

Fagg et al. find that Qk5 and Qk6 proteins have distinct functions in splicing and translation, respectively, enforced through differential subcellular localization. Qk5 and Qk6 regulate distinct target mRNAs in the cell and act in distinct ways on their own and each other's transcripts to create a network of autoregulatory and cross-regulatory feedback controls.
Keywords: Quaking, Qk, QKI, RNA-binding protein, autoregulation, RNA processing
Abstract
Quaking protein isoforms arise from a single Quaking gene and bind the same RNA motif to regulate splicing, translation, decay, and localization of a large set of RNAs. However, the mechanisms by which Quaking expression is controlled to ensure that appropriate amounts of each isoform are available for such disparate gene expression processes are unknown. Here we explore how levels of two isoforms, nuclear Quaking-5 (Qk5) and cytoplasmic Qk6, are regulated in mouse myoblasts. We found that Qk5 and Qk6 proteins have distinct functions in splicing and translation, respectively, enforced through differential subcellular localization. We show that Qk5 and Qk6 regulate distinct target mRNAs in the cell and act in distinct ways on their own and each other's transcripts to create a network of autoregulatory and cross-regulatory feedback controls. Morpholino-mediated inhibition of Qk translation confirms that Qk5 controls Qk RNA levels by promoting accumulation and alternative splicing of Qk RNA, whereas Qk6 promotes its own translation while repressing Qk5. This Qk isoform cross-regulatory network responds to additional cell type and developmental controls to generate a spectrum of Qk5/Qk6 ratios, where they likely contribute to the wide range of functions of Quaking in development and cancer.
Sequence-specific RNA-binding proteins (RBPs) regulate nearly every post-transcriptional step of gene expression. Recognition of cis-acting regulatory sequences by the RNA-binding domain (RBD) is followed by one or more consequences, including RNA refolding and folding or refolding of the protein itself, leading to altered interactions and functional impact on RNA polymerase, the spliceosome, the export and decay machineries, and the ribosome (Keene 2007; Fu and Ares 2014). Furthermore, many RBPs control their own levels (autogenous regulation) through the same mechanisms by which they regulate their targets (Wollerton et al. 2004; Lareau et al. 2007; Ni et al. 2007; Damianov and Black 2010; Sun et al. 2010). In many metazoans, multiple RBPs with the same or highly similar sequence-binding properties are present at the same time in the same cells but perform distinct functions (Boutz et al. 2007a; Makeyev et al. 2007; Spellman et al. 2007; Yeo et al. 2009; Gehman et al. 2011, 2012; Charizanis et al. 2012; Singh et al. 2014), suggesting that functional diversification is enforced by regulatory mechanisms that prevent cross-talk. These two circumstances—autogenous regulation through binding their own pre-mRNAs and mRNAs combined with functional diversification using the same recognition sequences—create a regulatory conundrum: How is the right amount of each distinctly functional RBP isoform established and maintained in cells?
Since all isoforms within a family recognize essentially the same RNA sequence, control of individual isoforms is unlikely to be mediated solely through their common RNA target sequences. Additional mechanisms for distinguishing isoforms include (1) isoform-specific localization that restricts function to processes within specific compartments (Caceres et al. 1998; Koizumi et al. 1999; Cazalla et al. 2002; Sanford et al. 2004) and (2) isoform-specific protein–protein interactions with different core machineries (e.g., spliceosome or ribosome) (Roscigno and Garcia-Blanco 1995; Jin et al. 2004; Cheever and Ceman 2009; Sharma et al. 2011). Because many RBPs are encoded by multigene families with complex alternative splicing (Underwood et al. 2005), whole menageries of functionally distinct RBP isoforms that recognize the same RNA target can be expressed within single cell types (Lee et al. 2009, 2016; Hamada et al. 2013). How does the cell recognize when each distinctly functional RBP family member is present in an appropriate amount for each process?
To examine this question, we focused on the single Quaking gene (QKI in humans, Qk in mice), which is required for a broad set of functions in diverse tissues (Ebersole et al. 1996; Zhao et al. 2010; Darbelli et al. 2016; de Bruin et al. 2016) through its contribution to RNA processing steps, including splicing (Hall et al. 2013; van der Veer et al. 2013; Darbelli et al. 2016), localization (Li et al. 2000; Larocque et al. 2002), stability/decay (Li et al. 2000; Larocque et al. 2005; Zearfoss et al. 2011; de Bruin et al. 2016), translation (Saccomanno et al. 1999; Zhao et al. 2010), and miRNA processing (Wang et al. 2013; Zong et al. 2014). These processes are regulated by dimeric Qk binding an RNA element that includes ACUAAY and a “half-site” (UAAY) separated by at least 1 nucleotide (nt) (Ryder and Williamson 2004; Galarneau and Richard 2005; Beuck et al. 2012; Teplova et al. 2013). Quaking gene transcription initiates primarily at a single major site, and, in most cell types, three alternatively spliced mRNAs encode three protein isoforms (Quaking-5 [Qk5], Qk6, and Qk7) that differ only in the C-terminal tail (Ebersole et al. 1996; Kondo et al. 1999). Although various cell types express different ratios of Qk protein isoforms (Ebersole et al. 1996; Hardy et al. 1996; Hardy 1998; van der Veer et al. 2013; de Bruin et al. 2016), it is unclear how the relative isoform ratios are maintained in order to support tissue-specific regulated RNA processing. Disruption of these ratios is associated with developmental defects (Ebersole et al. 1996; Cox et al. 1999), cancer (de Miguel et al. 2016; Sebestyen et al. 2016), and schizophrenia (Aberg et al. 2006).
