

Abstract
Paddy fields are important ecosystems, as rice is the primary food source for about half of the world’s population. Paddy fields are impacted by nitrogen fertilization and are a major anthropogenic source of methane. Microbial diversity and methane metabolism were investigated in the upper 60 cm of a paddy soil by qPCR, 16S rRNA gene amplicon sequencing and anoxic 13C-CH4 turnover with a suite of electron acceptors. The bacterial community consisted mainly of Acidobacteria, Chloroflexi, Proteobacteria, Planctomycetes, and Actinobacteria. Among archaea, Euryarchaeota and Bathyarchaeota dominated over Thaumarchaeota in the upper 30 cm of the soil. Bathyarchaeota constituted up to 45% of the total archaeal reads in the top 5 cm. In the methanogenic community, Methanosaeta were generally more abundant than the versatile Methanosarcina. The measured maximum methane production rate was 444 nmol gdwh-1, and the maximum rates of nitrate-, nitrite-, and iron-dependent anaerobic oxidation of methane (AOM) were 57 nmol, 55 nmol, and 56 nmol gdwh-1, respectively, at different depths. qPCR revealed a higher abundance of ‘Candidatus Methanoperedens nitroreducens’ than methanotrophic NC10 phylum bacteria at all depths, except at 60 cm. These results demonstrate that there is substantial potential for AOM in fertilized paddy fields, with ‘Candidatus Methanoperedens nitroreducens’ archaea as a potential important contributor.
Keywords: anaerobic oxidation of methane, paddy fields, 16S rRNA gene amplicon sequencing, Methanoperedens nitroreducens, NC10 phylum bacteria, Bathyarchaeota
Introduction
Methane, a significant greenhouse gas, has up to 34 times the global warming potential over 100 years compared to carbon dioxide (Myhre et al., 2013). Paddy fields contribute substantially to atmospheric methane concentrations and release 25–300 Tg of CH4 per annum (Bridgham et al., 2013), representing 10–20% of global methane emissions (Conrad, 2009; Bodelier, 2011). In the next decades, the land area designated for rice cultivation is predicted to increase even further. Without mitigation measures, this will result in elevated methane emission to the atmosphere.
The microbial community structure of paddy fields is influenced by several environmental and anthropogenic factors. Alteration in microbial community composition in paddy fields have been studied with respect to flooding (Rui et al., 2009), fertilization and straw application (Bao et al., 2016), temperature (Conrad et al., 2009; Noll et al., 2010), rice cultivar and soil type (Conrad et al., 2008), and plant growth stage (Breidenbach and Conrad, 2015). Paddy fields provide a habitat for both aerobic and anaerobic methanotrophs. Aerobic methanotrophs are found in the oxic layers of the soil and in oxic microhabitats of the rhizosphere. Methanogenic archaea, anaerobic methanotrophic archaea and/or bacteria thrive preferentially in the anoxic compartments of the waterlogged soil. The flux of methane to the atmosphere is the net result of production and consumption by methanogenic and methanotrophic microorganisms.
Since the discovery of “Bacillus methanicus” (Söhngen, 1906), aerobic methane-oxidizing bacteria (MOB) have been extensively studied. MOBs were long considered the only microbes capable of oxidizing methane. Currently, MOB belong to the phyla Proteobacteria and Verrucomicrobia (Op den Camp et al., 2009; Semrau et al., 2010). Proteobacterial aerobic methanotrophs inhabit a wide variety of environments, ranging from tundra soil (Dedysh et al., 2004) and arctic permafrost (Liebner et al., 2009) to sewage treatment sludge (Ho et al., 2013). Phylogenetic analyses of both 16S rRNA and the particulate methane mono-oxygenase subunit A (pmoA) gene have classified Proteobacteria into Gammaproteobacteria (Type I methanotrophs) and Alphaproteobacteria (Type II methanotrophs) (Trotsenko and Murrell, 2008; Semrau et al., 2010). Type I methanotrophs belong to the genera Methylosarcina, Methylobacter, Methylomonas, Methylomicrobium, Methylosoma, Methylosphera, and Methylovulum (Type Ia) and Methylococcus, Methylocaldum, Methylogaea, Methylohalobius, and Methylothermus (Type Ib). Alphaproteobacterial MOB belong to the genera Methylocystis and Methylosinus (Type IIa) and the genera Methylocella, Methylocapsa, and Methyloferula (Type IIb) (Dumont et al., 2014; Zheng et al., 2014; Knief, 2015). Aerobic methanotrophs have been detected in several paddy field soils (Ho et al., 2011; Lüke and Frenzel, 2011; Lee et al., 2014), and furthermore, it has been suggested that Type I methanotrophs can likely outcompete Type II methanotrophs for substrates in these nitrogen-loaded environments (Zheng et al., 2014). Compared to the proteobacterial aerobic methanotrophs, the more recently discovered Verrucomicrobia often inhabit more extreme environments with low pH values and/or high temperatures (Dunfield et al., 2007; Op den Camp et al., 2009; Sharp et al., 2014; van Teeseling et al., 2014).
Rice cultivation under waterlogged conditions creates anoxia in the majority of soil compartments and, consequently, provides a suitable habitat for methanogenic microorganisms. Rice maturation with the developed and decaying rhizosphere, releases root exudates that, together with dead roots, provide organic matter for an anaerobic food chain. Oxygen influx to soil occurs through diffusional transport via the aerenchyma and radial oxygen loss of the rice roots (Armstrong, 1971; Li and Wang, 2013). Although traditionally considered strict anaerobes, methanogens have been detected in the rhizosphere and on rice roots in several studies (Chin et al., 2004; Xu et al., 2012; Edwards et al., 2015; Lee et al., 2015). Lee et al. (2015) observed a higher abundance of methanogens in the rhizosphere than in bulk soil (Lee et al., 2015). The methanogens in the rhizosphere may live in non-active roots where no oxygen is released or, alternatively, may be oxygen tolerant and have mechanisms to counteract reactive oxygen radical species, as investigated for Rice Cluster I (RC I) (now known as Methanocella) methanogens (Erkel et al., 2006). The genomes of RC I harbor genes encoding catalase, three different superoxide anion scavengers, superoxide dismutase and two different super oxide reductase genes for oxygen detoxification (Erkel et al., 2006). The up-regulation of catalase genes in response to oxygen exposure has been observed in both Methanosarcina and Methanocella (Angel et al., 2011).
Both acetoclastic and hydrogenotrophic methanogens have been identified in paddy fields. Methanogenic archaea of the order Methanosarcinales derive methane from the methyl group of compounds such as methanol and methylamine, and until now, only Methanosarcina and Methanosaeta are known to use acetate for methane production (Jetten et al., 1992; Costa and Leigh, 2014; Welte and Deppenmeier, 2014). Hydrogenotrophic methanogens belonging to the orders Methanomicrobiales, Methanobacteriales and Methanocellales have been commonly found in paddy fields, with the exception of Methanococcales, which barely have been detected (Watanabe et al., 2010; Lee et al., 2015). Many of these hydrogenotrophic methanogens can use formate as a substrate but are unable to utilize acetate. Archaea belonging to RC I (Methanocella) (Kögel-Knabner et al., 2010), which forms a separate phylogenetic lineage branching between the orders Methanosarcinales and Methanomicrobiales, are considered key methanogens in rice fields. The reaction stoichiometry of methanogenesis (Conrad and Klose, 1999) indicates that acetoclastic methanogens could contribute approximately two-thirds to methane production, consistent with the dominance of acetoclastic over hydrogenotrophic methanogenesis in paddy fields (Krüger et al., 2001).
Previous theories suggesting a decrease in methane flux as a result of direct stimulation of methanotrophs after amendment with nitrogen fertilizers were unable to link observations to the activity of the denitrifying anaerobic methanotrophic bacteria and archaea as these microorganisms, were discovered only recently compared to the aerobic methanotrophs. Nitrite- and nitrate-dependent anaerobic oxidation of methane (AOM) were first described in 2006 in an enrichment culture consisting of archaea distantly related to ANME-2d and of bacteria that consume nitrite as an electron acceptor to oxidize methane anaerobically (Raghoebarsing et al., 2006). This novel denitrifying, methanotrophic bacterium of the candidate division NC10 was named ‘Candidatus Methylomirabilis oxyfera’ (Ettwig et al., 2010). Despite its preference for an anoxic habitat, it is postulated to have an intra-aerobic metabolism. The genome of the bacterium contains all genes of the aerobic methanotrophic pathway and encodes a particulate methane mono-oxygenase complex that can use the O2 released from nitric oxide for methane oxidation, similar to aerobic methanotrophs (Ettwig et al., 2010).
The genome of ANME-2d archaea was sequenced in 2013 and responsible organism named ‘Candidatus Methanoperedens nitroreducens’ (Haroon et al., 2013). This nitrate-reducing archaeon employs a reverse methanogenesis pathway to oxidize methane. The genomes of three different strains of ‘Candidatus Methanoperedens nitroreducens’ have been published, and the necessary genes for nitrate reduction and the methanogenic pathway have been identified (Haroon et al., 2013; Arshad et al., 2015; Vaksmaa et al., 2017a,b). Nitrite- and nitrate-dependent AOM microorganisms and/or activity have been detected in several freshwater environments, including paddy fields (Vaksmaa et al., 2016; Welte et al., 2016). Recently it was demonstrated that ‘Candidatus Methanoperedens nitroreducens’ can also oxidize methane using iron as electron acceptor (Ettwig et al., 2016).
Besides so far know methanotrophs and methanogens, recent investigations of microbial “dark matter” discovered key genes of the methane pathway to be present in phyla, which previously were not linked to the ability to produce or consume methane. Phylum Bathyarchaeota, renamed from Miscellaneous Crenarchaeotic Group is a deeply branching phylum consisting of 17 sub-groups (Kubo et al., 2012). It is abundant in marine environments but is also found in extreme habitats like hot springs, cold sulfur springs, Polar Regions and in mesophilic habitats like sewage waste, fresh water lakes and paddy fields. Though there are no pure isolates, based on culture independent methods, their function was speculated to be important in the global cycle of carbon (Parkes et al., 2005). To date there are eight different genomes annotated, out of which two BA1 and BA2 are hypothesized to be methane metabolizers (Evans et al., 2015; He et al., 2016).
