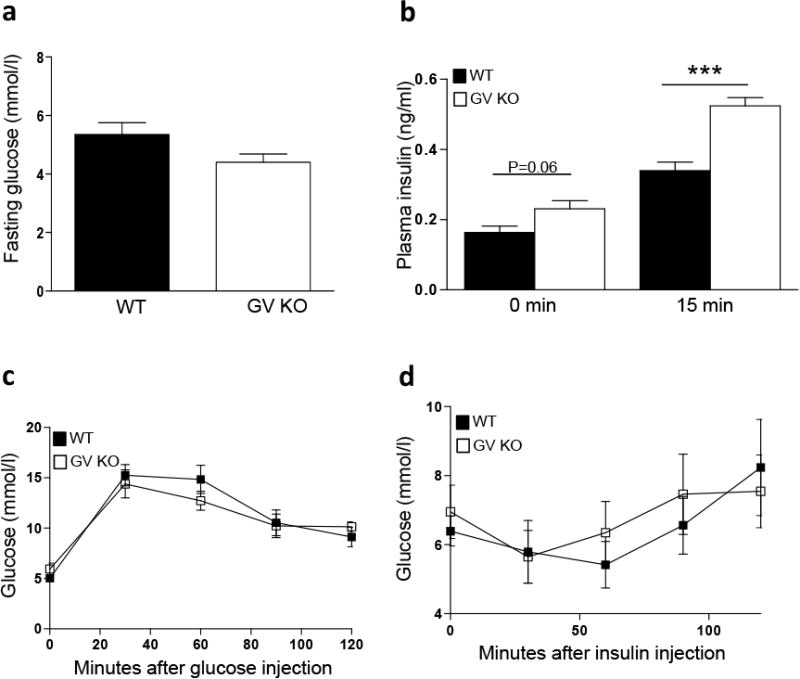
Enhanced in vivo GSIS in GV KO mice. a, Blood glucose levels after 16 h fast in WT and GV KO mice. b, Plasma insulin levels were determined before and 15 min after i.p. glucose injection (3 g/kg; n=4-5) in 6-month old WT and GV KO mice. c, 6- month old WT and GV KO mice were fasted for 6 h prior to i.p. injection of 1.5 mg glucose/g body weight, and glucose concentrations in the blood were determined at the indicated time after injection (n=5). d, 6- month old WT and GV KO mice were fasted for 4 h prior to i.p. glucose injection of human insulin (0.5 U/kg body weight), and glucose concentrations in the blood were determined at the indicated time after injection (n=4-5). e, body weight of WT and GV KO mice (n=4/strain). Data are presented as mean ± S.E; ***p<0.001.
GV KO mice have increased average islet size, β-cell mass and proliferation
Despite an apparent defect in GSIS in pancreatic islets that are deficient in GV sPLA2 (Fig. 3c), GV KO mice demonstrated increased GSIS in vivo (Fig. 4b). This discrepancy motivated us to investigate potential differences in pancreatic β islets between GV KO and WT mice. Extensive morphometric analysis of pancreatic tissue identified a significant 1.5-fold increase in β-islet mass (estimated by determining the portion of total pancreatic area in tissue sections corresponding to insulin-positive β-islets and the respective pancreatic weights) in GV KO compared to WT pancreas (Fig. 5a). There was no significant difference in pancreatic weights between the two strains (Fig. 5b). Therefore, an increase in β-islet mass must reflect an increase in islet number or an increase in islet size, or both. There was no difference in average number of islets in the pancreas of GV KO mice compared to WT mice (data not shown). However, average islet size was significantly larger for GV KO mice compared to WT mice (Fig. 5c). β-cell proliferation was assessed in pancreatic sections from 12-weeks old WT and GV KO (Fig. 5d shows representative images of GV KO pancreatic section) mice by determining the total number of insulin-positive cells (red staining) that were also positive for Ki67 (green staining) and DAPI nuclear stain. This analysis showed a significant increase in the number of proliferating cells in β-islets in GV KO mice compared to WT (Fig. 5e).