Many studies of Quaking function have used overexpression of Qk isoforms (Wu et al. 2002; Hafner et al. 2010; Wang et al. 2013) or depletion strategies and mutant models that do not distinguish which Qk isoform is functional (Hardy et al. 1996; Lu et al. 2003; van der Veer et al. 2013; Darbelli et al. 2016). Here we tested specific Qk isoforms for separate functions and identified in part how the appropriate balance of Qk isoforms is maintained. In mouse myoblasts, Qk5 and Qk6 are the predominantly expressed isoforms, and we found that Qk5, but not Qk6, regulates splicing, while Qk6 controls mRNA translation and decay. This functional specificity is mediated by subcellular localization encoded into the unique C-terminal amino acids of these isoforms. Furthermore, the relative expression of Qk protein isoforms is regulated in part by Qk protein isoforms themselves through both autoregulatory and cross-regulatory influences characteristic of the function of each isoform on its other RNA targets. These findings uncover unexpectedly complex isoform control within a single family of RBPs and suggest that the relative amounts of each isoform are set in a cell type-specific fashion and homeostatically controlled by Qk protein isoform levels themselves.
Results
Qk5 and Qk6 are the predominant isoforms in myoblasts
We analyzed the abundance and localization of Qk isoforms (Fig. 1A) in myoblasts and differentiated myotubes (Yaffe and Saxel 1977) using isoform-specific antibodies. Total Qk protein level increases during C2C12 myoblast differentiation (Fig. 1B; Hall et al. 2013), with Qk5 the most abundant, followed by Qk6 and then Qk7 (Fig. 1B). During differentiation, each isoform increases proportionately (Fig. 1B), and total Qk protein remains predominantly localized in nuclei (Supplemental Fig. S1A). Immunolocalization using isoform-specific antibodies shows that Qk5 is primarily nuclear, although some cytoplasmic localization is observed, whereas Qk6 and Qk7 are present in both the nuclear and cytoplasmic compartments (Fig. 1C). Cell-to-cell heterogeneity observed for nuclear Qk6 and Qk7 was sometimes evident (Fig. 1C) but was judged to be minor after quantification of nuclear/cytoplasmic ratios for many cells by high-throughput image analysis (Fig. 1D). Although the precise ratios using this two-dimensional method are subject to the cytoplasmic signal that overlays the nucleus, we conclude that the major isoforms in myoblasts are distributed in distinct nuclear/cytoplasmic ratios, with Qk5 being predominantly nuclear, and with Qk6 and Qk7 distributed throughout cells, but with Qk6 being more cytoplasmic than Qk7.
Figure 1.

Qk5 and Qk6 are the predominant isoforms and can activate splicing in C2C12 muscle cells. (A) Model depicting endogenous Qk protein isoforms (top), with the number of unique amino acids shown at the C terminus (right) and Myc epitope-tagged constructs (bottom) used in subsequent experiments; all Qk proteins share the same 311 amino acids, including Quaking homology 1 (Qua1), Qua2, and hnRNP-K homology (KH) domains. (B) Western blot of whole-cell extracts prepared from undifferentiated C2C12 myoblasts (UD) or C2C12 cells differentiated for 72 h (72h) simultaneously probed with infrared-conjugated secondary antibodies to Qk5 (top), Qk6 (middle), or Qk7 (bottom) in magenta (the notation at the right indicates the migration of specific isoforms) or PanQk in green, which recognizes all Qk isoforms. (C) Indirect immunofluorescence showing Qk isoform-specific (magenta), PanQk (green), and DAPI (blue) staining in C2C12 myoblasts, with overlay of Qk isoform and DAPI at the bottom and a higher magnification inset. The mean signal intensity for each Qk isoform and total Qk (PanQk) is shown at the bottom (±standard deviation), calculated for the total well (n = 9) (see the Materials and Methods for details). Bar, 100 µm. (D) Histogram of log2 nuclear/cytoplasmic signal intensity of Qk isoforms (X-axis) calculated for each cell imaged in C, with number of cells shown on the Y-axis. (E) Indirect immunofluorescence of C2C12 myoblasts transfected with either Myc-Qk5 (top) or Myc-Qk6 (bottom) stained using anti-Myc epitope antibody (in magenta; left), DAPI (in blue; middle), and both channels overlaid (right). Bar, 20 µm. (F) RT–PCR products from Dup-Capzb exon 9 splicing reporter analyzed on a BioAnalyzer from RNA extracted from C2C12 myoblasts either mock transfected or transfected with Myc-Qk5 or Myc-Qk6 plasmid. The mean values of the percentage included are shown below (±standard deviation) from three independent biological replicates.