Majority of previous studies of paddy field microbial communities have focused on either a specific group of microorganisms or environmental or anthropogenic effect on methane emissions or sampling had been carried out at a single depth, hindering direct comparison. The aim of the present study was to explore how the microbial communities in a paddy field are influenced by spatial factors along a depth gradient. The objectives of this study were (i) to characterize the bacterial and archaeal communities in a paddy field soil core by 16S rRNA gene amplicon sequencing with a focus on methane cycle-related organisms; (ii) determine the abundances of total bacteria, total archaea, ‘Candidatus Methanoperedens nitroreducens,’ NC10 phylum bacteria and Bathyarchaeota; and (iii) estimate the anaerobic methane oxidation potential using nitrate, nitrite and iron as electron acceptors at different soil depths.
Materials and Methods
Soil Sampling
Paddy field soil cores were sampled in August 2015 at the Italian Rice Research Unit in Vercelli, Italy (08°22’25.89”E; 45°19’26.98”N). The sampling fields were cultivated with the rice variety Oryza sativa temperate japonica Onice. The paddy fields were flooded for about 90 days, with fertilizer applied in April and twice in June. Soil cores were sampled in triplicate with 80-cm soil augers at approximately 5-m intervals. The porewater nitrate and ammonium concentrations were in average 0.6 μM and 6.8 μM throughout the 80 cm. Amorphous iron oxides over a 50 cm core were in top 25 cm in average 28.5 μmol per gram wet weight (gww) soil and in lower 25 cm 54.8 μmol per gww soil, with one maxima at 11 cm 68.6 μmol per gww soil and at 31 cm 76.0 μmol per gww soil (data obtained from the previous year), analysis was performed as described in Egger et al. (2015). For AOM and methanogenic activity incubation assays, the soil was sliced in the field and placed immediately in anaerobic jars. For DNA extraction, the samples were stored in 50-ml conical centrifuge tubes. All samples were stored at 4°C at the field site laboratory until transport on cool compresses by car. After transport to the lab, samples for DNA extraction were immediately frozen at -20°C, and samples for activity experiments were stored at 4°C.
Methane Measurements
To measure methane entrapped in the soil, three separate cores with lengths of 51, 58, and 68 cm were sampled. Immediately after sampling, while releasing the core from the auger, samples were taken with a 5-ml open-end syringe. These samples were then transferred to pre-weighed 120-ml bottles filled with saturated NaCl solution. The bottles were sealed with screw-caps with rubber stoppers. The CH4 concentration was quantified by gas chromatography (Hewlett Packard 5890, United States). Methane concentrations were calculated per gram dry weight (gdw) of the sampled soil at the respective depth.
DNA Extraction
For DNA extraction soil cores were divided to 13 different depths. Soil from the same depth of three cores was pooled. DNA was extracted from approximately 0.25 g of soil in duplicate using a PowerSoil DNA isolation Kit (MO BIO Laboratories Inc., Carlsbad, CA, United States) according to the manufacturer’s protocol. DNA was extracted from the following depths: 0 cm, 2.5 cm, 5 cm, 7.5 cm, 10 cm, 15 cm, 20 cm, 25 cm, 30 cm, 35 cm, 40 cm, 50 cm, and 60 cm. DNA quantity and quality were assessed by UV-VIS spectroscopy (NanoDrop, ND-1000, Isogen Life Science, Netherlands).
Quantification by qPCR
Quantification of the total bacterial and total archaeal communities using the 16S rRNA gene was performed in triplicate using the duplicate DNA extractions from each depth sample described above. For archaea, the following primers were used: forward Arch-349 (5′GYGCASCAGKCGMGAAW3′) (Takai and Horikoshi, 2000) and reverse Arch-807 (5′GGACTACVSGGGTATCTAAT3′) (Wang and Qian, 2009). For bacteria, the primers were forward Bact-341 (5′CCTACGGGNGGCWGCAG3′) and reverse Bact-785 (5′GACTACHVGGGTATCTAATCC3′) (Herlemann et al., 2011). Bathyarchaeota were targeted by primers amplifying 16S rRNA gene: MCG528 forward and MCG732 reverse (Kubo et al., 2012). ‘Candidatus Methanoperedens nitroreducens’ was targeted by primers amplifying the mcrA gene: McrA159F forward and McrA345R reverse (Vaksmaa et al., 2017a). The 16S rRNA gene of the NC10 phylum was amplified with the primers p2F_DAMO (5′GGGGAACTGCCAGCGTCAAG3′) and p2R_DAMO (5′CTCAGCGACTTCGAGTACAG3′) (Ettwig et al., 2009). All qPCR reactions were performed using PerfCTa Quanta master mix (Quanta Biosciences, United States) and 96-well optical plates (Bio-Rad, United States) on a Bio-Rad CFX96 Real-Time C1000 Touch Thermal Cycler (Bio-Rad, United States), as described in Vaksmaa et al. (2016, 2017a). Absolute quantification was performed by comparison to standard curves obtained using a 10-fold serial dilution of pGEM-T Easy plasmid DNA (Promega, United States) with an insert of the target gene obtained using the same primers as used for qPCR. Standard curve samples were used as a control for each qPCR run.
Amplicon Sequencing
The following primers were used for 16S rRNA gene amplification: forward Arch-0349 and reverse Arch-807 for archaea and forward Bact-0341 and reverse Bact-785 for bacteria. The amplicons were generated in a two-step reaction. DNA was pooled in equimolar amounts per depth to perform PCR under the following conditions: initial denaturation at 96°C for 3 min; 30–35 cycles of denaturation at 96°C for 40 s, primer annealing at 60°C (for archaea) or 61°C (for bacteria) for 30 s, and elongation at 72°C for 40 s; and a final elongation at 72°C for 2 min. Each PCR product was verified by 1% gel electrophoresis. The obtained PCR products were purified with a GeneJet PCR purification kit (Thermo Scientific, Netherlands). A second PCR was then performed with the same primers described above, which were extended with adapter sequences, specific barcodes and key sequences compatible with Ion Torrent sequencing at the 5′ end. The reaction conditions for this PCR were an initial denaturation at 96°C for 10 min; 10 cycles of denaturation at 96°C for 1 min, primer annealing at 60°C or 61°C for 1 min and elongation at 72°C for 2 min; and a final elongation step at 72°C for 10 min. The products were again pooled per depth and purified as described above. The DNA concentrations of the purified PCR products were then measured and diluted to a range of 0.2–0.4 ng/μl. The concentrations and fragment lengths of the libraries were determined with a Bioanalyzer 2100 and a High Sensitivity DNA kit (Agilent Technologies, United States). The obtained libraries were diluted to a final concentration of 100 pM, and the different barcoded libraries were pooled in equimolar amounts before sequencing. For Ion Torrent sequencing, the library fragments were attached to Ion Sphere particles using an Ion One Touch Instrument and Ion PGM Template OT2 400 Kit (Life Technologies, United States) according to the manufacturer’s instructions. After enrichment of the template-positive Ion Sphere Particles using the Ion One Touch ES (Life Technologies, United States), the samples were loaded on an Ion 316 v2 Chip. The DNA fragments were then sequenced using the Ion PGM Sequencing 400 Kit and 850 nucleotide flows according to the manufacturer’s instructions.
Analysis of 16S rRNA Gene Amplicon Data
The raw sequencing reads were automatically separated into clusters of each depth based on the unique barcodes. After sequencing, all raw reads were imported into CLC Genomics Workbench vs. 9 (QIAGEN Aarhus A/S, Denmark) for initial data analysis, including trimming of low-quality and short reads (cut-off value 200 nucleotides). After trimming, 6,661–11,785 reads were obtained per corresponding depth for archaea; the number of reads obtained per depth for bacteria was 4,477–7,198 reads. The exported reads were further processed using the automated pipeline of Silva NGS (Silva Next Generation Sequencing) of the SILVA rRNA gene database project (SILVAngs 1.2) (Quast et al., 2013). In this process, each read was aligned using the SILVA Incremental Aligner [SINA v1.2.10 for ARB SVN (revision 21008)] (Pruesse et al., 2012) against the SILVA SSUrRNA SEED and quality controlled (Quast et al., 2013). Reads shorter than 50 aligned nucleotides and reads with more than 2% ambiguities or 2% homopolymers were excluded from further processing. Putative contaminants, artifacts and reads with low alignment quality (50 alignment identity, 40 alignment score reported by SINA) were identified and excluded from downstream analysis. After these initial quality control steps, identical reads were identified (dereplication), unique reads were clustered (OTUs) on a per sample basis, and the reference read of each OTU was classified. Dereplication and clustering were performed using cd-hit-est (version 3.1.21) (Li and Godzik, 2006) running in accurate mode, ignoring overhangs, and applying identity criteria of 1.00 and 0.98, respectively. Classification was performed by local nucleotide BLAST search against the non-redundant version of the SILVA SSU Ref dataset (release 1192) using blastn3 (version 2.2.28+) with standard settings (Camacho et al., 2009). The classification of each OTU reference read was mapped onto all reads that were assigned to the respective OTU. This mapping yielded semi-quantitative information (number of individual reads per taxonomic path), within the limitations of PCR and sequencing technique biases, and multiple rRNA operons. Reads without any BLAST hits or reads with weak BLAST hits, in which the function ∖(% sequence identity + % alignment coverage)/2′′ did not exceed a value of 93, remained unclassified. These reads were assigned to the metagroup∖No Relative” in the SILVAngs fingerprint and Krona charts (Ondov et al., 2011). This method was first used in the publications (Ionescu et al., 2012; Klindworth et al., 2013). The amplicon sequencing data were deposited to the Short Read Archive under Bioproject ID PRJNA378333. Estimated quantities of individual taxa were calculated by multiplication of relative amplicon sequence data with qPCR data.