Fig 5.
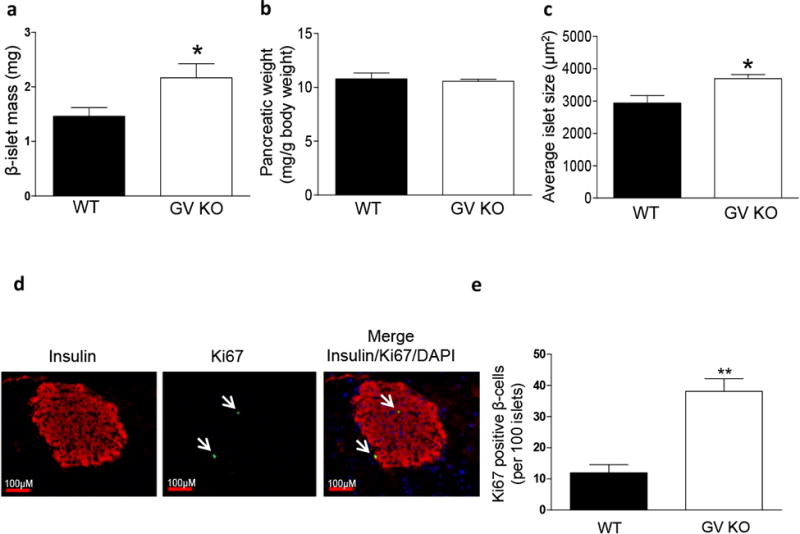
Increased β-islet mass, average islet size and β-cell proliferation in the pancreas of GV KO mice. a, Pancreatic β-islet mass of WT and GV KO mice (n=4/strain; 5-8 sections were analyzed from each mouse) were calculated by obtaining the fraction of the area of pancreatic tissue positive for insulin staining and multiplying this by the pancreatic weight as described in detail in the “Materials and Methods.” b, Pancreatic weight of WT and GV KO mice (n=4/strain). c, Average islet size of WT and GV KO pancreata (n=4/strain; 5-8 sections/mouse) was calculated by dividing the total area of pancreatic tissue positive for insulin staining by the total number of islets. d, Representative immunofluorescence images of pancreatic islets from GV KO mice at 20X magnification showing insulin (red) and Ki67 (indicated by arrows pointing to green staining). A merged image shows expression of Ki67 in insulin-producing cells of GV KO pancreas, co-localized with DAPI staining. e, Quantification of number of β-cells with Ki67-positive nuclei per 100 islets in WT and GV KO mice (n=3/group; 30-50 islets from 2 sections per mouse). Data are presented as mean ± S.E; *p<0.05; ***p<0.01.
Discussion
There is a large body of evidence pointing to arachidonic acid (AA) and its biologically active metabolites as key regulators of insulin secretion by β-cells [6, 25, 26, 28, 31, 32]. The important role of AA in β-cells is underscored by its relatively high abundance in islets, representing ∼30% of the total mass of fatty acyl glycerophospholipids [33]. The liberation of AA from cellular membranes is dependent on the action of PLA2 enzymes, which also represents the initial, rate-limiting step for the production of a myriad of AA-derived bioactive lipid mediators, including prostaglandins and leukotrienes. Despite the central role of PLA2's in AA metabolism, the relative contributions of the various intracellular and secreted forms of these enzymes in β-cells and their respective impact on the regulation of insulin secretion has not been fully defined. Thus, we have set out to investigate whether GV or GX sPLA2 play a role in regulating insulin secretion by β-cells. These two related enzymes have been shown to hydrolyze cell membrane phospholipids with much higher efficiency compared to other sPLA2 family members due to their high binding affinity to phosphatidylcholine (PC) [34, 35]. We previously reported that GX sPLA2 suppresses GSIS through a cyclooxygenase-2-dependent mechanism [19]. In this report, we provide evidence that GV sPLA2 regulates β-cell function in a manner that is distinct from GX sPLA2.