Qk-dependent alternative splicing regulation requires Qk5 but not Qk6
We showed previously that depletion of all Qk isoforms alters splicing of several hundred exons in myoblasts through Qk-responsive ACUAA sequence elements (Hall et al. 2013). To test which isoform is responsible for splicing regulation, we overexpressed a Myc-tagged cDNA construct for each isoform (Supplemental Fig. S1B) and measured splicing of the Qk-regulated Capzb exon 9 in a β-globin reporter (Dominski and Kole 1991; Hall et al. 2013). To our surprise, overexpression of either isoform efficiently activates splicing (Fig. 1F). Previous work shows that Qk proteins are stable only as a dimer in vivo (Beuck et al. 2012) and that overexpression of Qk isoforms can create ectopic Qk heterodimers (Pilotte et al. 2001). Consistent with this, we observed nuclear staining of Myc-Qk6 (Fig. 1E). To rigorously test isoform function in splicing, we used a depletion–replacement strategy in which endogenous Qk mRNAs and proteins are depleted using an siRNA to a common Qk region followed by expression of a single siRNA-resistant isoform (kind gift from Sean Ryder) to create a cellular pool of Qk dominated by a single isoform (Fig. 2A; Supplemental Fig. S2A). Under the depletion–replacement treatment, inclusion of the Capzb reporter exon is specifically increased by replacement with Myc-Qk5 but not with Myc-Qk6 (Fig. 2B). In addition, whereas reconstituted Myc-Qk5 accumulates efficiently in the nucleus, Myc-Qk6 remains predominantly cytoplasmic (Fig. 2C). Taken together, we conclude that Qk5 is predominantly nuclear and serves as a splicing factor, but Qk6 is not, consistent with this isoform regulating translation (Saccomanno et al. 1999). We suggest that overexpression of Qk6 in cells with high levels of Qk5 promotes formation of Qk5–Qk6 heterodimers that increase the amount of Qk in the nucleus, leading to an inappropriate change in splicing (cf. Figs. 1F and 2B). This suggests that isoform function might be enforced by nuclear localization.
Figure 2.

Nuclear localization is sufficient for Qk splicing function. (A) Model of Myc epitope-tagged Qk5 and Qk6 with the number of unique amino acids at the C terminus shown. (B, top) RT–PCR products from Dup-Capzb exon 9 splicing reporter analyzed on a Bioanalyzer from RNA extracted from C2C12 myoblasts transfected with either control siRNA (siNT) or siRNA targeting all Qk isoforms (siQk) or cotransfected with both siQk and Myc-Qk5 or siQk and Myc-Qk6. The mean percentage included is shown below with standard deviation from three independent biological replicates. (C) Indirect immunofluorescence using anti-Myc epitope antibody (magenta; top), DAPI (blue; middle), and channels overlaid (bottom) of C2C12 myoblasts transfected with siQk + Myc-Qk5 (left) or siQk + Myc-Qk6 (right). Bar, 20 µm. (D) Model of Qk mutant proteins tested for splicing function and localization: HA-QkBody consists of an N-terminal HA epitope tag and the 311 amino acids common to all Qk proteins (top), and HA-NLS-QkBody consists of an N-terminal HA tag and nuclear localization signal (NLS) from SV40 preceding the 311 amino acids common to all Qk isoforms (bottom). (E) RT–PCR products from Capzb exon 9 splicing reporter analyzed on a Bioanalyzer from RNA extracted from C2C12 myoblasts transfected with (from left to right) siNT, siNT and HA-QkBody, siNT and HA-NLS-QkBody, siQk, siQk and HA-QkBody, or siQk and HA-NLS-QkBody. The mean percentage included from three independent biological replicates with standard deviation is shown below. (F) Indirect immunofluorescence using HA antibody (magenta; top), DAPI staining (blue; middle), and overlaid images (bottom) of representative C2C12 myoblasts from (from left to right) siNT + HA-QkBody, siNT + HA-NLS-QkBody, siQk + HA-QkBody, and siQk + HA-NLS-QkBody transfections. Bar, 20 µm. (G) Graph showing nuclear/cytoplasmic signal intensity of Myc-Qk6 (left) or HA-QkBody (right) staining of C2C12 myoblasts transfected with siRNA targeting all Qk isoforms. n = 8 siQk. (**) P ≤ 0.01. (H, left) Representative images from which epitope tag and DAPI signal intensity were measured for data shown in G. Plots at the right show corresponding signal intensities (minus background; Y-axis) plotted over image coordinates (X-axis) based on the line drawn through representative images at the left.