Soil Incubations
Soil samples from the three cores were pooled at depths of 0–5 cm, 5–10 cm, 10–20 cm, 20–30 cm, 30–40 cm, 40–50 cm, and 50–60 cm. Soil slurries for each depth were prepared by mixing the soil with mineral salt medium as described by Ettwig et al. (2008). Activity assays were performed in 120-ml serum bottles with 60 ml of soil slurry. The wet and dry soil weight ratio of the slurry was determined in duplicate at each depth. The incubation bottles were sealed with red butyl rubber stoppers and crimp-caps. The headspace was exchanged with Ar/CO2 by five cycles of vacuum and gassing, with a final overpressure of 0.5 bar. Treatments at each of the depths were performed in duplicate and consisted of adding 5 mM NaNO3, 1 mM NaNO2, 20 mM iron nitrilotriacetic acid (FeNTA), or 20 mM ferrihydrite with 10% 13C-CH4 v/v (final concentrations) and controls in which either 10% CH4 v/v was added or no additions were made to the soil slurry. Each treatment was performed in duplicate with triplicate headspace measurements to quantify the CH4 concentration by gas chromatography (Hewlett Packard 5890, United States) as described previously (Ettwig et al., 2009). Headspace measurements were carried out over the period of 118 days, with methane concentration measured at day 0, 7, 14, 21, 46, 54, 85, 98, and 118 and the net production or consumption rates were calculated during the linear phase.
Results
Methane Measurements in the Soil Core
The highest methane concentration was measured in the top 15 cm of the soil (Figure 1). The highest peak was measured at 0 cm and 6.5 cm and corresponded to a methane concentration of approximately 165 μmol per gdw in two of the three cores. Below 15 cm, a rapid decrease in the methane concentration was observed; at a depth of 28 cm, methane concentrations were less than 7 μmol per gdw. At depths of 50 cm and below, the methane concentration was at the detection limit of 0.4–2 μmol per gdw.
FIGURE 1.
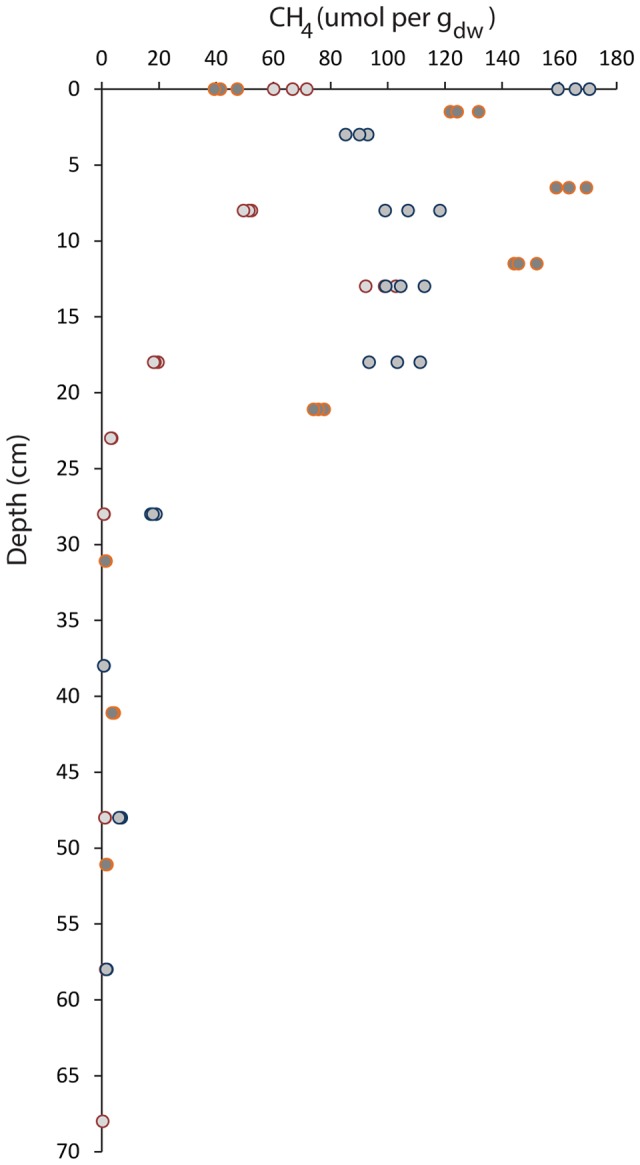
Vertical profile of methane concentrations along the depth gradient of the paddy field soil. The soil depth is depicted vertically, and the methane concentration in μmol per gdw is depicted horizontally. Three separate cores were sampled up to the maximum depth of 68 cm. Measurements of each sample were performed in triplicate by gas chromatography.
Quantification of Total Bacteria, Archaea, and Subgroups of Known Anaerobic Methanotrophs
The total abundance of bacteria, archaea and nitrate- and nitrite dependent anaerobic methanotrophs was quantified by qPCR. The total bacterial abundance was higher than the archaeal abundance at all depths of the soil core. As depicted in Figure 2, the highest copy number obtained with the archaeal primer combination was observed at a depth of 10 cm (1.0 ± 0.3∗109 16S rRNA gene copies per gdw). Below a depth of 20 cm (2.6 ± 0.4∗108 16S rRNA gene copies per gdw), the archaeal copy numbers decreased gradually until 60 cm, where 4.1 ± 2.2∗105 16S rRNA gene copies per gdw was observed. The highest amount of bacterial copies was observed at a depth of 10 cm (5.6 ± 1.4∗109 16S rRNA gene copies per gdw), and the lowest number was observed at a depth of 60 cm (1.4 ± 0.6∗107 16S rRNA gene copies per gdw). The known archaeal methanotroph ‘Candidatus Methanoperedens nitroreducens’ exhibited the highest abundance at 20 cm, with 1.8 ± 0.3∗107 mcrA gene copies per gdw, and lowest abundance at 60 cm, with 7.2 ± 1.5∗103 mcrA gene copies per gdw. The anaerobic methanotrophs belonging to NC10 phylum bacteria had two maxima at depths of 10 cm (2.3 ± 0.7∗105 16S rRNA gene copies per gdw) and 35 cm (2.3 ± 0.2∗105 16S rRNA gene copies per gdw). The lowest abundance was observed at a depth of 60 cm (1.5 ± 0.5∗104 16S rRNA gene copies per gdw). Among the targeted anaerobic methanotrophs, ‘Candidatus Methanoperedens nitroreducens’ had higher gene copy numbers than NC10 phylum bacteria at all depths except 60 cm, where NC10 phylum bacteria outnumbered ‘Candidatus Methanoperedens nitroreducens.’
FIGURE 2.

Depth profile of copy numbers of genes of interest obtained by qPCR. In all figures, the soil depth is depicted vertically. Horizontally, the copy numbers obtained by qPCR per gram dry weight are presented in log scale. (A) 16S rRNA gene copy numbers of total archaea amplified with Arch349F/Arch807R primers. (B) 16S rRNA gene copy numbers of Bathyarchaeota amplified with MCG528F/MCG732R primers. (C) mcrA gene copy numbers of Methanoperedens nitroreducens quantified with McrA159F/McrA345R primers. (D) 16S rRNA gene copy numbers of total bacteria quantified with Bac341F/Bac785R primers. (E) 16S rRNA gene copy numbers of NC10 phylum bacteria quantified with p2F_DAMO/p2R_DAMO primers.
Amplicon Sequencing of the 16S rRNA Gene in the Bacterial Community
At each depth, the 16S rRNA gene amplicon data were analyzed for both bacteria and archaea. In the bacterial community, a very large diversity was observed (Figure 3 and Supplementary Table S1), with most of the reads assigned to Acidobacteria, Chloroflexi, Proteobacteria, Planctomycetes, and Actinobacteria. Most of the phyla were observed throughout the soil core. However, at depths of 40 cm and below, Cyanobacteria, Bacteroidetes, and Chlorobi were hardly or not present at all. The opposite trend was observed for Latescibacteria, which increased gradually in relative abundance toward deeper layers. 16S rRNA gene reads assigned to NC10 phylum bacteria (classified into phylum Nitrospirae by Silva NGS) were recorded at all depths along the gradient of the soil core. The lowest relative abundance was recorded at the top layer of soil (0 cm). Thereafter, the copies increased gradually, with a maximum at a depth of 40 cm, where reads assigned to the NC10 phylum represented 2.4% of the total bacterial 16S rRNA gene reads. After 40 cm, a rapid decrease was observed in the relative abundance of reads assigned to the NC10 phylum to 50 cm (0.25%), followed by an increase at 60 cm (1.25%). The relative abundance of reads assigned to the NC10 phylum at all other depths, except 35 cm, 40 cm, and 60 cm, was less than 1% of the total bacterial reads (Figure 4).
FIGURE 3.
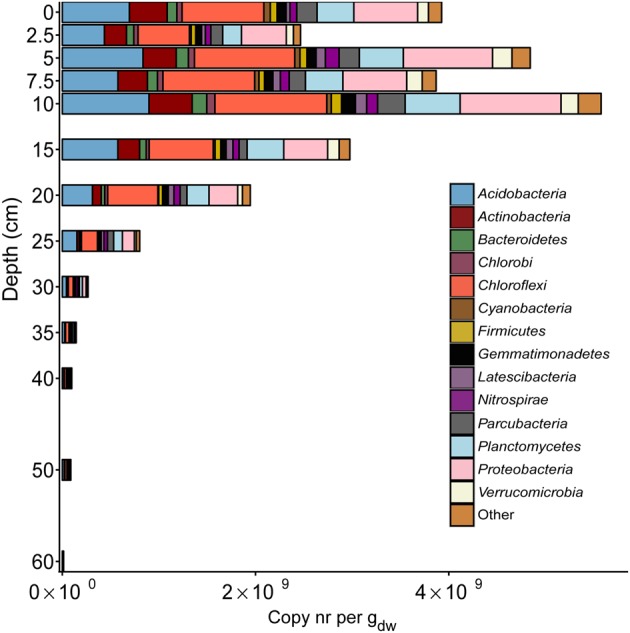
Distribution of 16S rRNA gene reads of major bacterial phyla along the depth profile of the paddy soil core. The soil depth in centimeters is depicted vertically, whereas the total amount of 16S rRNA gene amplicons per gram dry weight is depicted horizontally. The colored bars represent the relative amount of gene copies matching a bacterial phylum present in the soil at a particular depth.
FIGURE 4.