Like GX sPLA2, GV sPLA2 is expressed by insulin-producing cells in mouse pancreas and by MIN6 cells, a mouse pancreatic β-cell line. However, in contrast to GX sPLA2, gain and loss of function studies showed that GV sPLA2 enhances GSIS in MIN6 cells. Moreover, primary mouse islet cells deficient in GV sPLA2 exhibit significantly decreased GSIS ex vivo compared to pancreatic islets from wild-type mice. Taken together, our studies demonstrate the unexpected finding that whereas GX sPLA2 expressed by β-cells suppresses GSIS, the related GV sPLA2 enzyme serves to enhance GSIS. It is interesting to note we were unable to detect a significant increase in PGE2 production in MIN6 cells with forced overexpression of GV sPLA2, despite a significant 1.8-fold increase in PLA2 activity secreted by these cells. In comparable studies of GX sPLA2, a 2.2-fold increase in sPLA2 activity in transfected MIN6 cells was associated with a robust 2.5-fold increase in PGE2 generation [19]. The ability of GX sPLA2 to reduce GSIS was blocked in cells treated either with a cyclooxygenase-2 inhibitor or an EP3 receptor antagonist, demonstrating that the suppressive effect of GX sPLA2 is dependent on PGE2 synthesis and signaling through the EP3 receptor. The PGE2-EP3 receptor axis is thought to impact GSIS through decreased adenylyl cyclase activity and consequent reductions in cAMP, an established activator of insulin secretion [10, 11]. Our findings that overexpression of GX sPLA2 [19] but not GV sPLA2 (Fig. 2f), enhances intracellular cAMP concentrations in MIN6 cells is consistent with the conclusion that AA is coupled to increased PGE2 production only in the case of GX sPLA2. We speculate that increased GSIS in GV sPLA2-overexpressing MIN6 cells is a consequence of increased non-esterified AA in these cells, in line with previous studies showing that treatments that increase endogenous AA in β-cells leads to augmented insulin secretion [36]. The mechanisms for AA-mediated activation of GSIS are not totally understood. After glucose-induced depolarization of the plasma membrane and subsequent release of insulin, Kv2.1 channels present on the β-cell membrane serve to repolarize the membrane and consequently limit the duration of insulin secretion [37]. By attenuating Kv2.1 activity, AA prolongs the action potential, resulting in the amplification of GSIS for longer periods [38].
As noted above, our data indicate that AA liberated by GV sPLA2 versus GX sPLA2 has distinct metabolic fates in MIN6 cells, such that GX sPLA2 appears to be uniquely coupled to the prostaglandin synthesis pathway. Several in vitro studies point to differences in subcellular localization and actions for the two enzymes, which might account for our observations in β-cells. Though it is generally believed that GX sPLA2 hydrolyzes phospholipids in the outer leaflet of the plasma membrane, there is evidence that this enzyme may release AA prior to secretion [20]. It has also been suggested that GX sPLA2 is taken up by cells through its high affinity binding to the M-type receptor [39-41], whereas GV sPLA2 may be internalized and trafficked to intracellular compartments by heparin sulphate proteoglycans [42, 43]. Unlike GV sPLA2, which is expressed as the mature enzyme, GX sPLA2 is originally produced as a proenzyme that must be proteolytically cleaved by furin-like proprotein convertases in order to exert full catalytic activity [44, 45]. Thus, compartmentalization of GX sPLA2 activity may be dictated by the subcellular location of the activating convertase. It is also important to note that local Ca2+ concentrations may influence the relative hydrolytic activity of the two enzymes, since in vitro studies indicate the requirement for Ca2+ is 10-fold higher for GV sPLA2 compared to GX sPLA2 [46]. Clearly, additional studies are required to determine whether spatial segregation of the catalytic activities of Group V and Group X sPLA2 is responsible for the observed functional differences of these two enzymes in β-cells.
