

Abstract
The muscle regulatory transcription factor MyoD is a master regulator of skeletal myoblast differentiation. We have previously reported that MyoD is also necessary for the elevated expression of the pro-apoptotic Bcl2 family member PUMA, and the ensuing apoptosis, that occurs in a subset of myoblasts induced to differentiate. Herein, we report the identification of a functional MyoD binding site within the extended PUMA promoter. In silico analysis of the murine PUMA extended promoter revealed three potential MyoD binding sites within 2kb of the transcription start site. Expression from a luciferase reporter construct containing this 2kb fragment was enhanced by activation of MyoD in both myoblasts and fibroblasts and diminished by silencing of MyoD in myoblasts. Experiments utilizing truncated versions of this promoter region revealed that the potential binding site at position −857 was necessary for expression. Chromatin immunoprecipitation (ChIP) analysis confirmed binding of MyoD to the DNA region encompassing position −857. The increase in MyoD binding to the PUMA promoter as a consequence of culture in differentiation media (DM) was comparable to the increase in MyoD binding at the myogenin promoter and was diminished in myoblasts silenced for MyoD expression. Finally, ChIP analysis using an antibody specific for the transcription factor p53 demonstrated that, in myoblasts silenced for MyoD expression, p53 binding to the PUMA promoter was diminished in response to culture in DM. These data indicate that MyoD plays a direct role in regulating PUMA expression and reveal functional consequences of MyoD expression on p53 mediated transcription of PUMA.
Keywords: MyoD, p53, PUMA, apoptosis
Introduction
Treatment options relevant to the amelioration of muscle trauma or disease states associated with muscle degeneration include maximizing the regenerative potential of adult muscle stem cells as well as improving the efficacy of protocols utilizing skeletal myoblast transfer or skeletal muscle tissue engineering. For each option, a better understanding of the molecular events controlling skeletal myogenesis could identify additional targets for better therapeutic manipulation [1–8]. In many cell types, including skeletal myoblasts, apoptosis and differentiation are coordinately regulated. In skeletal myoblasts, these two processes result in mutually exclusive physiologically important endpoints. Upon induction of skeletal myoblast differentiation, apoptosis has been documented in the vertebrate models and in cultures of primary myoblasts and established muscle cell lines [9–13]. However, the molecular coordination of these two processes is not well understood [13,14].
We have previously reported that differentiation and apoptosis in response to culture in differentiation media (DM) are separable events in skeletal myoblasts, thus indicating the requirement for distinct signaling molecules [11]. We further determined that increased expression of the pro-apoptotic Bcl2 family member PUMA was required for, and distinct to, the process of apoptosis in skeletal myoblasts [15]. In contrast to later signaling events which are evidently unique to either apoptosis or differentiation, initial signaling events will have a commonality in that both differentiation and apoptosis are induced by culture in DM and abrogated by signaling events such as those initiated by oncogenic Ras [9, 11,12]. To this point, we have reported that shRNAi-mediated silencing of the muscle regulatory transcription factor MyoD in myoblasts abrogated apoptosis, as well as differentiation, in response to culture in DM. Further, ectopic expression and activation of MyoD in fibroblasts conferred the ability to undergo apoptosis, as well as differentiation. Under each condition, we determined that this apoptotic role for MyoD correlated with the induction of the pro-apoptotic Bcl2 family member PUMA. Moreover, this increase in PUMA expression was detected withing three hours of culture in DM. Thus, we identified MyoD as a molecule in common to both the induction of differentiation and apoptosis [16]. A direct role for MyoD in transcriptional regulation of a plethora of genes is well established. As genome-wide studies were aimed at understanding MyoD activity in proliferating or differentiating cells, the earliest time point investigated following culture in DM was six hours [17–20].
Herein, we report a direct role for MyoD in the elevated transcription of PUMA following only three hours of culture in DM. Using luciferase reporter analysis and chromatin immunoprecipitation (ChIP) analysis, we confirm recruitment of MyoD to the DNA region encompassing position −857 of the extended PUMA promoter in response to culture in DM. This recruitment of MyoD to the PUMA promoter is comparable to the recruitment of MyoD to the differentiation-associated myogenin promoter in response to culture in DM. Finally, in response to culture in DM, we report that the transcription factor p53 is also recruited to its well-established binding site in the PUMA promoter. However, in myoblasts silenced for MyoD expression, p53 is no longer recruited to the PUMA promoter. These data confirm a direct role for MyoD in regulating PUMA transcription and reveal functional consequences of MyoD expression on p53 mediated transcription of PUMA in skeletal myoblasts.
Methods
Cells and cell culture
The growth of 10T1/2 fibroblasts and ER-MyoD:10T1/2 fibroblasts, as well as the growth and differentiation properties 23A2 myoblasts and 23A2 myoblasts silenced for MyoD expression, have been reported previously [11, 16, 21].
Quantitative RT-PCR and PCR
Myoblasts were plated at equal density and the next day cultured as indicated in the figure and/or figure legend. For quantitative RT-PCR, total RNA was prepared using 1 mL of Trizol (Invitrogen) reagent per 100 mm plate for lysis and following the manufacturer’s instructions. Five hundred μg of RNA was then used for a 20 L SuperScript III RT (Invitrogen) reverse transcription reaction. Quantitative PCR (qPCR) was performed using a Bio-Rad DNA Engine Opticon 3 Real-Time PCR System using SYBR® Green Master PCR Mix according to the manufacturer’s instructions (Qiagen). qPCR for PUMA was performed as described [15]. Following chromatin immunoprecipitation and DNA purification, qPCR was performed for PUMA, myogenin and GAPDH using the indicated promoter-specific primers (given below) under the following conditions: 1 cycle at 95 for 15 min, 40 cycles of 94 1 min, 58 1 min, 72 1 min; and a final extension step at 72 for 5 minutes followed by analysis of the melting curve.