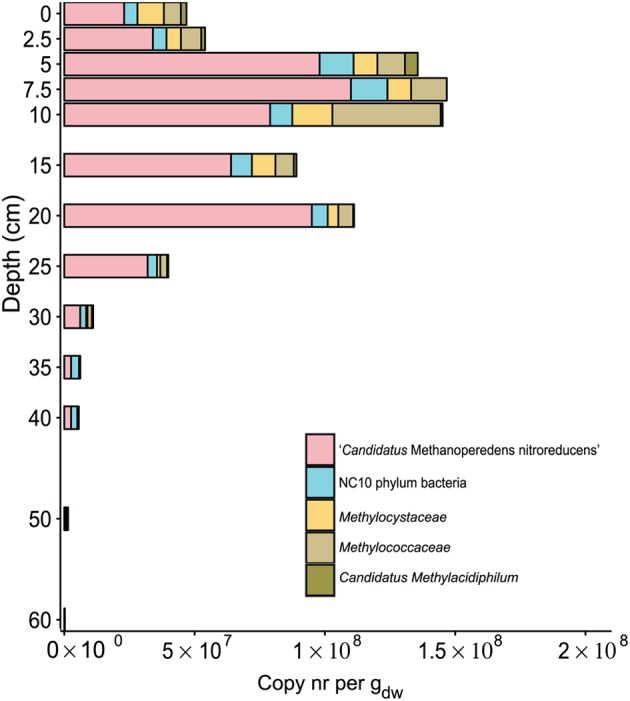
Distribution of sequence reads of proteobacterial (Methylococcaceae, Methylocystaceae) and verrucomicrobial (Candidatus Methylacidiphilum) aerobic methanotrophs together with anaerobic methanotrophs: ‘Candidatus Methanoperedens nitroreducens’ and NC10 phylum bacteria based on 16S rRNA gene amplification. Reads were assigned to phylogenetic groups based on the SILVA NGS pipeline. The soil depth in centimeters is depicted vertically, and the total amount of 16S rRNA gene amplicons per gram dry weight is depicted horizontally. The colored bars represent the relative amount of gene copies corresponding to aerobic and anaerobic methanotrophs present in the soil at a particular depth.
Among Proteobacteria, the relative abundance of Alphaproteobacteria was highest in the top 15 cm of the soil core and gradually decreased in the deeper layers of soil. Beta- and Gammaproteobacteria showed the lowest relative abundances, but their relative abundances exhibited little variation throughout the soil core. Deltaproteobacteria were the second most abundant in the top 15 cm. Their relative abundance peaked at 25 cm, corresponding to 8% of total bacterial reads, and decreased gradually thereafter. A detailed distribution of the proteobacterial classes is provided in Supplementary Table S2.
Among sequences assigned to aerobic methanotrophs, most of the reads were assigned to Methylococcaceae, except at depths of 0 cm and 15 cm, where more reads were assigned to Methylocystaceae. Surprisingly, we observed Verrucomicrobia methanotrophs in the paddy soil core, and reads assigned to Candidatus Methylacidiphilum were most abundant in the top 5 cm, constituting 20% of the total aerobic methanotrophic community. Overall, the relative abundance of aerobic methanotrophs was highest at a depth of 10 cm, representing 1.1% of the total bacterial community. The calculated abundance of aerobic methanotrophs in calculated copy numbers was on the order of 107 in the top 20 cm and then gradually decreased to 104 at a depth of 60 cm (Figure 4).
Phylogenetic Diversity of Verrucomicrobia and Candidatus Methylacidiphilum
Verrucomicrobial methanotrophs have rarely been observed outside acidic volcanic areas. Therefore, we extracted the 16S rRNA gene sequences from amplicon sequencing and analyzed sequences assigned to Verrucomicrobia in detail (Figure 5). Sequences clustering with Candidatus Methylacidiphilum were found at all depths except 7.5 cm, 50 cm, and 60 cm. The extracted sequences clustering with Candidatus Methylacidiphilum were 85% identical at the nucleotide level to cultivated strains of Candidatus Methylacidiphilum (van Teeseling et al., 2014).
FIGURE 5.

(A) Phylogenetic overview of verrucomicrobial 16S rRNA gene sequences. The phylogenetic position of Candidatus Methylacidiphilum (Incertae sedis Candidatus Methylacidiphilum) is marked in red. (B) Detailed presentation of the sequences of Candidatus Methylacidiphilum. Sequences of cultivated strains are shown in red, and sequences obtained from paddy field soil are shown in purple. The neighbor-joining phylogenetic tree was calculated using Jukes-Cantor correction and the Arctic 97B-4 marine group as outgroup.
Amplicon Sequencing of the 16S rRNA Gene of the Archaeal Community
In the archaeal community reads matching Euryarchaeota were more abundant than Thaumarchaeota in the top layers until a depth of 30 cm. At deeper depths of 35 cm, 50 cm, and 60 cm (Euryarchaeota were dominant at 40 cm), sequences matching Thaumarchaeota were the most abundant. Sequences matching the 16S rRNA gene of Bathyarchaeota (previously known as Miscellaneous Crenarchaeota Group (MCG)) were the most abundant in the top layers of the soil. At depths of 0–5 cm, 43–45% of the reads were assigned as Bathyarchaeota. This proportion decreased gradually throughout the soil core (Figure 6 and Supplementary Table S3).
FIGURE 6.
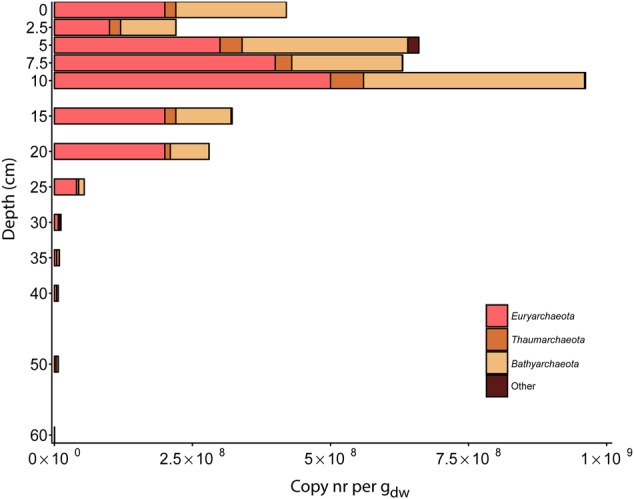
Distribution of 16S rRNA gene reads of major archaeal phyla along the depth profile of the paddy soil core. Reads were assigned to phylogenetic groups based on the SILVA NGS pipeline. The soil depth in centimeters is depicted vertically, and the total amount of 16S rRNA gene amplicons per gram dry weight is depicted horizontally. The colored bars represent the relative amount of gene copies matching an archaeal phylum present in the soil at a particular depth.
Analysis of the Methanomicrobia and Methanobacteria communities in greater detail revealed that sequencing reads assigned to Methanosaeta and Methanosarcina were most abundant among methanogens throughout the soil core (Figure 7). The top layer of soil had more diverse community than deeper layers. The methanogen community was largest at a depth of 10 cm, 52% of total archaea. The highest relative sequence abundance of the archaeal methanotroph ‘Candidatus Methanoperedens nitroreducens’ (GOM Arc I) was found at a depth of 25 cm, comprising 56.4% of the total archaeal reads. The estimated depth distribution of ‘Candidatus Methanoperedens nitroreducens’ calculated based on the sequencing read abundance and on the total archaeal copy numbers is depicted in Figure 4. The abundance based on amplicon data peaked at 1.1∗108 copies at a depth of 7.5 cm.
FIGURE 7.

Distribution of 16S rRNA gene reads of methanogens along the depth profile of the paddy soil core. Reads were assigned to phylogenetic groups based on the SILVA NGS pipeline. The soil depth in centimeters is depicted vertically, and the total amount of 16S rRNA gene amplicons per gram dry weight is depicted horizontally. The colored bars represent the relative amount of gene copies matching methanogens present in the soil at a particular depth.
Soil Slurry Incubations
Soil slurries of different depths were amended with methane and electron acceptors. Controls were prepared with and without addition of methane to detect methanogenic activity. The rates of potential methane oxidation with nitrate, nitrite and two forms of iron, FeNTA and ferrihydrite, were recorded. The potential methane oxidation or production rate was calculated based on methane concentration measurements in the headspace over a time course of 118 days (Figure 8).
FIGURE 8.

Methane oxidation and methanogenesis rates measured in soil slurries incubated with 10% v/v 13C-methane with the addition of 5 mM NaNO3, 1 mM NaNO2, 20 mM FeNTA or 20 mM ferrihydrite. As controls, soil slurries were incubated with 10% v/v 13C-methane and without added methane. GC measurements were performed in triplicate, and rates were calculated in the linear phase. Negative values stand for net oxidation of methane and positive values net production of methane.
The nitrate- and nitrite-dependent methane oxidation rates were highest in the top 20 cm. At depths of (0–5 cm) and (5–10 cm), higher AOM rates were measured in slurries amended with nitrate, 37 nmol gdw h-1 and 57 nmol gdw h-1, than in slurries amended with nitrite, 29 nmol gdw h-1 and 43 nmol gdw h-1, respectively. At a depth of 10–20 cm, the nitrite-amended samples exhibited the highest methane oxidation rate, 55 nmol gdw h-1, followed by 33 nmol gdw h-1 in the nitrate-amended samples. Methane oxidation was measured in the top 20-cm slurries in samples amended with FeNTA, with a peak of 48 nmol gdw h-1 at a depth of 5–10 cm.
In the top layers up to 20 cm, addition of ferrihydrite did not stimulate methane oxidation. In the deeper layers, the pattern was the same as that for the addition of FeNTA. The highest methane oxidation rate was observed at a depth of 40–50 cm in slurries amended with FeNTA, 25 nmol gdw h-1, followed by ferrihydrite, 20 nmol gdw h-1. At a depth of 50–60 cm, the respective rates for FeNTA and ferrihydrite were 56 nmol gdw h-1 and 29 nmol gdw h-1.
In the control samples amended with methane, initial methane oxidation was monitored for a maximum time period of 21 days, after which methane production prevailed, with production of 277 nmol gdw h-1, 369 nmol gdw h-1 and 85 nmol gdw h-1 at depths of 0–5 cm, 5–10 cm, and 10–20 cm, respectively. A similar pattern of methanogenesis in soil slurry incubations with no additions was observed. After a lag phase of approximately 21 days, the methane production rate increased. The highest methane production rate, 444 nmol gdw h-1, was observed at a depth of 5–10 cm. At a depth of 10–20 cm, methanogenesis was still observed, with a rate of 69 nmol gdw h-1, which decreased to less than 1 nmol gdw h-1 in deeper layers. At depths of 20–50 cm, oxidation prevailed over methane production in slurries amended with methane.