Intriguingly, our in vivo results show that whole-body ablation of GV sPLA2 results in enhanced GSIS in mice (Fig. 4b), contrary to the suppressive effect of GV sPLA2 deficiency observed in isolated islets ex vivo (Fig. 3c). Enhanced GSIS in GV sPLA2-deficient mice was associated with a significant increase in islet mass as well as markers of β -cell proliferation, which are likely to at least partially overcome the reduction in GSIS observed for pancreatic islets isolated from GV sPLA2-deficient mice. Recently, Sato et al. reported that diet-induced obesity induces a robust increase in adipocyte expression of GV sPLA2. Additionally, GV sPLA2-deficient mice had significantly increased body weight and greater insulin resistance, mainly in white adipose tissue, compared to wild-type littermates after high fat diet feeding [47]. Interestingly, high fat diet-induced hyperinsulinemia was greater in GV sPLA2-deficient mice compared to wild-type mice, whereas glucose disposal and fasting blood glucose levels were similar for the two strains. The authors also showed that GSIS was significantly more robust in obese GV sPLA2-deficient mice compared to control, although the mechanism for increased GSIS was not investigated. While it is difficult to tease out the systemic versus local effects of GV sPLA2 in β-cell function, our studies in mice fed a normal rodent diet indicate that whole-body GV sPLA2 deficiency is associated with increased β-cell proliferation, an increase in islet mass, and overall capacity to secrete insulin, which may ultimately serve a protective effect in the setting of profound insulin resistance. The observed effects on β-cell proliferation and islet mass in the GV KO mice may be the consequence of GV sPLA2 deficiency in tissues other than the pancreas. Additional studies using tissue-specific knock-out mice are necessary to fully understand the biology of these intriguing PLA2's.
Conclusions
Our results demonstrate that GV sPLA2 plays multiple roles in regulating β-cell function in mice. Based on in vitro studies in MIN6 cells and isolated pancreatic islets, it is clear that GV sPLA2 acts in an autocrine and/or paracrine manner to enhance GSIS, possibly by mediating the release of AA from membrane glycerophospholipids. This activity is in marked contrast to GX sPLA2, which we previously showed suppresses GSIS by providing AA substrate for PGE2 generation in β-cells [19]. The opposing roles of GV and GX sPLA2 in regulating insulin secretion underscore the fact that these two related enzymes are not functionally redundant. Additional in depth studies are necessary to fully understand the biology of these intriguing PLA2's. Despite the capacity of GV sPLA2 to enhance insulin secretion by β-cells, the aggregate effect of whole-body GV sPLA2 deficiency was to augment GSIS in mice, an outcome that was associated with increased β-cell proliferation and pancreatic islet mass. These data are consistent with a previous report that GV sPLA2-deficient mice are partially protected from impaired GSIS associated with severe obesity-induced insulin resistance [47]. Thus, GV sPLA2 may provide a potential target for improving β-cell compensation in the setting of insulin resistance.
Acknowledgments
We thank Dr. Sabire Ozcan, University of Kentucky for providing MIN6 cells and Dr. M. Gelb, University of Washington for supply of anti-mouse GV sPLA2 antibody. We also thank Dr. Wendy Katz for helping us with paraffin embedding and tissue sectioning.
Funding: This work was supported in whole or in part by National Institutes of Health Grant R01 DK082419 (to N. R. W.) and the NIGMS Grant P20 GM103527 (to P. S.).
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
Ethical approval: All procedures performed in studies involving mice were in accordance with the guidelines of the University of Kentucky Institutional Animal Care and Use Committees. The article does not contain any studies with human participants performed by any of the authors.