PUMA (−2000/−1643) forward 5′-GGGTCCCTGAACCCCGAGGA-3′
reverse 5′-CCAAGCCCATTTTTGAGCACAGCA-3′,
PUMA (−1170/−856) forward 5′-TGACACCCTTTCACAGCGGGC-3′
reverse 5′GGCCCGCCTGGCGTAATACC-3′,
PUMA (−936/−557) forward 5′-CGGGCATGTCTGTGCCAGA-3′
reverse 5′-ACGCACAAACCCGTGTCCCC-3′
PUMA (−558/+225) forward 5′-TGGGGACACGGGTTTGTGCG-3′
reverse 5′-CACCCCGGGGGCATGAACAC-3′
myogenin promoter forward 5′-GAATCACATGTAATCCACTGGA-3′
reverse 5′-ACGCCAACTGCTGGGTGCCA-3′
GAPDH forward 5′-GGGGGTTTGGTGCCCTCTGGT-3′
reverse 5′-TCCTTGGAGGCCATGTAGGCCAT-3′
Reporter construct creation
The 5′ promoter region of the PUMA gene (−2000/+225) was amplified by PCR using the primers noted below from fosmid (WI1-216 M21 from CHORI) containing a partial murine chromosome 7 including the PUMA gene. The PCR products were inserted into the TOPO TA gateway vector pCR2.1 TOPO per manufacturer’s instructions (Invitrogen Life Technologies). TOPO vectors containing inserted fragments, as determined by visualization following agarose gel electrophoresis, and the luciferase reporter vector pGL4.11 were digested using EcoRV and Hind III or KpnI, depending on the orientation of the insert into the TOPO vector as determined by digestion and visualization following agarose gel electrophoresis. Digested products from pCR2.1 TOPO and linearized pGL4.11 were resolved by agarose gel electrophoresis, and bands were gel purified (5 PRIME) followed by ligation using T4 DNA ligase (New England Biolabs). All vectors were sequenced to confirm the fidelity of all inserts. Plasmid pPUMA −2000/+225 was constructed by amplifying genomic region of the PUMA promoter corresponding to −2000 to +225 base pairs from the transcriptional start site using forward 5′-GGGTCCCTGAACCCCGAGGA-3′ primer and reverse 5′-CACCCCGGGGGCATGAACAC-3′ primer. Plasmid pPUMA −1170/+225 was constructed by amplifying genomic region of the PUMA promoter corresponding to −1170 to +225 base pairs from the transcriptional start site using forward primer 5′-TGACACCCTTTCACAGCGGGC-3′ and reverse primer 5′-CACCCCGGGGGCATGAACAC-3′. Plasmid pPUMA −558/+225 was constructed by amplifying a genomic region of the PUMA promoter corresponding to −558 to +225 base pairs from the transcriptional start site using forward primer 5′-TGGGGACACGGGTTTGTGCG-3′ and reverse primer 5′CACCCCGGGGGCATGAACAC-3′. Plasmid pPUMA −2000/−557 was constructed by amplifying genomic region of the PUMA promoter corresponding to −2000 to −557 base pairs from the transcriptional start site using forward primer 5′ - GGGTCCCTGAACCCCGAGGA-3′ and reverse primer 5′ - ACGCACAAACCCGTGTCCCC-3′.
Transient transfection and Luciferase reporter assays
An equal number of cells were plated on gelatin-coated 6 well plates 24 hours prior to transfection. PUMA promoter-luciferase reporter plasmids or empty pGL4.11 plasmid, as well as pcMyoD or empty vector were co-transfected with a pRK-TK plasmid (Promega 10ng/sample) using Lipofectamine and PLUS reagent (Invitrogen). Dual luciferase assays were performed using the Dual-Luciferase Assay Kit (E1910, Promega) according to manufacturers instructions. Experiments were performed within the linear range of the assay, and a background (no lysate) measurement was taken and subtracted from each experiment. The activity of Photinus pyralis luciferase encoded by the reporter plasmid was normalized in each transfection to the activity of Renilla reniformus luciferase. All experiments were performed at least two times and each in triplicate.
Chromatin immunoprecipitation
ChIP was performed following the protocol provided in the EZ-ChIP™ kit (Millipore/Upstate) and as described in [22]. Cells were plated on 150mm plates and next day cultured in GM or DM for 3 or 8 hours as indicated. Cells were fixed in 0.5%formaldehyde for 10 minutes at room temperature. Formaldehyde was inactivated by the addition of .125M glycine to the cells for 5 minutes at room temperature. Cells were then washed with ice-cold PBS containing 5mM Na Butyrate and 0.5mM PMSF and pelleted by centrifugation at 1500 rpm for 5 minutes and then resuspended in 5ml cold Cell Lysis Buffer (CLB: 60 mM KCl, 15 mM NaCl, 5mM MgCl, 10 mM Tris pH 7.4, 300mM sucrose, 0.1 mM EGTA, 0.1% NP-40, 5 mM Na Butyrate, 0.5 mM PMSF). Cells were sonicated once for 10 sec to ensure lysis of the plasma membrane. Isolated nuclei were washed once in 30 ml of CLB and once in 1 ml of cold Nuclei Digestion Buffer (Cell Lysis Buffer without NP-40 and PMSF). For MNase digestion, intact nuclei were resuspended in 125 μl of Nuclei Lysis Buffer (prewarmed to 37° C), digested with MNase (50 units/ml) at 37° C for 5 minutes, and terminated by 5 mM EDTA. An aliquot from each sample was assessed for sufficient chromatin fragmentation (500–1000bp) by gel electrophoresis. Samples were sonicated twice to ensure lysis of the nuclei prior to immunoprecipitation. The remaining steps of the immunoprecipitation were performed using the EZ ChIP™ Chromatin Immunoprecipitation Kit (Upstate) per manufacturer’s instructions. Subsequently, anti-MyoD (M-318: sc-760 Santa Cruz Biotechnology), anti-p53 (OP03: Calbiochem) or appropriate IgG control (Sigma-Aldrich) were added for immunoprecipitation. For each immunoprecipitation, 5 g of the appropriate antibody was incubated with a precleared chromatin aliquot overnight at 4 C with rotation. The next day, protein A/G sepharose beads were added and incubated for 1 hour at 4 with rotation. The immunoprecipitates were pelleted, washed and the antibody-protein-DNA complex was eluted from bead by incubation in 100mM NaHCO3 and 1%SDS. Following immunoprecipitation and elution, the eluent was treated with RNase A followed by reverse crosslinking by incubation at 65 C overnight. Protein was removed by addition of proteinase K and incubation at 45 for 2 hours. DNA was purified using mini columns provided in the kit. qPCR was performed as described in the “Quantitative RT-PCR and PCR” section. Data was normalized to the signal detected from the input of each sample. The fold enrichment of each target site was calculated as 2 to the power of the cycle threshold (cT) difference between input chromatin and ChIP samples.