Discussion
Paddy fields are a major source of methane emitted to the atmosphere. The flux of methane is controlled by the microbial community present in the soil, particularly by methanogens and methanotrophs.
The vertical profile of the methane gradient included a higher methane concentration in the top 15 cm of the soil core, followed by a drastic drop. At a depth of approximately 28 cm, methane was nearly undetectable. This depth correlates with the interface of annual plowing and undisturbed soil as well as the rice root penetration depth.
The profile of the total abundance of microorganisms along the depth gradient followed the same trend as methane. The highest copy numbers of both bacteria and archaea were detected at a depth of 10 cm, followed by a decrease to 25 cm, after which the microbial population size was a few orders of magnitude smaller. The total bacterial and archaeal population sizes correlate well with previous reports. The total bacterial and archaeal 16S rRNA gene copy numbers in a Chinese paddy field ranged from 1.4∗1010 to 2.9∗1010 per gdw and 5.4∗108-1.7∗109 per gdw, respectively (Ahn et al., 2012). In paddy fields in the Philippines, the total bacterial copy numbers and archaeal copy numbers were on the order of 1010 and 108 per gdw, respectively (Breidenbach and Conrad, 2015). In paddy fields in India, 9.6∗109-1.4∗1010 bacterial 16S rRNA copies per gdw and 7.13∗107-3.02∗108 archaeal 16S rRNA copies per gdw were reported (Singh et al., 2012). We recorded maximum bacterial and archaeal abundances of 5.6 ± 1.4∗109 and 1.0 ± 0.3∗109 16S rRNA gene copies per gdw, respectively.
The rice root system has been described as the key determinant in shaping the microbial community via release of root exudates, decaying roots and organic matter as well as oxygen (Kuzyakov and Blagodatskaya, 2015). Diffusion of oxygen to the soil creates micro-oxic niches for oxygen-dependent microorganisms. We detected sequences belonging to aerobic methanotrophs throughout the soil core. The relative abundance of aerobic methanotrophs was highest at a depth of 10 cm and was twice as high as of that in the surface layer. Along the entire depth gradient, MOB were dominated by Type I Methylococcaceae, followed by Type II Methylocystaceae. Methylococcaceae have been detected in several environments with low oxygen concentration, even tolerating periods of hypoxia (Hernandez et al., 2015). The presence of these aerobic methanotrophs in low oxygen environments, such as the investigated paddy field, could possibly be explained by their denitrifying ability as has been demonstrated for Methylomonas denitrificans, which during hypoxia carries out nitrate reduction and methane oxidation (Kits et al., 2015). Other Methylococcaceae, such as Methylobacter contain in their genome besides respiratory nitrate and nitrite reductases as well genes necessary for dinitrogen fixation (Kalyuzhnaya et al., 2015).
In addition to detecting sequences of well-known proteobacterial aerobic methane oxidizers, sequences belonging to methanotrophic Verrucomicrobia were identified in this study. Detailed phylogenetic analysis revealed the presence of aerobic methanotrophs distantly related (85% nucleotide identity of the 16S rRNA gene) to cultured members of Candidatus Methylacidiphilum. Only a very small number of verrucomicrobial methanotrophs have been detected in ecosystems other than acidic volcanic areas, including paddy field soil (GenBank JF984005.1), forest soil (GenBank JF420089), lake sediment (GenBank GU305773) arctic soil (GenBank HQ462506) and a few other environments. The reported cultivated strains originate exclusively from extreme hot or acidic environments in Italy, Kamchatka or New Zealand. Further studies are needed to determine if the microbes found in less extreme environments also contain pmoA genes in their genome and have the capability to oxidize methane. We hypothesize that there is a niche for these aerobic verrucomicrobial methane oxidizers in less acidic methane-rich environments such as paddy fields.
The translation of 16S rRNA gene sequencing read numbers to copy numbers indicated that the methanogen population abundance was highest at a depth of 10 cm, with 4.5∗108 copies per gdw, followed by a decline in abundance to 60 cm, with 2.7∗104 copies per gdw. The methanogenic population size determined previously in the same Italian paddy field was 107–108 copies per gdw (Conrad and Klose, 2006). Compared to other sampling sites, our observed abundances are slightly higher than the previously reported methanogen abundances of 1.1∗107 or 1.4∗107 copies per gdw (Singh et al., 2012) or 104–105 copies per gdw (Hou et al., 2000). A previous vertical profile study of methanogens identified the highest abundance based on mcrA gene copy numbers at a depth of approximately 20 cm in three Japanese paddy fields, peaking at 107 (Watanabe et al., 2010). Together, these results suggest that the methanogenic zone is located approximately 10–20 cm below the soil surface and co-occurs with the end of the main root system in soil.
The community analysis of methanogens revealed a diverse composition throughout the soil core. The methanogenic community was dominated by Methanosaeta, Methanosarcina, Methanobacterium, Methanoregulaceae, and the RC I cluster (Methanocella), which have also been found previously in temperate climate paddy fields (Conrad and Klose, 2006; Watanabe et al., 2010). The community throughout the core was dominated by the strictly acetoclastic Methanosaeta, followed by more versatile Methanosarcina spp. The sampling time of the soil at the end of the growing season, when most root exudates are released (Aulakh et al., 2001) and the ammonia concentration is highest, may explain the methanogen community structure (Singh et al., 2012). Methanosarcina spp. have been shown to be present during the rice-growing season, whereas during pre-planting, tilling or post-harvest, Methanosaeta were present in lower numbers (Singh et al., 2012), correlating with the lower concentrations of acetate available in the soil (Kruger et al., 2002). In paddy field soil, acetate-dependent methanogenesis (acetoclastic) generally dominates over hydrogen-dependent methanogenesis (hydrogenotrophic), as demonstrated by 13C-labeling experiments (Conrad, 1999, 2005; Conrad et al., 2002; Zhang et al., 2016).
The total methane concentration in soil over the course of rice maturation peaks at the flowering and ripening stage (Singh et al., 2012). Previous studies in Italian paddy fields have demonstrated that methane emission rates reach approximately 400 nmol CH4 per gdw d-1 70–80 days after flooding (Kruger et al., 2005) or even approximately 600 nmol per gdw d-1 (Conrad and Klose, 2006). We previously observed methanogenic activity of the same paddy field soil of 432 nmol and 358 nmol per gdw d-1 without and with the addition of methane, respectively, in incubation assays (Vaksmaa et al., 2016). In the current soil core, the highest methanogenic activity was recorded at a depth of 5–10 cm, with rates of 369 and 444 nmol per gdw d-1 with and without the addition of methane. In control incubations in which methane was added, methanotrophic activity was initially observed. After 3 weeks, methanogenesis became the dominant process, with methane oxidation rates identical to those observed in the control treatment without the addition of methane. Furthermore, ferrihydrite added to slurry incubations seemed to stimulate methanogens in the top 20 cm. In those treatments, no initial methanotrophic activity was observed; only stimulation of methanogenic activity was recorded, even when both methane and possible electron donors were supplied. Previous studies in wetlands and paddy fields have demonstrated that the addition of poorly crystalline iron, such as ferrihydrite, has an inhibitory effect on methanogenic activity (Achtnich et al., 1995; Lueders and Friedrich, 2002). In contrast, the addition of highly crystalline iron oxide species of hematite or magnetite stimulates methanogens enriched from paddy field soil via a positive effect on either direct interspecies electron transfer or the availability of diffusive carriers such as hydrogen or formic acid (Kato et al., 2012; Holmes et al., 2017).
Methanotrophic bacteria of the NC10 phylum have been previously detected in paddy field soil based on the 16S rRNA gene or pmoA genes and activity assays with nitrite and methane. Conflicting results regarding the vertical distribution of NC10 phylum bacteria in soil have been reported. Zhou et al. (2014) indicated that the highest abundance of 1.0∗108 copies per gdw occurred at a depth of 100–120 cm (Zhou et al., 2014). This finding was supported by a study by Hu et al. (2014) of a Chinese paddy field, in which the highest copy number abundance of 1.5 ± 0.2∗106 to 4.5 ± 0.3∗106 copies per gdw was observed at a depth of 50–60 cm. However, the methane-oxidizing potential of soil slurries amended with nitrite was highest at a depth of 90–100 cm, with values of 1.68 ± 0.03 to 2.04 ± 0.06 nmol of CO2 per gdw (Hu et al., 2014). By contrast, in a subtropical paddy field soil core sampled to 100 cm, the abundance of NC10 phylum bacteria was highest at the 0–10 cm depth, with 1.0 ± 0.1∗105 copies per gdw, followed by 7.5 ± 0.4∗104 at 30–40 cm and a subsequent gradual decrease, with no detection at depths of 70 cm and beyond (Wang et al., 2012).
The phylogenetic comparison of the 16S rRNA gene reads obtained from amplicon sequencing revealed that, in the top layers, the 16S rRNA gene reads were assigned exclusively to group B (Ettwig et al., 2009; Welte et al., 2016), with nucleotide identities of 95.6–96.7% to ‘Candidatus Methylomirabilis oxyfera.’ Sequences belonging to Group A of NC10 phylum bacteria were found only at depths of 40 cm and below. This distribution is consistent with previous reports in which 16S rRNA gene sequencing and relative read abundance indicated that these nitrite-dependent AOM bacteria formed the largest subset of sequencing reads among total bacterial reads at depths of 50 cm and 100 cm (Ding et al., 2015). In our activity assays with nitrite, we observed the highest methane oxidation potential of 55 nmol per gdw h-1 in samples from 10 to 20 cm, which correlates with the first peak of high abundance of NC10 phylum bacteria. However, all the sequences at 10–20 cm all belonged to group B, for which no methane-oxidizing ability has been demonstrated thus far and needs further investigation.