References
- 1.Prentki M, Nolan CJ. Islet β cell failure in type 2 diabetes. JClin Invest. 2006;116:1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramanadham S, Gross RW, Han X, Turk J. Inhibition of arachidonate release by secretagogue-stimulated pancreatic islets suppresses both insulin secretion and the rise in beta-cell cytosolic calcium ion concentration. Biochemistry. 1993;32:337–346. doi: 10.1021/bi00052a042. [DOI] [PubMed] [Google Scholar]
- 3.Ramanadham S, Song H, Bao S, Hsu FF, Zhang S, Ma Z, Jin C, Turk J. Islet complex lipids: involvement in the actions of group VIA calcium-independent phospholipase A(2) in beta-cells. Diabetes. 2004;53(Suppl 1):S179–185. doi: 10.2337/diabetes.53.2007.s179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson RP, Tsai P, Little SA, Zhang HJ, Walseth TF. Receptor-mediated adenylate cyclase-coupled mechanism for PGE2 inhibition of insulin secretion in HIT cells. Diabetes. 1987;36:1047–1053. doi: 10.2337/diab.36.9.1047. [DOI] [PubMed] [Google Scholar]
- 5.Sjoholm A. Prostaglandins inhibit pancreatic beta-cell replication and long-term insulin secretion by pertussis toxin-insensitive mechanisms but do not mediate the actions of interleukin-1 beta. Biochim Biophys Acta. 1996;1313:106–110. doi: 10.1016/0167-4889(96)00058-4. [DOI] [PubMed] [Google Scholar]
- 6.Kimple ME, Keller MP, Rabaglia MR, Pasker RL, Neuman JC, Truchan NA, Brar HK, Attie AD. Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes. 2013;62:1904–1912. doi: 10.2337/db12-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seaquist ER, Walseth TF, Nelson DM, Robertson RP. Pertussis toxin-sensitive G protein mediation of PGE2 inhibition of cAMP metabolism and phasic glucose-induced insulin secretion in HIT cells. Diabetes. 1989;38:1439–1445. doi: 10.2337/diab.38.11.1439. [DOI] [PubMed] [Google Scholar]
- 8.Tran PO, Gleason CE, Poitout V, Robertson RP. Prostaglandin E2 Mediates Inhibition of Insulin Secretion by Interleukin-1β. J Biol Chem. 1999;274:31245–31248. doi: 10.1074/jbc.274.44.31245. [DOI] [PubMed] [Google Scholar]
- 9.Fujita H, Kakei M, Fujishima H, Morii T, Yamada Y, Qi Z, Breyer MD. Effect of selective cyclooxygenase-2 (COX-2) inhibitor treatment on glucose-stimulated insulin secretion in C57BL/6 mice. Biochem Biophys Res Commun. 2007;363:37–43. doi: 10.1016/j.bbrc.2007.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. JLipid Res. 2009;50:S423–S428. doi: 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yajima H, Komatsu M, Schermerhorn T, Aizawa T, Kaneko T, Nagau M, Sharp GW, Hashizume K. cAMP enhances insulin secretion by an action on the ATP-sensitive K+ channel-independent pathway of glucose signaling in rat pancreatic islets. Diabetes. 1999;48:1006–1012. doi: 10.2337/diabetes.48.5.1006. [DOI] [PubMed] [Google Scholar]
- 12.Oshima H, Taketo MM, Oshima M. Destruction of Pancreatic β-Cells by Transgenic Induction of Prostaglandin E2 in the Islets. J Biol Chem. 2006;281:29330–29336. doi: 10.1074/jbc.M602424200. [DOI] [PubMed] [Google Scholar]