Western Analysis
Lysates were prepared and 50 g were denatured and electrophoresed through denaturing polyacrylamide gels (10%) followed by electrophoretic transfer as previously described (16,22). Membranes were blocked for one hour in 1× TBS/0.1%NP40 with 10% newborn calf serum and 5% dry milk and incubated at 4C overnight with anti-p53 (Oncogene:OP03, diluted 1:1000) or anti-MyoD or anti-hsp70 for loading and transfer control of each Western analysis (BD Biosciences, each diluted 1:1000). Appropriate HRP-conjugated secondary antibodies, diluted 1:10,000, were incubated with the membranes for one hour. After each incubation with antibody and prior to the addition of chemiluminescent substrate, membranes were washed five times in 1xTBS (Tris- buffered saline pH 7.4) with 1% Tween 20. Membranes were then incubated with (SuperSignal West Pico Chemiluminescent Substrate: Thermo Scientific: #34078) for 60 seconds and bands were visualized using (Li-Cor Phospho-imager: Image Studio Ver. 2.1).
Results
Identification of a MyoD responsive element in the PUMA extended promoter
We have previously reported that increased levels of PUMA were required for a subset of myoblasts to undergo apoptosis rather than differentiation in response to culture in differentiation media (DM) [15]. Further, we reported that activation of MyoD correlated with increased PUMA mRNA and protein levels following three hours of culture in DM [16]. Since MyoD is a transcription factor, this effect could be either as a consequence of direct binding to a responsive element in the PUMA gene or the increased expression of another factor. As a preliminary experiment to assess the possibility that MyoD might be directly responsible for driving the increase in PUMA mRNA levels as a consequence of culture in DM, we cultured either 23A2 or C2C12 myoblasts in DM supplemented with cycloheximide (CHX) sufficient to block new protein synthesis. Doing so did not prevent the increase in PUMA mRNA (Figure 1A). To demonstrate further the specificity of this increase in PUMA mRNA to the expression and activation of MyoD, we analyzed the effect of CHX on the increase in PUMA mRNA in 10T1/2 fibroblasts stably expressing an estrogen receptor:MyoD fusion (ER-MyoD:10T1/2) and in 10T1/2 fibroblasts transiently transfected with MyoD [21]. In neither case, either in response to MyoD expression or activation by estradiol, did CHX diminish the increase in PUMA mRNA (Figure 1B and C).
Figure 1.
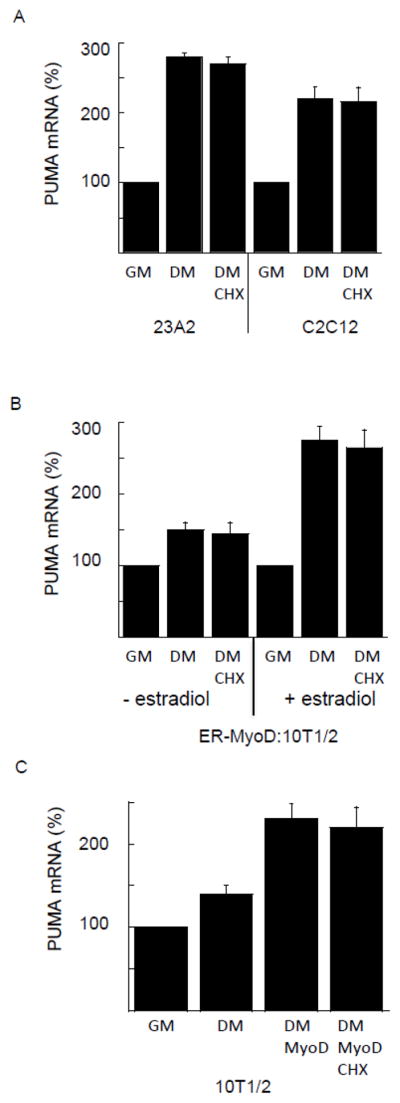
New protein synthesis is not required for increased PUMA mRNA in response to serum withdrawal. In (A) equal cell numbers of 23A2 or C2C12 myoblasts were plated and the next day switched to fresh GM or DM with or without 10 g/ml cycloheximide (CHX) for three hours. In (B) equal cell numbers of ER-MyoD:10T1/2 fibroblasts were plated and the next day pretreated with estradiol (1 M) before switching to fresh GM or DM with or without CHX with or without estradiol as indicated for three hours. In (C), equal cell numbers of 10T1/2 fibroblasts were plated and the next day transfected with 1 g pcMyoD. Two days later, cultures were switched to fresh GM or DM with or without CHX for three hours. Quantitative RT-PCR was performed as described in Methods. Shown is an average of 3 experiments (mean +/− SEM).