The nuclear presence of the common Qk protein sequence is sufficient for splicing function
If localization in the nucleus of the common (i.e., non-isoform-specific) parts of Qk is sufficient to activate splicing, then localization of just the common Qk sequences by other means should also serve. The C-terminal 30 amino acids of Qk5 encode a noncanonical nuclear localization signal (NLS) (Wu et al. 1999); however, this domain could be required for other functions specifically necessary for splicing. To test this, we made two Qk constructs lacking any isoform-specific C-terminal amino acids and comprising only the common or “body” 311 amino acids shared by all natural Qk isoforms (Fig. 2D). One construct encoded an N-terminal HA epitope tag (HA-QkBody), and the other encoded an N-terminal HA tag followed by the canonical SV40 NLS (HA-NLS-QkBody). Upon expression in myoblasts (Supplemental Fig. S2B), these mutant Qk proteins promote inclusion of Capzb exon 9 regardless of whether endogenous Qk protein is depleted (Fig. 2E). This shows that all necessary residues required for Qk splicing functions reside in the body of the Qk protein and not in the Qk5 isoform-specific tail. The HA-QkBody protein is distributed throughout the cell, whereas the HA-NLS-QkBody protein is concentrated in nuclei (Fig. 2F), reinforcing the conclusion that nuclear localization is sufficient for splicing function, mediated by the noncanonical NLS-containing Qk5-specific tail. By comparing the subcellular distribution of the HA-QkBody protein with that of Myc-Qk6 in this reconstituted cell system, the contribution of the Qk6 isoform-specific tail to cytoplasmic localization can be measured (Fig. 2G,H). This result indicates that the isoform-specific Qk6 tail contains a cytoplasmic retention or nuclear export signal that limits its contributions to splicing and directs it to cytoplasmic functions such as translation, decay, and localization.
Qk5 is required for expression of Qk6 and Qk7
To refine the understanding of Qk isoform-specific functions obtained from cells expressing single Qk protein isoforms, we selectively targeted individual protein isoforms using isoform-specific siRNAs (siQk5 or siQk6/7) (see the Supplemental Material). Treatment with siQk5 produces the expected alternative splicing changes (Hall et al. 2013) in the endogenous Rai14 cassette exon 11 (loss of repression) (Fig. 3A) and the Capzb exon 9 splicing reporter (loss of activation) (Fig. 3B), whereas treatment with siQk6/7 causes slight but statistically significant responses in the opposite direction. Analysis of the samples for protein depletion reveals that treatment with siQk5 unexpectedly depletes all Qk protein isoforms (Fig. 3C), whereas treatment with siQk6/7 leads to the loss of only Qk6 and Qk7 (Fig. 3C; Supplemental Fig. S3A). We specifically targeted Qk5 mRNA with two additional siRNAs leading to the same loss of all Qk proteins (Supplemental Fig. S3B). Depletion of Qk6 and Qk7 proteins by Qk5-specific siRNA is as efficient as that observed with a Qk6/7-specific siRNA (Supplemental Fig. S3A,B). We interpret these data to mean that cells cannot express Qk6 or Qk7 RNA or protein unless they also or first express Qk5 protein. We conclude that Qk5 is required to promote efficient expression of all of the major proteins produced by the Qk gene.
Figure 3.
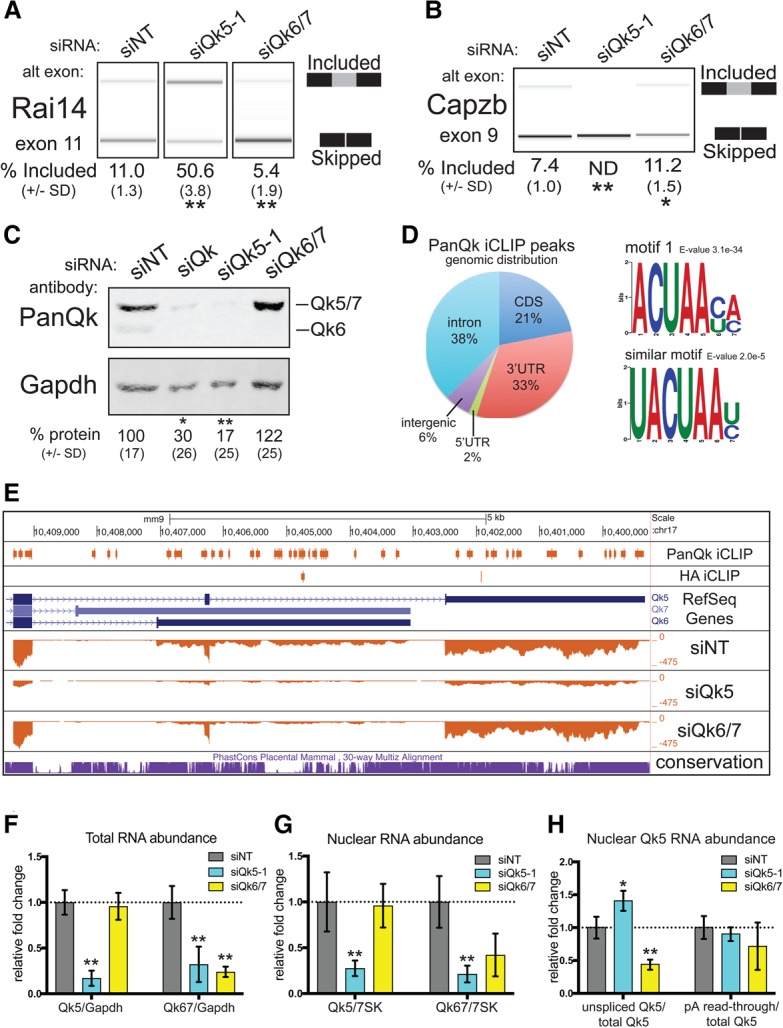
Qk5 is required for splicing and Qk5 and Qk6 accumulation. (A) RT–PCR products from endogenous Rai14 exon 11 analyzed on a Bioanalyzer from RNA extracted from C2C12 myoblasts transfected with siNT, siQk5-1, and siQk6/7. The mean percentage included from three biological replicates ± standard deviation is shown below. (**) P < 0.01. (B) RT–PCR products from the Dup-Capzb exon 9 splicing reporter analyzed on a Bioanalyzer from RNA extracted from C2C12 myoblasts transfected with siNT, siQk5-1, and siQk6/7. The mean percentage included from three biological replicates ± standard deviation is shown below. (**) P < 0.01; (*) P < 0.05. (C) Representative Western blot of protein extracted from C2C12 myoblasts transfected with siNT, siQk, siQk5-1, and siQk6/7 probed simultaneously with anti-PanQk and anti-Gapdh (the notation at the right shows the migration pattern of Qk protein isoforms). The mean