- 13.Song K, Zhang X, Zhao C, Ang NT, Ma ZA. Inhibition of Ca2+-independent phospholipase A2 results in insufficient insulin secretion and impaired glucose tolerance. Mol Endocrinol. 2005;19:504–515. doi: 10.1210/me.2004-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bao S, Bohrer A, Ramanadham S, Jin W, Zhang S, Turk J. Effects of stable suppression of Group VIA phospholipase A2 expression on phospholipid content and composition, insulin secretion, and proliferation of INS-1 insulinoma cells. J Biol Chem. 2006;281:187–198. doi: 10.1074/jbc.M509105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juhl K, Hoy M, Olsen HL, Bokvist K, Efanov AM, Hoffmann EK, Gromada J. cPLA2alpha-evoked formation of arachidonic acid and lysophospholipids is required for exocytosis in mouse pancreatic beta-cells. Am J Physiol Endocrinol Metab. 2003;285:E73–81. doi: 10.1152/ajpendo.00086.2003. [DOI] [PubMed] [Google Scholar]
- 16.Milne HM, Burns CJ, Squires PE, Evans ND, Pickup J, Jones PM, Persaud SJ. Uncoupling of nutrient metabolism from insulin secretion by overexpression of cytosolic phospholipase A(2) Diabetes. 2005;54:116–124. doi: 10.2337/diabetes.54.1.116. [DOI] [PubMed] [Google Scholar]
- 17.Jones PM, Burns CJ, Belin VD, Roderigo-Milne HM, Persaud SJ. The role of cytosolic phospholipase A2 in insulin secretion. Diabetes. 2005;53:S172–S178. doi: 10.2337/diabetes.53.2007.s172. [DOI] [PubMed] [Google Scholar]
- 18.Ramanadham S, Ma Z, Arita H, Zhang S, Turk J. Type IB secretory phospholipase A2 is contained in insulin secretory granules of pancreatic islet β-cells and is co-secreted with insulin from glucose-stimulated islets. Biochim Biophys Acta. 1998;1390:301–312. doi: 10.1016/s0005-2760(97)00189-6. [DOI] [PubMed] [Google Scholar]
- 19.Shridas P, Zahoor L, Forrest KJ, Layne JD, Webb NR. Group X secretory phospholipase A2 regulates insulin secretion through a cyclooxygenase-2-dependent mechanism. J Biol Chem. 2014;289:27410–27417. doi: 10.1074/jbc.M114.591735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mounier CM, Ghomashchi F, Lindsay MR. Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A(2) occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A(2)-alpha. J Biol Chem. 2004;279:25024–25038. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- 21.Wooton-Kee CR, Boyanovsky BB, Nasser MS, de Villiers WJS, Webb NR. Group V sPLA2 Hydrolysis of Low-Density Lipoprotein Results in Spontaneous Particle Aggregation and Promotes Macrophage Foam Cell Formation. Arterioscler Thromb Vasc Biol. 2004;24:762–767. doi: 10.1161/01.ATV.0000122363.02961.c1. [DOI] [PubMed] [Google Scholar]
- 22.Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J Biol Chem. 2004;279:16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoemaker R, Yiannikouris F, Thatcher S, Cassis L. ACE2 deficiency reduces beta-cell mass and impairs beta-cell proliferation in obese C57BL/6 mice. Am J Physiol Endocrinol Metab. 2015;309:E621–631. doi: 10.1152/ajpendo.00054.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinohara H, Balboa MA, Johnson CA, Balsinde J, Dennis EA. Regulation of delayed prostaglandin production in activated P388D1 macrophages by group IV cytosolic and group V secretory phospholipase A2s. J Biol Chem. 1999;274:12263–12268. doi: 10.1074/jbc.274.18.12263. [DOI] [PubMed] [Google Scholar]
- 25.Meng ZX, Sun JX, Ling JJ, Lv JH, Zhu DY, Chen Q, Sun YJ, Han X. Prostaglandin E2 regulates Foxo activity via the Akt pathway: implications for pancreatic islet beta cell dysfunction. Diabetologia. 2006;49:2959–2968. doi: 10.1007/s00125-006-0447-5. [DOI] [PubMed] [Google Scholar]