To search for a MyoD responsive element in the PUMA extended promoter, we created a luciferase reporter construct containing −2000/+225 of the PUMA gene. In silico analysis using Mat Inspector revealed three canonical E-boxes within this region (E1 at position −1987, E2 at position −857 and E3 at position −320). Both the E2 and E3 box motifs (CAGGTG) were specific for MyoD [23]. Transfection of this reporter construct (Figure 2A) into 23A2 myoblasts followed by luciferase activity analysis to measure promoter activity revealed induction of luciferase expression in a dose responsive manner, ranging from 130 fold induction when 125 ng of the reporter construct was transfected to 1,300 fold induction when 1 g of the reporter construct was transfected (Figure 2B). To determine the contribution of MyoD to this induction, we utilized several approaches. Firstly, we utilized cell lines that we had previously created to demonstrate a role for MyoD in PUMA mRNA and protein induction, and the ensuing apoptosis, in response to culture in DM [16]. 23A2 parental myoblasts, 23A2 myoblasts mock silenced for MyoD expression (A2:mock), or 23A2 myoblasts silenced for MyoD expression (clones A2:13 and A2:15) were transfected with the luciferase reporter construct containing −2000/+225 of the PUMA gene followed by luciferase analysis to measure promoter activity. Again (see Figure 2B), transfection of 500ng of the reporter construct yielded a nearly 500 fold induction of luciferase and this was significantly reduced in the myoblast cell lines silenced for MyoD expression (Figure 2C; p=0.015 for A2:13 and p=0.001 for A2:15). Next, we transfected the luciferase reporter construct with and without MyoD into 10T1/2 fibroblasts. The expression of MyoD increased the induction of luciferase from roughly 200 fold to roughly 500 fold (p=0.02) when cells were cultured in DM (Figure 2D). Finally, we utilized 10T1/2 fibroblasts which stably express an estrogen receptor (ER):MyoD fusion protein [21]. We have previously reported that treatment of ER-MyoD:10T1/2 cells with estradiol in DM is sufficient to induce differentiation, or PUMA mRNA and protein expression and the ensuing apoptosis, similar to that observed in 23A2 myoblasts and that estradiol has no effect on these parameters in parental 10T/12 fibroblasts [16]. We now report that estradiol-mediated activation of the ER:MyoD fusion protein also induces an increase in the induction of luciferase driven by the extended PUMA promoter similar to that observed following transfection of MyoD in 10T1/2 fibroblasts, from roughly 200 fold to roughly 500 fold (p=0.03) and that estradiol has no such effect in parental 10T1/2 fibroblasts (Figure 2E). To localize the MyoD responsive element, we created reporter contracts with various deletions in the PUMA extended promoter. Following transfection into 23A2 myoblasts and subsequent culture in DM for three hours, luciferase activity measurements revealed that E1 was not required for MyoD responsiveness (Figure 2F). However, a deletion mutant also lacking E2 resulted in a significant decrease in luciferase activity (Figure 2E, p=0.008). This result indicated that E2 was required for MyoD responsiveness. Consistent with these results, sequence alignment indicated that only E2 is conserved between humans and mice. Western analysis confirmed elevation of endogenous MyoD levels indicative of ER-MyoD activation (Figure 3A and (21)). We also performed Western analysis to confirm expression of transiently transfected MyoD (Figure 3B) and to confirm silencing of MyoD in 23A2 myoblasts (Figure 3C and (16)).
Figure 2.


Localization of MyoD responsive element in the 5′ extended promoter of PUMA. (A) Schematic representation of construct used in (B–E). In (B), equal cell numbers of 23A2 myoblasts were plated and the next day transfected with the indicated amount of a luciferase reporter construct containing −2000/+225 of the PUMA gene. The next day, cells were switched to DM for three hours prior to the determination of luciferase activity as described in Methods. In (C), equal cell numbers of 23A2 myoblasts, 23A2 myoblasts mock silenced for MyoD expression (A2:mock), or 23A2 myoblasts silenced for MyoD expression (clones A2:13 and A2:15) were plated and the next day transfected with 500ng of the luciferase reporter construct containing −2000/+225 of the PUMA gene. The next day, cells were switched to DM for three hours prior to the determination of luciferase activity as described in Methods. In (D), equal cell numbers of 10T1/2 fibroblasts were plated and the next day transfected as described in Methods with 500 ng of the luciferase reporter construct containing −2000/+225 of the PUMA gene with or without pcMyoD. The next day, cultures were switched to DM for three hours prior to the determination of luciferase activity as described in Methods. In (E), equal cell numbers of ER-MyoD:10T1/2 fibroblasts or 10T/12 fibroblasts were plated and the next day transfected as described in Methods with 500 ng of the luciferase reporter construct containing −2000/+225 of the PUMA gene. The next day, cultures were pretreated with estradiol (1 M) as indicated before switching to DM with or without estradiol as indicated for three hours prior to the determination of luciferase activity as described in Methods. In (F), equal cell numbers of 23A2 myoblasts were plated and the next day transfected with the luciferase reporter construct containing −2000/+225 of the PUMA gene or the indicated deletion mutant (500 ng). The next day, cells were switched to DM for three hours prior to the determination of luciferase activity as described in Methods. Shown for each (B–F) is an average of at least 2 experiments (mean +/− SEM).
Figure 3.

Confirmation of elevated or silenced MyoD levels. For (A), equal cell numbers of ER-MyoD:10T1/2 cells were plated and the next day cultured with or without estradiol (1 M) as indicated. In (B), equal cell numbers of 10T1/2 cells were plated and the next day transfected as described in Figure legend 1. In (C), equal cell numbers of 23A2 myoblasts, 23A2 myoblasts mock silenced for MyoD expression (A2:mock), or 23A2 myoblasts silenced for MyoD expression (clones A2:13 and A2:15) were plated. In (A–C), after treatment or transfection as indicated, lysates were prepared and Western analysis to measure MyoD levels was performed as described in Methods.