relative percentage protein abundance from three biological replicates ± standard deviation is shown below. (**) P < 0.01; (*) P < 0.05. (D) The percentage of iCLIP (individual nucleotide-resolution cross-linking immunoprecipitation) peaks for PanQk that did not overlap with control HA peaks is shown mapped to genome region (left) and motifs found from unique PanQk iCLIP peaks (right). (E) University of California at Santa Cruz (UCSC) Genome Browser screen shot showing Qk 3′ end with (from top to bottom) PanQk and HA iCLIP peaks; RefSeq genes; coverage tracks for siNT, siQk5-1, and siQk6/7; and conservation. (F) RT-qPCR analysis of RNA extracted from C2C12 myoblast cultures transfected with siNT, siQk5-1, or siQk6/7 for Qk5 (left) or Qk6/7 (right) RNA normalized to Gapdh RNA and displayed as fold change relative to siNT. Error bars show standard deviation from the mean using three independent biological replicates. (**) P < 0.01. (G) RT-qPCR analysis of nuclear RNA extracted from C2C12 myoblasts cultures transfected with siNT, siQk5-1, or siQk6/7 for Qk5 (left) or Qk6/7 (right) RNA normalized to 7SK RNA and displayed as fold change relative to siNT. Error bars show standard deviation from the mean using three independent biological replicates. (**) P < 0.01. (H) RT-qPCR analysis of samples described in G but measuring unspliced Qk5 or nonpolyadenylated Qk5 normalized to total Qk5 and displated as fold change relative to siNT. Error bars show standard deviation from the mean using three independent biological replicates. (**) P < 0.01 or P < 0.05.
Qk5 and Qk6 are required for expression of distinct sets of mRNAs enriched for Qk recognition sequences
The results above suggest at a minimum that Qk5 expression is required for expression of Qk6, probably through its RNA-binding activity. To explore the impact of loss of Qk5 and Qk6 broadly on the myoblast transcriptome and map Qk binding to its RNA targets, we performed RNA sequencing (RNA-seq) on cells treated with siQk5-1, siQk6/7, or the nontargeting siNT and also performed individual nucleotide-resolution cross-linking immunoprecipitation (iCLIP) with sequencing (iCLIP-seq) (Huppertz et al. 2014) on unperturbed myoblasts. We mapped and compared CLIP peaks generated using the pan-Qk antibody that recognizes all Qk isoforms as well as an HA antibody control (Supplemental Table 1). The distribution of Qk-specific peaks indicates that Qk binds mostly introns and 3′ untranslated regions (UTRs) in myoblasts and less frequently in coding regions (Fig. 3D). The top motif identified in the Pan-Qk CLIP peaks is related to the known Qk-responsive element (Fig. 3D). Consistent with the idea of cross-isoform regulation and with previous observations in human cell lines, the distinct 3′ UTRs of both the Qk5 and Qk6 mRNAs bind Qk protein at numerous sites (Fig. 3E).
RNA-seq libraries were mapped to the mouse genome (mm9) and analyzed by DESeq2 (Love et al. 2014) to extract gene-specific changes due to each isoform (see the Supplemental Material; Supplemental Table 2). We identified two sets of genes: one whose mRNAs change significantly up or down upon loss of Qk5 and another whose mRNAs change significantly upon loss of Qk6. Although there is no overlap between these two gene sets (Supplemental Table 2G), we did observe other genes whose expression changed significantly in opposite directions when Qk5 (and Qk6) are depleted by siQk5 compared with when only Qk6 is depleted, indicating that there are genes that respond in more complex ways to loss of both isoforms. Quantitative PCR (qPCR) measurement of expression changes for several genes from different classes validated this approach. Loss of Qk5, but not Qk6, causes Hmga2 mRNA to go down while Vcan mRNA goes up, showing that these are Qk5-responsive, whereas the Qk6-specific gene Fbn1 goes up when either siQk5 or siQk6 is used for depletion, as expected for a Qk6-responsive gene (Supplemental Fig. S3E). Some genes, such as Celf2, change expression upon Qk6 depletion but behave differently when both Qk5 and Qk6 are lost upon siQk5 treatment. Based on these data, we conclude that each isoform has distinct roles in the expression of large nonoverlapping sets of genes and that some genes may depend on both proteins in different ways for their expression.