In addition to detecting nitrite-dependent AOM bacteria of the NC10 phylum, we observed high numbers nitrate-dependent AOM archaea ‘Candidatus Methanoperedens nitroreducens’ throughout the soil core. However compared to NC10 phylum bacteria, these archaea were more abundant at all depths, except 60 cm, where NC10 phylum bacteria outnumbered ‘Candidatus Methanoperedens nitroreducens.’ Sequences classified as ‘Candidatus Methanoperedens nitroreducens’ have been detected previously in paddy fields, including fields in Vercelli, Italy (Lueders et al., 2001; Conrad et al., 2008), Chinese paddy fields (Xu et al., 2012), and Korean paddy fields (Lee et al., 2015) as well as in natural wetlands (Narrowe et al., 2017). We previously quantified and detected ‘Candidatus Methanoperedens nitroreducens’ in an Italian paddy field based on 16S rRNA gene (Vaksmaa et al., 2016) and mcrA gene sequences in high abundance (Vaksmaa et al., 2017a). High relative sequence abundance has also been observed in other paddy fields based on the 16S rRNA gene, with 60% of all archaeal reads classified as ‘Candidatus Methanoperedens nitroreducens’ (GOM Arc I) at a depth of 60 cm in bulk soil (Lee et al., 2015). In a study by Lee et al. (2015), the soil core depth profile exhibited the same trend observed in the current study (Lee et al., 2015). The abundance of ‘Candidatus Methanoperedens nitroreducens’ increased with depth, peaking at 20 cm with 1.8 ± 0.3∗107copies per gdw. The activity assays performed with nitrate and methane indicated that the activity was highest at a depth of 5–10 cm (57 nmol per gdw d-1), followed by a depth of 10–20 cm (33 nmol per gdw d-1). We previously observed a methane-oxidizing potential of 80 nmol methane per gdw d-1 in mixed and sieved soil slurry from a depth of 10–20 cm (Vaksmaa et al., 2016). The present study is the first to evaluate potential methane oxidation rates utilizing nitrate as an electron acceptor in different depths of a paddy soil core. Recent research has revealed that ‘Candidatus Methanoperedens nitroreducens’ not only can couple nitrate reduction to methane oxidation but is also able to reduce oxidized metals (Ettwig et al., 2016) and may play in important role in both methane and iron cycling in natural and man-made wetlands (Narrowe et al., 2017).
Finally the large number of Bathyarchaeota observed in this paddy field and other wetland systems is intriguing, and their potential role in methane cycling needs further investigation (Evans et al., 2015; Narrowe et al., 2017). For the microbial community members, which the function is still unknown, we detected Bathyarchaeota to be present throughout soil core with highest abundance at 5 cm with 2.1 ± 1.1∗108 16S rRNA gene copies per gdw. From the total archaeal community, these account for almost 50%. Albeit their unknown function, their high abundance and wide distribution indicates that though the function is unknown they might be relevant microorganisms. Up to date, the members of phylum Bathyarchaeota have been detected in a wide range of habitats from terrestrial to marine, cold and hot temperatures or surface and subsurface environments. Generally they are known to be abundant in marine environments (Teske et al., 2002; Lipp et al., 2008; Kubo et al., 2012). Similarly, other studies showed Bathyarchaeota to be present in freshwater environments (Porat et al., 2010; Li et al., 2012). Previous studies have detected Bathyarchaeota in paddy fields as well (Lee et al., 2014, 2015) with abundance of Bathyarchaeota increasing from 17 to 23% in three different phases of rice cultivation (Breidenbach and Conrad, 2015) and with relative abundance up to 42% in paddy field sub-soils (Bai et al., 2017).
In summary, we observed high diversity of the archaeal and bacterial microbial communities throughout the soil core and determined the methane-oxidation potential with various electron acceptors at several soil depths. This study highlights the usage of various electron acceptors for the AOM process. Our findings provide support for the significant role of ‘Candidatus Methanoperedens nitroreducens’ carrying out nitrate-dependent and/or iron-dependent AOM in paddy fields. NC10 phylum bacteria seem to play a less significant role in AOM in paddy fields. The as-yet unknown functions of members of the Candidatus Methylacidiphilum genus and Bathyarchaeota in paddy field soil will hopefully be explained in studies in the near future. We acknowledge that the small sample size of our study does have its limitations, and future studies should include more samples in order to more accurately estimate the contribution of AOM in paddy fields on a larger scale.
Author Contributions
AV and CL designed the research and carried out the fieldwork. EL and GV provided the access to the sampling station, and supported the design and fieldwork. AV carried out the experiments in the laboratory. TvA and AV collected and interpreted the sequencing data. AV, CL, KE, and MJ drafted and finalized the manuscript with input from all authors. The manuscript was checked by a professional peerwith.com editor.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sonja Volman (Radboud University, Nijmegen, NL) for performing initial amplicon sequencing during her internship, Matthias Egger (Utrecht University, Utrecht, NL) for the iron measurements in soil, Mohammad Ghashghavi (Radboud University, Nijmegen, NL) and Gabriele Orasen from the CREA-Rice Research Unit of Vercelli (Italy) for assistance during sampling.
Funding. This work was supported by the Netherlands Organization for Scientific Research [VENI 863.13.007 to KE], the European Research Council [ERC AG 339880 Eco_MoM to MJ, AV, and CL], a Gravitation grant [024002002 Soehngen Institute of Anaerobic Microbiology to MJ; 024002001 NESSC], and the Spinoza prize to MJ.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02127/full#supplementary-material
References
- Achtnich C., Bak F., Conrad R. (1995). Competition for electron donors among nitrate reducers, ferric iron reducers, sulfate reducers, and methanogens in anoxic paddy soil. Biol. Fertil. Soils 19 65–72. 10.1007/BF00336349 [DOI] [Google Scholar]
- Ahn J. H., Song J., Kim B. Y., Kim M. S., Joa J. H., Weon H. Y. (2012). Characterization of the bacterial and archaeal communities in rice field soils subjected to long-term fertilization practices. J. Microbiol. 50 754–765. 10.1007/s12275-012-2409-6 [DOI] [PubMed] [Google Scholar]
- Angel R., Matthies D., Conrad R. (2011). Activation of methanogenesis in arid biological soil crusts despite the presence of oxygen. PLOS ONE 6:e20453. 10.1371/journal.pone.0020453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong W. (1971). Radial oxygen losses from intact rice roots as affected by distance from the apex, respiration and waterlogging. Physiol. Plant. 25 192–197. 10.1111/j.1399-3054.1971.tb01427.x [DOI] [Google Scholar]
- Arshad A., Speth D. R., De Graaf R. M., Op Den Camp H. J., Jetten M. S., Welte C. U. (2015). A metagenomics-based metabolic model of nitrate-dependent anaerobic oxidation of methane by Methanoperedens-like archaea. Front. Microbiol. 6:1423. 10.3389/fmicb.2015.01423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulakh M. S., Wassmann R., Bueno C., Kreuzwieser J., Rennenberg H. (2001). Characterization of root exudates at different growth stages of ten rice (Oryza sativa L.) cultivars. Plant Biol. 3 139–148. 10.1055/s-2001-12905 [DOI] [Google Scholar]
- Bai R., Wang J.-T., Deng Y., He J.-Z., Feng K., Zhang L.-M. (2017). Microbial community and functional structure significantly varied among distinct types of paddy soils but responded differently along gradients of soil depth layers. Front. Microbiol. 8:945. 10.3389/fmicb.2017.00945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Q., Huang Y., Wang F., Nie S., Nicol G. W., Yao H., et al. (2016). Effect of nitrogen fertilizer and/or rice straw amendment on methanogenic archaeal communities and methane production from a rice paddy soil. Appl. Microbiol. Biotechnol. 100 5989–5998. 10.1007/s00253-016-7377-z [DOI] [PubMed] [Google Scholar]
- Bodelier P. L. (2011). Toward understanding, managing, and protecting microbial ecosystems. Front. Microbiol. 2:80. 10.3389/fmicb.2011.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenbach B., Conrad R. (2015). Seasonal dynamics of bacterial and archaeal methanogenic communities in flooded rice fields and effect of drainage. Front. Microbiol. 5:572. 10.3389/fmicb.2014.00752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgham S. D., Cadillo-Quiroz H., Keller J. K., Zhuang Q. (2013). Methane emissions from wetlands: biogeochemical, microbial, and modeling perspectives from local to global scales. Glob. Change Biol. 19 1325–1346. 10.1111/gcb.12131 [DOI] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K. J., Lueders T., Friedrich M. W., Klose M., Conrad R. (2004). Archaeal community structure and pathway of methane formation on rice roots. Microb. Ecol. 47 59–67. 10.1007/s00248-003-2014-7 [DOI] [PubMed] [Google Scholar]
- Conrad R. (1999). Contribution of hydrogen to methane production and control of hydrogen concentrations in methanogenic soils and sediments. FEMS Microbiol. Ecol. 28 193–202. 10.1111/j.1574-6941.1999.tb00575.x [DOI] [Google Scholar]
- Conrad R. (2005). Quantification of methanogenic pathway using stable carbon isotopic signatures: a review and a proposal. Org. Geochem. 36 739–752. 10.1016/j.orggeochem.2004.09.006 [DOI] [Google Scholar]
- Conrad R. (2009). The global methane cycle: recent advances in understanding the microbial processes involved. Environ. Microbiol. Rep. 1 285–292. 10.1111/j.1758-2229.2009.00038.x [DOI] [PubMed] [Google Scholar]
- Conrad R., Klose M. (1999). How specific is the inhibition by methyl fluoride of acetoclastic methanogenesis in anoxic rice field soil? FEMS Microbiol. Ecol. 30 47–56. 10.1111/j.1574-6941.1999.tb00634.x [DOI] [Google Scholar]