Binding of MyoD to a region in the extended PUMA promoter containing E2
Having used transactivation assays to confirm the transcriptional activity of MyoD on the PUMA extended promoter, we next sought to measure MyoD recruitment using chromatin immunoprecipitation analysis. We first assayed for MyoD recruitment over the entire 2.5 kb region in response to culture in DM. Confirming the results of our experiments utilizing the deletion mutant reporter construct, we determined a roughly four-fold increase in MyoD recruitment to the region encompassing the E2 site after three hours of culture in DM, relative to that detected in GM. Further, after eight hours of culture in DM, the level of MyoD recruited to this region had returned to the level detected in GM (Figure 4A). We confirmed the specificity of this recruitment of MyoD to the region encompassing the E2 site by comparing the 23A2 parental myoblasts and 23A2 myoblasts mock silenced for MyoD expression (A2:mock) to the 23A2 myoblasts silenced for MyoD expression (clones A2:13 and A2:15). Again, in both the 23A2 parental myoblasts and 23A2 myoblasts mock silenced for MyoD expression (A2:mock), we detected a roughly four-fold increase in MyoD recruitment after three hours of culture in DM and that returned to GM levels after eight hours of culture in DM. We detected no recruitment above GM levels in either of the clones silenced for MyoD expression (clones A2:13 and A2:15) (Figure 4B). To assess the significance of a four-fold recruitment of MyoD to the PUMA promoter, we compared the level of recruitment of MyoD to the differentiation-associated myogenin promoter [17–20]. After three hours of culture in DM, we also detected a roughly four-fold increase in MyoD recruitment relative to GM in both 23A2 parental myoblasts and 23A2 myoblasts mock silenced for MyoD expression (A2:mock). However, a three-fold increase in MyoD could still be detected at the myogenin promoter in both of these cell lines after eight hours of culture in GM (Figure 4C). As predicted, we detected no recruitment of MyoD to the myogenin promoter above GM levels in either of the clones silenced for MyoD expression (clones A2:13 and A2:15) (Figure 4C).
Figure 4.


Binding of MyoD to a region in the extended PUMA promoter containing E2. For (A–C), equal cell numbers were plated and the next day cultured as indicated. Chromatin Immunoprecipitation was performed on each cell sample using EZ ChIP™ Chromatin Immunoprecipitation Kit (Upstate) per manufacturer’s instructions using 5 g anti-MyoD (M-318) (sc-760 Santa Cruz Biotechnology) or IgG control (Sigma-Aldrich). Quantitative PCR was used to assay for the relative levels of the indicated DNA as described in Methods. Data was normalized to the signal detected from the input of each sample. Error bars represent mean +/− SEM of triplicates.
Effect of MyoD silencing on recruitment of p53 to its binding site in the PUMA promoter
Transcription driven by MyoD typically requires two E-boxes or an E-box and a cooperative co-activator [24]. PUMA (p53 upregulated modulator of apoptosis) was originally identified as a transcriptional target of the tumor suppressor p53 [25, 26]. Thus, we next assessed the recruitment of p53 to its binding site in the PUMA promoter. Similar to that determined for MyoD binding, the recruitment of p53 to its site was increased four-fold after 3 hours of culture in DM when compared to GM (Figure 5A) in both parental 23A2 and 23A2 myoblasts mock silenced for MyoD expression (A2:mock). Again similar to MyoD binding, this recruitment returned to levels detected in GM after eight hours of culture in DM (Figure 5A). Surprisingly, p53 was not recruited to the PUMA promoter in either of the clones silenced for MyoD expression (clones A2:13 and A2:15) (Figure 5A). Since the expression of a dominant negative (dn) p53, as well as shRNA-mediated knockdown of p53, has been reported to decrease the expression of MyoD [27, 28], we next assessed the effect of silencing MyoD on the expression of p53. Consistent with previous reports, p53 expression is increased in response to culture in DM [29]. However, silencing MyoD did not affect the expression of p53 in either GM or DM (Figure 5B).
Figure 5.

Increased binding of p53 to the extended PUMA promoter in response to culture in DM does not occur in the absence of MyoD binding. For all, equal cell numbers were plated and the next day cultured as indicated. In (A) Chromatin Immunoprecipitation was performed on each cell sample using EZ ChIP™ Chromatin Immunoprecipitation Kit (Upstate) per manufacturer’s instructions using 5 g anti-p53 (OP03-Calbiochem) or IgG control (Sigma-Aldrich). Quantitative PCR was used to assay for the relative levels of the PUMA (−936/−556). Data was normalized to the signal detected from the input of each sample. Error bars represent mean +/− SEM of triplicates. In (B) Western analysis to measure p53 levels was performed as described in Methods. Shown are results from one experiment that are representative of three independent experiments.
Discussion
Improving the efficacy of protocols utilizing skeletal myoblast transfer or skeletal muscle tissue engineering, as well as maximizing the regenerative potential of adult muscle stem cells, requires a thorough understanding of myogenesis. Since the discovery of the MyoD family of basic-helix-loop-helix (bHLH) muscle regulatory transcription factors, skeletal myogenesis has served as the paradigm for understanding lineage specification and differentiation [30, 31]. Further, the MyoD family is capable of activating the myogenic program in lieu of other cell fates initiating the concept of transdifferentiation, a major emphasis of the stem cell field of regenerative medicine. MyoD is uniquely responsible for the differentiation of the adult myoblast stem cells [30]. Thus, a thorough understanding of the varied and extensive ability of MyoD to regulate gene expression is critical to our understanding of muscle regeneration, skeletal myoblast transfer, skeletal muscle tissue engineering as well as cell transdifferentiation.