We analyzed splicing using DEXSeq and found that splicing changes are due primarily to loss of Qk5, as indicated from the reconstruction experiments in Figure 2, A–C, and as observed previously (Hall et al. 2013). We compared the extent of splicing change in these samples with those generated by a pan-Qk siRNA analyzed on microarrays (Supplemental Table 3; Hall et al. 2013) and found these to be in good agreement (Supplemental Fig. S3F), confirming that Qk splicing functions are executed by Qk5.
We next asked whether Qk-binding sites are enriched in the mRNA sequences of genes affected by loss of Qk by examining the distribution of CLIP peaks among genes with different responses to loss of Qk5 or Qk6 (Supplemental Table 2H). Genes up-regulated after loss of either Qk5 or Qk6 are significantly enriched for Qk-binding sites (Supplemental Fig. S3G), suggesting that, in many cases, Qk5 or Qk6 binding to mRNA represses mRNA levels. Genes down-regulated after loss of Qk6 are not enriched for Qk-binding sites, and those down-regulated after loss of Qk5 are actually slightly depleted of Qk-binding sites. This result suggests that at least some of the gene expression changes caused by loss of Qk5 or Qk6 may be direct; however, others may be indirect and could be mediated by other RBPs that Qk may control (Supplemental Fig. S3E; Zhao et al. 2010; Zearfoss et al. 2011; Mandler et al. 2014). Gene ontology analysis (Supplemental Table 2I) shows no enrichment of functional gene classes among Qk5-affected genes; however, Qk6-affected genes are enriched for those involved in multicellular life. The extensive binding of Qk to its own 3′ UTRs (Fig. 3E) suggests that Qk expression and control of appropriate amounts of the differently functioning Qk isoforms are regulated directly by Qk isoforms that bind isoform-specific regions of each mRNA.
Qk5 and Qk6 nuclear RNA accumulation requires Qk5 protein
Coverage tracks of RNA-seq reads from siQk5, siQk6, and siNT libraries were examined to determine how siRNA depletion affected expression of Qk isoform mRNAs. Consistent with observation that loss of Qk5 results in loss of Qk6 protein, siQk5 causes a reduction in the levels of spliced RNA for both Qk5 and Qk6 isoforms, whereas siQk6/7 treatment results in the loss of only the Qk6-specific RNA (Fig. 3E). This suggests that Qk5 is required for accumulation of RNA from the Qk locus and explains the depletion of Qk6 protein after treatment with siRNA specific for Qk5. To validate this observation and test whether the nuclear protein Qk5 is required for nuclear RNA accumulation, we fractionated siRNA-treated cells and compared levels of Qk5 and Qk6/7 RNA in whole cells and in isolated nuclei (Fig. 3F,G). As expected from the coverage tracks, siQk5 causes a reduction of Qk5 and Qk6 total RNA; however, siQk6 causes a reduction of only Qk6 total RNA. Measurement of RNA from isolated nuclei shows that siQk5 reduces nuclear Qk5 and nuclear Qk6 RNA, whereas siQk6/7 has no effect on nuclear Qk5 RNA but slightly (although not significantly) reduces nuclear Qk6 RNA (Fig. 3G).
Qk5 might promote the stability of Qk nuclear RNA by one or more of several mechanisms—including, most obviously, splicing (Figs. 2A,B, 3A,B)—but also by promoting polyadenylation or other stabilizing events. To test this, we examined splicing at the Qk5-specific 3′ splice site for the last intron of the Qk5 mRNA and evaluated readthrough at the Qk5 poly(A) site relative to total nuclear Qk5 (Fig. 3H). After treatment with siQk5, total Qk5 (Fig. 3F) and nuclear Qk5 (Fig. 3G) RNA are reduced, but the remaining RNA shows a relative increase in unspliced Qk5, suggesting that Qk5 promotes splicing of its own mRNA (Fig. 3H). Depletion of Qk6 appears to activate splicing of Qk5, as the fraction of nuclear Qk5 that is unspliced is reduced upon loss of Qk6 (Fig. 3H), indicating that one response of Qk6 depletion includes activating splicing of Qk5 mRNA, an effect consistent with other observations that loss of Qk6 promotes Qk5 through relief of translational repression by Qk6 (see below). There is no significant change in the fraction of RNA that is cleaved at the poly(A) site for Qk5 mRNA (Fig. 3H).
To exclude the possibility that the dramatic RNA accumulation defect is a unique effect of siQk5-1, we tested two other Qk5 siRNAs that target distant regions of the Qk5 isoform mRNA that do not overlap with Qk6 mRNA and found that each also reduces both Qk5 RNA and Qk6/7 RNA (Supplemental Fig. S3I). Absent a large contribution of this dramatic reduction by direct action of siQk5 on nuclear RNA, these results support a hierarchical model in which initial production of small amounts of Qk5 promote increased expression of Qk5 mRNA through increased Qk5-specific 3′ splice site usage, leading to increased Qk5 protein, which then increasingly promotes Qk6 and Qk7 RNA expression.