- 26.Meng Z, Lv J, Luo Y, Lin Y, Zhu Y, Nie J, Yang T, Sun Y, Han X. Forkhead Box O1/Pancreatic and Duodenal Homeobox 1 Intracellular Translocation Is Regulated by c-Jun N-Terminal Kinase and Involved in Prostaglandin E2-Induced Pancreatic β-Cell Dysfunction. Endocrinology. 2009;150:5284–5293. doi: 10.1210/en.2009-0671. [DOI] [PubMed] [Google Scholar]
- 27.Landt M, Easom RA, Colca JR, Wolf BA, Turk J, Mills LA, McDaniel ML. Parallel effects of arachidonic acid on insulin secretion, calmodulin-dependent protein kinase activity and protein kinase C activity in pancreatic islets. Cell calcium. 1992;13:163–172. doi: 10.1016/0143-4160(92)90044-s. [DOI] [PubMed] [Google Scholar]
- 28.Metz SA. Exogenous arachidonic acid promotes insulin release from intact or permeabilized rat islets by dual mechanisms. Putative activation of Ca2+ mobilization and protein kinase C. Diabetes. 1988;37:1453–1469. doi: 10.2337/diab.37.11.1453. [DOI] [PubMed] [Google Scholar]
- 29.Balboa MA, Balsinde J, Winstead MV, Tischfield JA, Dennis EA. Novel group v phospholipase A2 involved in arachidonic acid mobilization in murine P388D1 macrophages. J Biol Chem. 1996;271:32381–32384. doi: 10.1074/jbc.271.50.32381. [DOI] [PubMed] [Google Scholar]
- 30.Reaven EP, Gold G, Walker W, Reaven GM. Effect of variations in islet size and shape on glucose-stimulated insulin secretion. Horm Metab Res. 1981;13:673–674. doi: 10.1055/s-2007-1019372. [DOI] [PubMed] [Google Scholar]
- 31.Jones PM, Persaud SJ. Arachidonic acid as a second messenger in glucose-induced insulin secretion from pancreatic β-cells. J Endocrinol. 1993;137:7–14. doi: 10.1677/joe.0.1370007. [DOI] [PubMed] [Google Scholar]
- 32.Luo P, Wang MH. Eicosanoids, β-cell function, and diabetes. Prostaglandins Other Lipid Mediat. 2011;95:1–10. doi: 10.1016/j.prostaglandins.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramanadham S, Bohrer A, Mueller M, Jett P, Gross RW, Turk J. Mass spectrometric identification and quantitation of arachidonate-containing phospholipids in pancreatic islets: Prominence of plasmenylethanolamine molecular species. Biochemistry. 1993;32:5339–5351. doi: 10.1021/bi00071a009. [DOI] [PubMed] [Google Scholar]
- 34.Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, Sadilek M, Nguyen E, Lazdunski M, Lambeau G, Gelb MH. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J Biol Chem. 2002;277:48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 35.Valentin E, Ghomashchi F, Gelb MH, Lazdunski M, Lambeau G. On the diversity of secreted phospholipases A(2). Cloning, tissue distribution, and functional expression of two novel mouse group II enzymes. J Biol Chem. 1999;274:31195–31202. doi: 10.1074/jbc.274.44.31195. [DOI] [PubMed] [Google Scholar]
- 36.Persaud SJ, Muller D, Belin VD, Kitsou-Mylona I, Asare-Anane H, Papadimitriou A, Burns CJ, Huang GC, Amiel SA, Jones PM. The Role of Arachidonic Acid and Its Metabolites in Insulin Secretion From Human Islets of Langerhans. Diabetes. 2007;56:197–203. doi: 10.2337/db06-0490. [DOI] [PubMed] [Google Scholar]