The mutually exclusive biological endpoints of differentiation and apoptosis are coordinately induced in skeletal myoblasts by culture in differentiation media (DM). The fact that these events are mutually induced by culture in DM necessitates signaling events in common, while the fact that these events are mutually exclusive necessitates a bifurcation in that signaling pathway. We have previously reported that silencing of MyoD impairs both differentiation and apoptosis of skeletal myoblasts, implicating MyoD as a signaling molecule in common to both pathways, while we have identified PUMA as a signaling molecule unique to the apoptotic pathway [15, 16]. We also correlated the role for MyoD in the apoptotic pathway with the expression of PUMA [16] and herein identify the MyoD responsive element in the PUMA promoter and confirm MyoD binding to a canonical E-box at position −857. Since MyoD is clearly a master regulator of genes required for skeletal myoblast differentiation, our data indicates that a distinction in the MyoD molecules (such as a unique post-translational modification) and/or binding partner(s) must exist in cells where MyoD induces PUMA and apoptosis rather than differentiation (Figure 6). Our discovery of the MyoD responsive element in the PUMA gene will facilitate future studies to test this hypothesis and to elucidate the mechanism whereby MyoD drives PUMA expression and apoptosis rather than differentiation.
Figure 6.
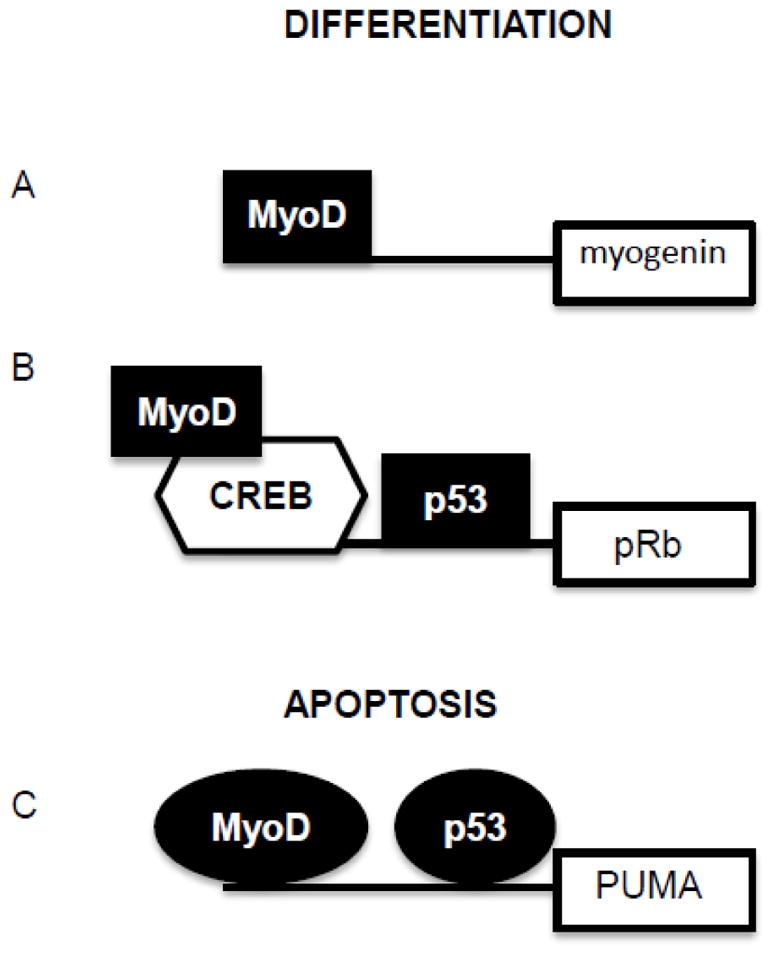
Proposed model for the coordinated regulation of differentiation and apoptosis by MyoD and p53. MyoD is well known as a pioneer transcription factor responsible for the differentiation of skeletal myoblasts. MyoD drives transcription necessary for differentiation through both direct (A) and indirect (B) binding to DNA. p53 is well known for its role in tumor suppression as a pivotal transcription factor responsible for interpreting the extent of DNA damage into either cell cycle arrest or apoptosis (C). Shown are two examples where both p53 and MyoD sites have been confirmed. We propose that post-translational modification(s) or distinct binding partners, portrayed herein as shape changes, could explain the mutually exclusive, dual, biological roles in differentiation or apoptosis for both of these key transcription factors.
MyoD mediated transcription requires binding to two E-boxes or one E-box and a cooperative co-activator [24]. Since only the E-box at position 857 is conserved among species and we detected binding only to this E-box, we speculated that the transcription factor p53 might also be involved [25, 26]. The timing and fold recruitment of MyoD to the PUMA promoter was similar to that observed for p53 in that the level of each was four-fold that detected in GM after three hours of culture in DM but equal to that detected in GM after eight hours of culture in DM. The fold recruitment of MyoD to the PUMA promoter was comparable to the fold recruitment of MyoD to the differentiation-associated myogenin promoter in response to culture in DM. However, unlike recruitment of MyoD to the PUMA promoter, the recruitment of MyoD to the myogenin promoter was still three-fold after eight hours of culture in DM. Thus, the persistence of MyoD at the PUMA promoter mimics the persistence of p53 at the PUMA promoter, and both are distinct from the persistence of MyoD at the myogenin promoter in response to culture in DM. A role for p53 in the induction of PUMA and apoptosis presents a similar situation to that of MyoD in that p53 has likewise been shown to be required for differentiation, for example by virtue of the participation of both MyoD and p53 in the induction of pRb as well as other differentiation associated genes [27, 29, 32]. Thus, we again hypothesize that a distinction in the p53 molecules (such as a unique post-translational modification) must exist in cells where p53 participates in the induction of PUMA and apoptosis rather than the induction of pRb and differentiation (Figure 5). Many studies have documented molecular distinctions in signaling in response to DNA damage that allows p53 to either induce genes responsible for growth arrest or genes responsible for apoptosis [33] and we speculate that the scenario in skeletal myoblasts in response to culture in DM may be analogous. We acknowledge that solid data has been published suggesting that p53 is not required for the differentiation associated apoptosis of skeletal myoblasts [34]. These studies were performed using reporter constructs to assess p53 mediated transcription. However, expression from these reporter constructs did not mimic the expression of endogenous Rb known to be p53-dependent in skeletal myoblasts [27, 34]. Further, these studies were performed in myoblasts isolated from p53 null mice or in established myoblasts expressing dnp53. For the former, we speculate that compensatory mechanisms could have evolved to substitute for p53 and for the latter we speculate that the level of dnp53 expression may not have been sufficient to block apoptosis. This study pre-dated our discovery of the importance of PUMA expression in skeletal myoblast apoptosis and did not assess the level of PUMA. Moreover, we published that the p53 pharmacological inhibitor Pifithrin, at a concentration sufficient to block differentiation, did not impair PUMA induction [16]. We have since repeated those experiments with a higher concentration of Pifithrin and detected inhibition of PUMA induction (data not shown).