Testing isolated segments of the Qk gene to identify cross-isoform regulatory controls
Our attempts to understand Qk isoform regulation have been challenged by the very regulatory elements that we sought to study, in particular the effect of endogenous Qk5 on the assessment of Qk6 function (Figs. 1, 2) and the requirement of Qk5 for nuclear RNA production of Qk5 and Qk6 (Fig. 3). To address this, we isolated segments of the Qk gene into reporters and studied expression of these segments after manipulation of levels of different isoforms (Fig. 4A). First, we wanted to confirm the effect of Qk5 loss on expression and splicing at the Qk5-specific 3′ splice site. We cloned this segment of the gene into the DUP reporter (Fig. 4A, middle) and also created a version with a mutated 3′ splice site. Depletion of Qk5 (and Qk6/7) with siQk5 causes a reduction in both total RNA from the reporter gene (Fig. 4B, left panel) and spliced Qk5 RNA (Fig. 4B, right panel) relative to control siRNA, indicating that Qk5 is required for the accumulation of Qk5 RNA. Mutation of the 3′ splice site abolishes the accumulation of spliced Qk5 RNA as expected and leads to a nearly 50% reduction in the accumulation of RNA from the reporter, suggesting that splicing provides some but not all of the stabilization of RNA provided by Qk5. Since RNA from the reporter does not contain the target site for siQk5, it is unlikely that direct destabilization by siRNA explains this result. We conclude that Qk5 promotes accumulation of RNA from the Qk locus through splicing as well as other unknown mechanisms.
Figure 4.

Autoregulation and cross-isoform regulation of Qk isoforms. (A) UCSC Genome Browser screen shot showing the 3′ end of Qk isoforms, including PanQk iCLIP, RefSeq genes, siNT RNA-seq coverage, and conservation tracks with regions of the Qk sequence cloned into the respective reporter gene constructs tested below. (B) RT-qPCR performed on nuclear RNA extracted from C2C12 myoblasts transfected with the Qk5 3′ intron-CDS reporter (wild-type [WT] or 3′ splice site mutant [MT]) or wild-type reporter plus siNT and siQk5-1, measuring total reporter RNA (left) and spliced reporter RNA (right) normalized to Gapdh RNA and reported on a scatter plot as fold change relative to the respective control. Error bars represent standard deviation from the mean of six (wild-type vs. mutant) or three (siNT vs. siQk5) biological replicates. (**) P < 0.01. (C, top) Western blot of protein extracted from C2C12 myoblasts cotransfected with siNT, siQk, or siQk6/7 and renilla luciferase Qk5 3′ UTR reporter. (Bottom) Firefly luciferase as well as renilla protein were normalized to firefly protein relative to control siNT. The mean values of three biological replicates ± standard deviation are shown. (**) P < 0.01. (D) Scatter plot showing renilla/firefly values of C2C12 cells transfected with either wild-type or mutant Qk5 3′ UTR reporter. n = 6. (**) P < 0.01. (E) Summary of endogenous Qk6 expression analysis during mock, Myc-Qk5, or Myc-Qk6 transfection of C2C12 myoblasts. The top shows Western blot using Qk6 (top) and Gapdh (bottom) antibodies with the observed migration pattern of Qk6 noted at the right. The table below shows relative quantities of endogenous Qk6 protein (normalized to Gapdh; top), RNA (normalized to Gapdh; middle), and translational efficiency (relative protein/relative RNA; bottom). Mean values of three independent replicates are reported ± standard deviation. (*) P < 0.05. (F) Measurement of renilla/firefly protein from C2C12 myoblasts cotransfected with the Qk6 3′ UTR reporter and control tdTomato vector (left) or Myc-Qk6 (right). The mean of three independent replicates ± standard deviation is reported. (**) P < 0.01. (G) Western blot of whole-cell protein extracted from C2C12 myoblasts transfected with siNT, siQk5-1, and siQk6/7 probed with Qk5 or Gapdh antibody; Qk5 protein level was normalized to Gapdh protein. The mean percentage protein abundance relative to siNT was calculated from three independent biological replicates ± standard deviation and is shown at the bottom.
Qk5 inhibits its own expression by 3′ UTR binding
Here we provided evidence that Qk5 is required for Qk gene expression, especially when Qk5 levels are greatly reduced (Figs. 3E–H, 4A). Something must feed back to limit Qk5 expression when sufficiently high levels of Qk5 are reached. Previous studies of mammalian Qk5 (Larocque et al. 2002) or its fly homolog, HowL (Nabel-Rosen et al. 2002), suggest a general role for Qk5 as a repressor of gene expression through binding to 3′ UTRs, consistent with the observation that Qk5-repressed genes are enriched for Qk-binding sites (Supplemental Fig S3G; Supplemental Table 2H). One attractive site for such regulation occurs where we (Fig. 3E, CLIP track) and others (Hafner et al. 2010; Van Nostrand et al. 2016) found Qk binding at a conserved computationally predicted binding site (Paz et al. 2014) near the end of the Qk5 3′ UTR. We cloned the entire Qk5 3′ UTR into a renilla luciferase reporter and compared expression of this reporter with expression of a cotransfected firefly luciferase reporter carrying the bovine growth hormone 3′ UTR in cells depleted of either all Qk proteins (siQk) or Qk6 and Qk7 (siQk6/7; N.B. siQk5 targets this reporter and was not used). Depletion of all Qk protein forms, but not Qk6/7 alone, leads to a statistically significant 1.8-fold increase in renilla protein expression relative to firefly luciferase (Fig. 4C). This suggests that Qk5 negatively regulates its own expression through its 3′ UTR. To test whether the binding site near the end of the Qk5 3′ UTR mediates this repression by Qk5, we made a reporter mutation to substitute UACUAAC (wild type) with UGGUACC in the RNA (Fig. 4A, right). This mutation leads to a nearly fivefold increase in expression compared with wild type (Fig. 4D). We noted previously that overexpression of Myc-Qk5 leads to a reduction in endogenous Qk5 mRNA and protein (Supplemental Fig. S4A), so we tested whether overexpression of Myc-Qk5 would significantly down-regulate the expression of the Qk5 3′ UTR reporter, and it does (Supplemental Fig. S4B,C).