- 37.Jacobson DA, Weber CR, Bao S, Turk J, Philipson LH. Modulation of the pancreatic islet beta-cell-delayed rectifier potassium channel Kv2.1 by the polyunsaturated fatty acid arachidonate. J Biol Chem. 2007;282:7442–7449. doi: 10.1074/jbc.M607858200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammala CE, Philipson LH. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell metab. 2007;6:229–235. doi: 10.1016/j.cmet.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rouault M, Le Calvez C, Boilard E, Surreal F, Singer A, Ghomashchi F, Bezzine S, Scarzello S, Gelb MH, Lambeau G. Recombinant production and properties of binding of the full set of mouse secreted phospholipases A2 to the mouse M-type receptor. Biochemistry. 2007;46:1647–1662. doi: 10.1021/bi062119b. [DOI] [PubMed] [Google Scholar]
- 40.Morioka Y, Saiga A, Yokota Y, Suzuki N, Ikeda M, Ono T, Nakano K, Fujiii N, Ischizaki J, Arita H, Hanasaki K. Mouse group x secretory phospholipase A2 induces a potent release of arachidonic acid from spleen cells and acts as a ligand for the phospholipase A2 Receptor. Arch Biochem Biophy. 2000;381:31–42. doi: 10.1006/abbi.2000.1977. [DOI] [PubMed] [Google Scholar]
- 41.Yokota Y, Higashino K, Nakano K, Arita H, Hanasaki K. Identification of group X secretory phospholipase A(2) as a natural ligand for mouse phospholipase A(2) receptor. FEBS Lett. 2000;478:187–191. doi: 10.1016/s0014-5793(00)01848-2. [DOI] [PubMed] [Google Scholar]
- 42.Rosengren B, Peilot H, Umaerus M, Jonsson-Rylander AC, Mattsson-Halten L, Hallberg C, Cronet P, Rodriquez-Lee M, Hurt-Camejo E. Secretory phospholipase A2 group V: lesion distribution, activation by arterial proteoglycans, and induction in aorta by a western diet. Arterioscler Thromb Vasc Biol. 2006;26:1579–1585. doi: 10.1161/01.ATV.0000221231.56617.67. [DOI] [PubMed] [Google Scholar]
- 43.Murakami M, Koduri RS, Enomoto A, Shimbara S, Seki M, Yoshihara K, Singer A, Valentin E, Ghomashchi F, Lambeau G, Gelb MH, Kudo I. Distinct arachidonate-releasing functions of mammalian secreted phospholipase A2s in human embryonic kidney 293 and rat mastocytoma RBL-2H3 cells through heparan sulfate shuttling and external plasma membrane mechanisms. J Biol Chem. 2001;276:10083–10096. doi: 10.1074/jbc.M007877200. [DOI] [PubMed] [Google Scholar]
- 44.Layne JD, Shridas P, Webb NR. Ectopically expressed pro-group X secretory phospholipase A2 is proteolytically activated in mouse adrenal cells by furin-like proprotein convertases: implications for the regulation of adrenal steroidogenesis. J Biol Chem. 2015;290:7851–7860. doi: 10.1074/jbc.M114.634667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jemel I, Ii H, Oslund RC, Payre C, Dabert-Gay AS, Douquet D, Charqui K, Scarzello S, Gelb MH, Lambeau G. Group x secreted phospholipase A2 proenzyme is matured by a furin-like proprotein convertase and releases arachidonic acid inside of human HEK293 cells. J Biol Chem. 2011;286:36509–36521. doi: 10.1074/jbc.M111.268540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamitani S, Yamada K, Yamamoto S, Ishimoto Y, Ono T, Saiga A, Hanasaki K. Differences between group x and group v secretory phospholipase A(2) in lipolytic modification of lipoproteins. Cell Mol Biol Lett. 2012;17:459–478. doi: 10.2478/s11658-012-0019-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sato H, Taketomi Y, Ushida A, Isogai Y, Kojima T, Hirabayashi T, Miki Y, Yamamoto K, Nishito Y, Kobayashi T, Ikeda K, Taguchi R, Hara S, Ida S, Miyamoto Y, Watanabe M, Baba H, Miyata K, Oike Y, Gelb MH, Murakami M. The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell metab. 2014;20:119–132. doi: 10.1016/j.cmet.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]