We also report herein that increased binding of p53 to the PUMA promoter in response to culture in DM does not occur in the absence of MyoD. Since MyoD is known to extensively modify the epigenome by binding to thousands of sites in addition to those responsible for driving the expression of muscle-specific genes [18], myoblasts silenced for MyoD expression are not the equivalent of cells that have never expressed MyoD. For instance, non-myoblasts do not elevate the level of p21WAF1 in response to culture in DM, but myoblasts lacking MyoD do elevate the level of p21WAF1 in response to culture in DM [35–37]. Moreover, the ability of p53 to directly regulate Rb expression is unique to myoblasts and perhaps unique to the presence of MyoD [38]. Thus, this MyoD-dependent binding of p53 to the PUMA promoter in response to culture in DM may be myoblast specific and could be a consequence of either direct MyoD binding, MyoD induced genes, and/or MyoD induced changes to the epigenome [18]. In non-muscle cell types like 10T1/2 fibroblasts, serum withdrawal (culture in DM) results in cell cycle exit rather than apoptosis, despite a modest increase in PUMA induction [16]. Nonetheless, p53 binding to its promoter site in the PUMA gene and the subsequent induction of PUMA transcription in non-muscle cell types in response to DNA damage is clearly sufficient to elicit an apoptotic response [37]. It remains to be determined if p53 binding to the PUMA promoter requires MyoD in myoblasts in response to DNA damage. Experiments are underway to determine if this MyoD-dependent binding of p53 to the PUMA promoter is stimuli specific or if p53 binding to the Rb promoter requires MyoD.
Acknowledgments
This work was supported by NIH grant AR053857-S1 and funds awarded by Cleveland State and the Center for Gene Regulation in Health and Disease to C. M. Weyman.
References
- 1.Ceafalan LC, Popescu BO, Hinescu ME. Cellular players in muscle regeneration. Biomed Res Int. 2014 doi: 10.1155/2014/957014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ten Broek RW, Grefte S, Von den Hoff JW. Regulatory factors and cell populations involved in skeletal muscle regeneration. J Cell Physiol. 2010;224(1):7–16. doi: 10.1002/jcp.22127. [DOI] [PubMed] [Google Scholar]
- 3.Partridge TA, Morgan JE. Multiple insights from myogenic cell transplants. Hum Gene Ther. 2014;25(5):404–5. doi: 10.1089/hum.2014.035. [DOI] [PubMed] [Google Scholar]
- 4.Grenier G, Rudnicki MA. The potential use of myogenic stem cells in regenerative medicine. Handb Exp Pharmacol. 2006;174:299–317. [PubMed] [Google Scholar]
- 5.Skuk D, Caron NJ, Goulet M, Roy B, Tremblay JP. Resetting the problem of cell death following muscle-derived cell transplantation: detection, dynamics and mechanisms. J Neuropathol Exp Neurol. 2003;62:951–67. doi: 10.1093/jnen/62.9.951. [DOI] [PubMed] [Google Scholar]
- 6.Skuk D, Tremblay JP. Clarifying Misconceptions About Myoblast Transplantation in Myology. Molecular Therapy. 2014;22:897–898. doi: 10.1038/mt.2014.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercuri E, Muntoni F. Muscular dystrophy: new challenges and review of the current clinical trials. Curr Opin Pediatr. 2013;25(6):701–7. doi: 10.1097/MOP.0b013e328365ace5. [DOI] [PubMed] [Google Scholar]
- 8.Cheng CS, Davis BN, Madden L, Bursac N, Truskey GA. Physiology and metabolism of tissue-engineered skeletal muscle. Exp Biol Med. 2014;239:1203–1214. doi: 10.1177/1535370214538589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandri MC, Massimino ML, Geromel V, Arslan P. Myoblasts and myotubes in primary cultures deprived of growth factors undergo apoptosis. Basic Appl Myol. 1996;6:257–260. [Google Scholar]
- 10.Miller JB, Stockdale FE. Developmental regulation of the multiple myogenic cell lineages of the avian embryo. J Cell Biol. 1986;103:2197–208. doi: 10.1083/jcb.103.6.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dee K, Freer M, Mei Y, Weyman CM. Apoptosis coincident with the differentiation of skeletal myoblasts is delayed by caspase 3 inhibition and abrogated by MEK-independent constitutive Ras signaling. Cell Death Differ. 2002;9:209–18. doi: 10.1038/sj.cdd.4400930. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Walsh K. Resistance to apoptosis conferred by Cdk inhibitors during myocyte differentiation. Science. 1996;273:359–61. doi: 10.1126/science.273.5273.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandi M, Carraro U. Apoptosis of skeletal muscles during development and disease. Int J Biochem Cell Biol. 1999;12:1373–1390. doi: 10.1016/s1357-2725(99)00063-1. [DOI] [PubMed] [Google Scholar]
- 14.Fernando P, Megeney LA. Is caspase-dependent apoptosis only cell differentiation taken to the extreme? FASEB J. 2006;21(1):8–17. doi: 10.1096/fj.06-5912hyp. [DOI] [PubMed] [Google Scholar]
- 15.Shaltouki A, Freer M, Mei Y, Weyman CM. Increased expression of the pro-apoptotic Bcl(2) family member PUMA is required for mitochondrial release of cytochrome C and the apoptosis associated with skeletal myoblast differentiation. Apoptosis. 2007;12:2143–54. doi: 10.1007/s10495-007-0135-z. [DOI] [PubMed] [Google Scholar]