- Conrad R., Klose M. (2006). Dynamics of the methanogenic archaeal community in anoxic rice soil upon addition of straw. Eur. J. Soil Sci. 57 476–484. 10.3389/fmicb.2012.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad R., Klose M., Claus P. (2002). Pathway of CH4 formation in anoxic rice field soil and rice roots determined by C-13-stable isotope fractionation. Chemosphere 47 797–806. 10.1016/S0045-6535(02)00120-0 [DOI] [PubMed] [Google Scholar]
- Conrad R., Klose M., Noll M. (2009). Functional and structural response of the methanogenic microbial community in rice field soil to temperature change. Environ. Microbiol. 11 1844–1853. 10.1111/j.1462-2920.2009.01909.x [DOI] [PubMed] [Google Scholar]
- Conrad R., Klose M., Noll M., Kemnitz D., Bodelier P. L. E. (2008). Soil type links microbial colonization of rice roots to methane emission. Glob. Change Biol. 14 657–669. 10.1111/j.1365-2486.2007.01516.x [DOI] [Google Scholar]
- Costa K. C., Leigh J. A. (2014). Metabolic versatility in methanogens. Curr. Opin. Biotechnol. 29 70–75. 10.1016/j.copbio.2014.02.012 [DOI] [PubMed] [Google Scholar]
- Dedysh S. N., Berestovskaya Y. Y., Vasylieva L. V., Belova S. E., Khmelenina V. N., Suzina N. E., et al. (2004). Methylocella tundrae sp. nov., a novel methanotrophic bacterium from acidic tundra peatlands. Int. J. Syst. Evol. Microbiol. 54 151–156. 10.1099/ijs.0.02805-0 [DOI] [PubMed] [Google Scholar]
- Ding J., Ding Z. W., Fu L., Lu Y. Z., Cheng S. H., Zeng R. J. (2015). New primers for detecting and quantifying denitrifying anaerobic methane oxidation archaea in different ecological niches. Appl. Microbiol. Biotechnol. 99 9805–9812. 10.1007/s00253-015-6893-6 [DOI] [PubMed] [Google Scholar]
- Dumont M. G., Lüke C., Deng Y., Frenzel P. (2014). Classification of pmoA amplicon pyrosequences using BLAST and the lowest common ancestor method in MEGAN. Front. Microbiol. 5:34 10.3389/fmicb.2014.00034 [DOI] [Google Scholar]
- Dunfield P. F., Yuryev A., Senin P., Smirnova A. V., Stott M. B., Hou S., et al. (2007). Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 450 879–882. 10.1038/nature06411 [DOI] [PubMed] [Google Scholar]
- Edwards J., Johnson C., Santos-Medellin C., Lurie E., Podishetty N. K., Bhatnagar S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112 E911–E920. 10.1073/pnas.1414592112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger M., Rasigraf O., Sapart C. J., Jilbert T., Jetten M. S., Rockmann T., et al. (2015). Iron-mediated anaerobic oxidation of methane in brackish coastal sediments. Environ. Sci. Technol. 49 277–283. 10.1021/es503663z [DOI] [PubMed] [Google Scholar]
- Erkel C., Kube M., Reinhardt R., Liesack W. (2006). Genome of Rice Cluster I archaea - the key methane producers in the rice rhizosphere. Science 313 370–372. 10.1126/science.1127062 [DOI] [PubMed] [Google Scholar]
- Ettwig K. F., Butler M. K., Le Paslier D., Pelletier E., Mangenot S., Kuypers M. M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464 543–548. 10.1038/nature08883 [DOI] [PubMed] [Google Scholar]
- Ettwig K. F., Shima S., van de Pas-Schoonen K. T., Kahnt J., Medema M. H., op den Camp H. J. M., et al. (2008). Denitrifying bacteria anaerobically oxidize methane in the absence of Archaea. Environ. Microbiol. 10 3164–3173. 10.1111/j.1462-2920.2008.01724.x [DOI] [PubMed] [Google Scholar]
- Ettwig K. F., Van Alen T., Van De Pas-Schoonen K. T., Jetten M. S., Strous M. (2009). Enrichment and molecular detection of denitrifying methanotrophic bacteria of the NC10 phylum. Appl. Environ. Microbiol. 75 3656–3662. 10.1128/AEM.00067-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettwig K. F., Zhu B., Speth D., Keltjens J. T., Jetten M. S., Kartal B. (2016). Archaea catalyze iron-dependent anaerobic oxidation of methane. Proc. Natl. Acad. Sci. U.S.A. 10.1073/pnas.1609534113 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. N., Parks D. H., Chadwick G. L., Robbins S. J., Orphan V. J., Golding S. D., et al. (2015). Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350 434–438. 10.1126/science.aac7745 [DOI] [PubMed] [Google Scholar]
- Haroon M. F., Hu S., Shi Y., Imelfort M., Keller J., Hugenholtz P., et al. (2013). Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500 567–570. 10.1038/nature12375 [DOI] [PubMed] [Google Scholar]
- He Y., Li M., Perumal V., Feng X., Fang J., Xie J., et al. (2016). Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat. Microbiol. 1:16035. 10.1038/nmicrobiol.2016.35 [DOI] [PubMed] [Google Scholar]
- Herlemann D. P. R., Labrenz M., Jürgens K., Bertilsson S., Waniek J. J., Andersson A. F. (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5 1571–1579. 10.1038/ismej.2011.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez M. E., Beck D. A. C., Lidstrom M. E., Chistoserdova L. (2015). Oxygen availability is a major factor in determining the composition of microbial communities involved in methane oxidation. PeerJ 3:e801. 10.7717/peerj.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A., Luke C., Cao Z., Frenzel P. (2011). Ageing well: methane oxidation and methane oxidizing bacteria along a chronosequence of 2000 years. Environ. Microbiol. Rep. 3 738–743. 10.1111/j.1758-2229.2011.00292.x [DOI] [PubMed] [Google Scholar]
- Ho A., Vlaeminck S. E., Ettwig K. F., Schneider B., Frenzel P., Boon N. (2013). Revisiting methanotrophic communities in sewage treatment plants. Appl. Environ. Microbiol. 79 2841–2846. 10.1128/AEM.03426-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes D. E., Shrestha P. M., Walker D. J., Dang Y. (2017). Metatranscriptomic evidence for direct interspecies electron transfer between Geobacter and Methanothrix species in methanogenic rice paddy soils. Appl. Environ. Microbiol. 83:e00223-17. 10.1128/AEM.00223-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou A. X., Wang Z. P., Chen G. X., Patrick W. H. (2000). Effects of organic and N fertilizers on methane production potential in a Chinese rice soil and its microbiological aspect. Nutr. Cycling Agroecosyst. 58 333–338. 10.1023/A:1009875509876 [DOI] [Google Scholar]
- Hu B. L., Shen L. D., Lian X., Zhu Q., Liu S., Huang Q., et al. (2014). Evidence for nitrite-dependent anaerobic methane oxidation as a previously overlooked microbial methane sink in wetlands. Proc. Natl. Acad. Sci. U.S.A. 111 4495–4500. 10.1073/pnas.1318393111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu D., Siebert C., Polerecky L., Munwes Y. Y., Lott C., Hausler S., et al. (2012). Microbial and chemical characterization of underwater fresh water springs in the Dead Sea. PLOS ONE 7:e38319. 10.1371/journal.pone.0038319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetten M. S. M., Stams A. J. M., Zehnder A. J. B. (1992). Methanogenesis from acetate: a comparison of the acetate metabolism in Methanothrix soehngenii and Methanosarcina spp. FEMS Microbiol. Lett. 88 181–198. 10.1111/j.1574-6968.1992.tb04987.x [DOI] [Google Scholar]
- Kalyuzhnaya M. G., Lamb A. E., Mctaggart T. L., Oshkin I. Y., Shapiro N., Woyke T., et al. (2015). Draft genome sequences of gammaproteobacterial methanotrophs isolated from Lake Washington sediment. Genome Announc. 3:e00103-15. 10.1128/genomeA.00103-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S., Kikuchi S., Kashiwabara T., Takahashi Y., Suzuki K., Itoh T., et al. (2012). Prokaryotic abundance and community composition in a freshwater iron-rich microbial mat at circumneutral pH. Geomicrobiol. J. 29 896–905. 10.1080/01490451.2011.635763 [DOI] [Google Scholar]
- Kits K. D., Klotz M. G., Stein L. Y. (2015). Methane oxidation coupled to nitrate reduction under hypoxia by the Gammaproteobacterium Methylomonas denitrificans, sp. nov. type strain FJG1. Environ. Microbiol. 17 3219–3232. 10.1111/1462-2920.12772 [DOI] [PubMed] [Google Scholar]
- Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41 e1. 10.1093/nar/gks808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knief C. (2015). Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front. Microbiol. 6:1346. 10.3389/fmicb.2015.01346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kögel-Knabner I., Amelung W., Cao Z., Fiedler S., Frenzel P., Jahn R., et al. (2010). Biogeochemistry of paddy soils. Geoderma 157 1–14. 10.1016/j.geoderma.2010.03.009 [DOI] [Google Scholar]
- Kruger M., Eller G., Conrad R., Frenzel P. (2002). Seasonal variation in pathways of CH4 production and in CH4 oxidation in rice fields determined by stable carbon isotopes and specific inhibitors. Glob. Change Biol. 8 265–280. 10.1046/j.1365-2486.2002.00476.x [DOI] [Google Scholar]
- Krüger M., Frenzel P., Conrad R. (2001). Microbial processes influencing methane emission from rice fields. Glob. Change Biol. 7 49–63. 10.1046/j.1365-2486.2001.00395.x [DOI] [Google Scholar]
- Kruger M., Frenzel P., Kemnitz D., Conrad R. (2005). Activity, structure and dynamics of the methanogenic archaeal community in a flooded Italian rice field. FEMS Microbiol. Ecol. 51 323–331. 10.1016/j.femsec.2004.09.004 [DOI] [PubMed] [Google Scholar]
- Kubo K., Lloyd K. G., F Biddle J., Amann R., Teske A., Knittel K. (2012). Archaea of the miscellaneous crenarchaeotal group are abundant, diverse and widespread in marine sediments. ISME J. 6 1949–1965. 10.1038/ismej.2012.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzyakov Y., Blagodatskaya E. (2015). Microbial hotspots and hot moments in soil: concept & review. Soil Biol. Biochem. 83 184–199. 10.1016/j.soilbio.2015.01.025 [DOI] [Google Scholar]
- Lee H. J., Jeong S. E., Kim P. J., Madsen E. L., Jeon C. O. (2015). High resolution depth distribution of Bacteria, Archaea, methanotrophs, and methanogens in the bulk and rhizosphere soils of a flooded rice paddy. Front. Microbiol. 6:639. 10.3389/fmicb.2015.00639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. J., Kim S. Y., Kim P. J., Madsen E. L., Jeon C. O. (2014). Methane emission and dynamics of methanotrophic and methanogenic communities in a flooded rice field ecosystem. FEMS Microbiol. Ecol. 88 195–212. 10.1111/1574-6941.12282 [DOI] [PubMed] [Google Scholar]