- 16.Harford TJ, Shaltouki A, Weyman CM. Increased expression of the pro-apoptotic Bcl2 family member PUMA and apoptosis by the muscle regulatory factor MyoD in response to a variety of stimuli. Apoptosis. 2010;15(1):71–82. doi: 10.1007/s10495-009-0428-5. [DOI] [PubMed] [Google Scholar]
- 17.Bergstrom DA, Penn BH, Strand A, Perry RL, Rudnicki MA, Tapscott SJ. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol Cell. 2002;9(3):587–600. doi: 10.1016/s1097-2765(02)00481-1. [DOI] [PubMed] [Google Scholar]
- 18.Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, Gentleman RC, Tapscott SJ. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell. 2010;18(4):662–74. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mousavi K, Zare H, Dell’orso S, Grontved L, Gutierrez-Cruz G, Derfoul A, Hager GL, Sartorelli V. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell. 2013;51(5):606–17. doi: 10.1016/j.molcel.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soleimani VD, Yin H, Jahani-Asl A, Ming H, Kockx CE, van Ijcken WF, Grosveld F, Rudnicki MA. Snail regulates MyoD binding site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol Cell. 2012;47(3):457–68. doi: 10.1016/j.molcel.2012.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollenberg SM, Cheng PF, Weintraub H. Use of a conditional MyoD transcription factor in studies of MyoD trans-activation and muscle determination. Proc Natl Acad Sci U S A. 1993;90:8028–32. doi: 10.1073/pnas.90.17.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freer-Prokop M, O’Flaherty J, Ross J, Weyman CM. Non-canonical role for the TRAIL receptor DR5/FADD/caspase pathway in the regulation of MyoD expression and skeletal myoblast differentiation. Differentiation. 2009;78(4):205–212. doi: 10.1016/j.diff.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fong AP, Yao Z, Zhong JW, Cao Y, Ruzzo WL, Gentleman RC, Tapscott SJ. Genetic and epigenetic determinants of neurogenesis and myogenesis. Dev Cell. 2012;22(4):721–35. doi: 10.1016/j.devcel.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fong AP, Tapscott SJ. Skeletal muscle programming and reprogramming. Curr Opin Genet Dev. 2013;23(5):568–573. doi: 10.1016/j.gde.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakano K, Vousden KH. PUMA, a novel pro-apoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 26.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 27.Cerone MA, Coen S, Gurtner A, Fontemaggi G, Cimino L, Piaggio G, Sacchi A, Soddu S. p53 regulates myogenesis by triggering the differentiation activity of pRb. J Cell Biol. 2000;151(6):1295–1304. doi: 10.1083/jcb.151.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Couture O, Lombardi E, Davis K, Hays E, Chandar N. Gene expression profiles resulting from stable loss of p53 mirrors its role in tissue differentiation. PLoS One. 2013;8(11):e82494. doi: 10.1371/journal.pone.0082494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamir Y, Bengal E. p53 protein is activated during muscle differentiation and participates with MyoD in the transcription of muscle creatine kinase gene. Oncogene. 1998;17(3):347–356. doi: 10.1038/sj.onc.1201929. [DOI] [PubMed] [Google Scholar]
- 30.Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;2(12):2685–95. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- 31.Rudnicki MA, Le Grand F, McKinnell I, Kuang S. The molecular regulation of muscle stem cell function. Cold Spring Harb Symp Quant Biol. 2008;73:323–31. doi: 10.1101/sqb.2008.73.064. [DOI] [PubMed] [Google Scholar]
- 32.Magenta A, Cenciarelli C, De Santa F, Fuschi P, Martelli F, Caruso M, Felsani A. MyoD stimulates RB promoter activity via the CREB/p300 Nuclear Transduction Pathway. Mol Cell Biol. 2003;23(8):2893–2906. doi: 10.1128/MCB.23.8.2893-2906.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida K, Miki Y. The cell death machinery governed by the p53 tumor suppressor in response to DNA damage. Cancer Sci. 2010;101(4):831–5. doi: 10.1111/j.1349-7006.2010.01488.x. [DOI] [PubMed] [Google Scholar]
- 34.Cerone MA, Marchetti A, Bossi G, Blandino G, Sacchi A, Soddu S. p53 is involved in the differentiation but not in the differentiation-associated apoptosis of myoblasts. Cell Death Differ. 2000;7(5):506–8. doi: 10.1038/sj.cdd.4400676. [DOI] [PubMed] [Google Scholar]
- 35.Parker SB, Eichele G, Zhang P, Rawls A, Sands AT, Bradley A, Olson EN, Harper JW, Elledge SJ. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science. 1995;267:1024–1027. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- 36.Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- 37.Guo K, Wang J, Andrés V, Smith RC, Walsh K. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol. 1995;15(7):3823–3829. doi: 10.1128/mcb.15.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osifchin NE, Jiang D, Ohtani-Fujita N, Fujita T, Carroza M, Kim SJ, Sakai T, Robbins PD. Identification of a p53 binding site in the human retinoblastoma susceptibility gene promoter. J Biol Chem. 1994;269(9):6383–9. [PubMed] [Google Scholar]