- Li Q., Wang F., Chen Z., Yin X., Xiao X. (2012). Stratified active archaeal communities in the sediments of Jiulong River estuary, China. Front. Microbiol. 3:311. 10.3389/fmicb.2012.00311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Godzik A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22 1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- Li Y., Wang X. (2013). Root-induced changes in radial oxygen loss, rhizosphere oxygen profile, and nitrification of two rice cultivars in Chinese red soil regions. Plant Soil 365 115–126. 10.1007/s11104-012-1378-1 [DOI] [Google Scholar]
- Liebner S., Rublack K., Stuehrmann T., Wagner D. (2009). Diversity of aerobic methanotrophic bacteria in a permafrost active layer soil of the Lena Delta, Siberia. Microb. Ecol. 57 25–35. 10.1007/s00248-008-9411-x [DOI] [PubMed] [Google Scholar]
- Lipp J. S., Morono Y., Inagaki F., Hinrichs K. U. (2008). Significant contribution of Archaea to extant biomass in marine subsurface sediments. Nature 454 991–994. 10.1038/nature07174 [DOI] [PubMed] [Google Scholar]
- Lueders T., Chin K. J., Conrad R., Friedrich M. (2001). Molecular analyses of methyl-coenzyme M reductase alpha-subunit (mcrA) genes in rice field soil and enrichment cultures reveal the methanogenic phenotype of a novel archaeal lineage. Environ. Microbiol. 3 194–204. 10.1046/j.1462-2920.2001.00179.x [DOI] [PubMed] [Google Scholar]
- Lueders T., Friedrich M. W. (2002). Effects of amendment with ferrihydrite and gypsum on the structure and activity of methanogenic populations in rice field soil. Appl. Environ. Microbiol. 68 2484–2494. 10.1128/AEM.68.5.2484-2494.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüke C., Frenzel P. (2011). Potential of pmoA amplicon pyrosequencing for methanotroph diversity studies. Appl. Environ. Microbiol. 77 6305–6309. 10.1128/AEM.05355-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhre G., Shindell D., Bréon F. M., Collins W., Fuglestvedt J., Huang J., et al. (2013). “Anthropogenic and natural radiative forcing,” in Proceedings of the Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (Cambridge: Cambridge University Press; ). [Google Scholar]
- Narrowe A. B., Angle J. C., Daly R. A., Stefanik K. C., Wrighton K. C., Miller C. S. (2017). High-resolution sequencing reveals unexplored archaeal diversity in freshwater wetland soils. Environ. Microbiol. 19 2192–2209. 10.1111/1462-2920.13703 [DOI] [PubMed] [Google Scholar]
- Noll M., Klose M., Conrad R. (2010). Effect of temperature change on the composition of the bacterial and archaeal community potentially involved in the turnover of acetate and propionate in methanogenic rice field soil. FEMS Microbiol. Ecol. 73 215–225. 10.1111/j.1574-6941.2010.00883.x [DOI] [PubMed] [Google Scholar]
- Ondov B. D., Bergman N. H., Phillippy A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. 10.1186/1471-2105-12-385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Op den Camp H. J. M., Islam T., Stott M. B., Harhangi H. R., Hynes A., Schouten S., et al. (2009). Environmental, genomic and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ. Microbiol. Rep. 1 293–306. 10.1111/j.1758-2229.2009.00022.x [DOI] [PubMed] [Google Scholar]
- Parkes R. J., Webster G., Cragg B. A., Weightman A. J., Newberry C. J., Ferdelman T. G., et al. (2005). Deep sub-seafloor prokaryotes stimulated at interfaces over geological time. Nature 436 390–394. 10.1038/nature03796 [DOI] [PubMed] [Google Scholar]
- Porat I., Vishnivetskaya T. A., Mosher J. J., Brandt C. C., Yang Z. K., Brooks S. C., et al. (2010). Characterization of archaeal community in contaminated and uncontaminated surface stream sediments. Microb. Ecol. 60 784–795. 10.1007/s00248-010-9734-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E., Peplies J., Glockner F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28 1823–1829. 10.1093/bioinformatics/bts252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41 D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghoebarsing A. A., Pol A., Van De Pas-Schoonen K. T., Smolders A. J., Ettwig K. F., Rijpstra W. I., et al. (2006). A microbial consortium couples anaerobic methane oxidation to denitrification. Nature 440 918–921. 10.1038/nature04617 [DOI] [PubMed] [Google Scholar]
- Rui J., Peng J., Lu Y. (2009). Succession of bacterial populations during plant residue decomposition in rice field soil. Appl. Environ. Microbiol. 75 4879–4886. 10.1128/AEM.00702-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semrau J. D., Dispirito A. A., Yoon S. (2010). Methanotrophs and copper. FEMS Microbiol. Rev. 34 496–531. 10.1111/j.1574-6976.2010.00212.x [DOI] [PubMed] [Google Scholar]
- Sharp C. E., Smirnova A. V., Graham J. M., Stott M. B., Khadka R., Moore T. R., et al. (2014). Distribution and diversity of Verrucomicrobia methanotrophs in geothermal and acidic environments. Environ. Microbiol. 16 1867–1878. 10.1111/1462-2920.12454 [DOI] [PubMed] [Google Scholar]
- Singh A., Singh R. S., Upadhyay S. N., Joshi C. G., Tripathi A. K., Dubey S. K. (2012). Community structure of methanogenic archaea and methane production associated with compost-treated tropical rice-field soil. FEMS Microbiol. Ecol. 82 118–134. 10.1111/j.1574-6941.2012.01411.x [DOI] [PubMed] [Google Scholar]
- Söhngen N. L. (1906). Über bakterien, welche methan als kohlenstoffnahrung und energiequelle gebrauchen. Z. Bakteriol. Parasitenkund. Infektionskr. II Abt. 15 513–517. [Google Scholar]
- Takai K., Horikoshi K. (2000). Rapid detection and quantification of members of the archaeal community by quantitative PCR using fluorogenic probes. Appl. Environ. Microbiol. 66 5066–5072. 10.1128/AEM.66.11.5066-5072.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teske A., Hinrichs K. U., Edgcomb V., De Vera Gomez A., Kysela D., Sylva S. P., et al. (2002). Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Appl. Environ. Microbiol. 68 1994–2007. 10.1128/AEM.68.4.1994-2007.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotsenko Y. A., Murrell J. C. (2008). Metabolic aspects of aerobic obligate methanotrophy. Adv. Appl. Microbiol. 63 183–229. 10.1016/S0065-2164(07)00005-6 [DOI] [PubMed] [Google Scholar]
- Vaksmaa A., Guerrero-Cruz S., van Alen T. A., Cremers G., Ettwig K. F., Lüke C., et al. (2017b). Enrichment of anaerobic nitrate-dependent methanotrophic ‘Candidatus Methanoperedens nitroreducens’ archaea from an Italian paddy field soil. Appl. Microbiol. Biotechnol. 101 7075–7084. 10.1007/s00253-017-8416-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaksmaa A., Jetten M. S. M., Ettwig K. F., Lüke C. (2017a). McrA primers for the detection and quantification of the anaerobic archaeal methanotroph ‘Candidatus Methanoperedens nitroreducens’. Appl. Microbiol. Biotechnol. 101 1631–1641. 10.1007/s00253-016-8065-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaksmaa A., Luke C., Van Alen T., Vale G., Lupotto E., Jetten M. S., et al. (2016). Distribution and activity of the anaerobic methanotrophic community in a nitrogen-fertilized Italian paddy soil. FEMS Microbiol. Ecol. 92:fiw181. 10.1093/femsec/fiw181 [DOI] [PubMed] [Google Scholar]
- van Teeseling M. C. F., Pol A., Harhangi H. R., Van Der Zwart S., Jetten M. S. M., Op Den Camp H. J. M., et al. (2014). Expanding the verrucomicrobial methanotrophic world: description of three novel species of Methylacidimicrobium gen. nov. Appl. Environ. Microbiol. 80 6782–6791. 10.1128/AEM.01838-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Qian P. Y. (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLOS ONE 4:e7401. 10.1371/journal.pone.0007401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zhu G., Harhangi H. R., Zhu B., Jetten M. S. M., Yin C., et al. (2012). Co-occurrence and distribution of nitrite-dependent anaerobic ammonium and methane-oxidizing bacteria in a paddy soil. FEMS Microbiol. Lett. 336 79–88. 10.1111/j.1574-6968.2012.02654.x [DOI] [PubMed] [Google Scholar]
- Watanabe T., Wang G., Taki K., Ohashi Y., Kimura M., Asakawa S. (2010). Vertical changes in bacterial and archaeal communities with soil depth in Japanese paddy fields. Soil Sci. Plant Nutr. 56 705–715. 10.1111/j.1747-0765.2010.00511.x [DOI] [Google Scholar]
- Welte C., Deppenmeier U. (2014). Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochim. Biophys. Acta 1837 1130–1147. 10.1016/j.bbabio.2013.12.002 [DOI] [PubMed] [Google Scholar]
- Welte C., Rasigraf O., Vaksmaa A., Versantvoort W., Arshad A., Op Den Camp H., et al. (2016). Nitrate-and nitrite-dependent anaerobic oxidation of methane. Environ. Microbiol. Environ. Microbiol. Rep. 8 941–955. 10.1111/1758-2229.12487 [DOI] [PubMed] [Google Scholar]
- Xu Y., Ma K., Huang S., Liu L., Lu Y. (2012). Diel cycle of methanogen mcrA transcripts in rice rhizosphere. Environ. Microbiol. Rep. 4 655–663. 10.1111/j.1758-2229.2012.00392.x [DOI] [PubMed] [Google Scholar]
- Zhang G., Yu H., Fan X., Ma J., Xu H. (2016). Carbon isotope fractionation reveals distinct process of CH4 emission from different compartments of paddy ecosystem. Sci. Rep. 6:27065. 10.1038/srep27065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Huang R., Wang B. Z., Bodelier P. L. E., Jia Z. J. (2014). Competitive interactions between methane- and ammonia-oxidizing bacteria modulate carbon and nitrogen cycling in paddy soil. Biogeosciences 11 3353–3368. 10.5194/bg-11-3353-2014 [DOI] [Google Scholar]
- Zhou L., Wang Y., Long X. E., Guo J., Zhu G. (2014). High abundance and diversity of nitrite-dependent anaerobic methane-oxidizing bacteria in a paddy field profile. FEMS Microbiol. Lett. 360 33–41. 10.1111/1574-6968.12567 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.