Qk proteins also bind the Qk6 mRNA 3′ UTR (Fig. 3E; Hafner et al. 2010; Van Nostrand et al. 2016), and phylogenetically conserved Qk-binding motifs are also found in this sequence (Paz et al. 2014). To test whether Qk5 similarly controls Qk6 through binding to the Qk6 3′ UTR, we made a reporter gene with the Qk6 3′ UTR fused to the renilla luciferase coding region and cotransfected it with a firefly luciferase control reporter and either control, siQk, or siQk5-1 siRNAs (Supplemental Fig. S4D). We observed no change in reporter protein relative to control (Supplemental Fig. S4E), suggesting that Qk proteins do not repress expression through the Qk6 3′ UTR reporter as they do through the Qk5 3′ UTR.
Together, these results suggest that Qk5 binds the UACUAAC element in its own 3′ UTR to negatively regulate its expression. Thus, Qk5 protein at low levels promotes additional Qk5 mRNA accumulation through splicing and stabilization of nuclear RNA but at high levels acts by binding its own 3′ UTR to limit expression through an as yet unknown mechanism. Our analysis of the distribution of Qk-binding sites in genes specifically up-regulated after loss of Qk5 indicates that many such genes have binding sites for Qk protein, suggesting that Qk5 mRNA is not the only mRNA whose expression is repressed by Qk5.
Qk6 positively regulates its own translation through its 3′ UTR
During overexpression experiments with Myc-Qk6, we noticed a reproducible increase in levels of endogenous Qk6. We repeated the overexpression of Myc-Qk6 in myoblasts and performed quantitative Western blotting to measure both ectopic and endogenous Qk6 (Fig. 4E). We observed a 2.5-fold increase in endogenous Qk6 protein without a significant change in RNA level, indicating a 2.43-fold increase in translational efficiency (P ≤ 0.05) (Fig. 4E). No significant change in endogenous Qk6 protein levels is observed when Myc-Qk5 is overexpressed, suggesting that increasing the level of Qk5 has little ability to further increase Qk6 RNA accumulation in these cells (Fig. 4E). It is possible that the slight and statistically insignificant increase in translation of Qk6 that was observed is due to increased levels of Qk5 in the cytoplasm, where it may contribute to normal Qk6 protein function by mislocalization.
To determine whether Qk6 autogenous control of protein abundance is mediated by translational control through its 3′ UTR, we tested a luciferase reporter carrying the Qk6 3′ UTR (Fig. 4A, left) and compared its expression with the control firefly luciferase reporter with the bovine growth hormone 3′ UTR after cotransfection with a Myc-Qk6-expressing plasmid or a control plasmid expressing tdTomato (Fig. 4F; Supplemental Fig. S4F). Overexpression of Myc-Qk6, but not tdTomato protein, leads to a significant increase in renilla expression mediated by the Qk6 3′ UTR as compared with control (Fig. 4F), indicating that the Qk6 3′ UTR contains an autoregulatory element through which Qk6 activates its own translation. We conclude that Qk6 promotes translation of its own mRNA.
Qk6 negatively regulates Qk5 protein expression
If Qk5 protein promotes expression of both its own and Qk6 mRNA (Fig. 3), then reciprocal influences may exist whereby Qk6 protein feeds back to control Qk5 levels. We routinely observed a 15%–25% increase in endogenous Qk5 protein levels when Qk6 is depleted using either of two Qk6 siRNAs (Figs. 3C, 4G; Supplemental Fig. S4G,H). In other experiments where Qk6 is depleted, small but measurable effects in the direction opposite the effect of Qk5 depletion are often observed, such as statistically significant additional splicing repression (Rai14) (Fig. 3A) or activation (Capzb) of exons repressed or activated by Qk5, as would be expected when Qk5 levels increase. This increase in Qk5 protein level upon loss of Qk6 is accompanied by a consistent but modest reduction in Qk5 mRNA level relative to control (Fig. 3F) to produce a 37% increase in translational efficiency (Vasudevan and Steitz 2007) of Qk5 mRNA when Qk6 is depleted. This opposing relationship between RNA stability and translational efficiency has been described previously (Kawai et al. 2004), and, in this case, suggests that Qk6 protein binding to the Qk5 mRNA stabilizes the transcript as it represses translation. Although not statistically significant, these data suggest that Qk6 represses translation of Qk5 mRNA, consistent with the function of Qk6 on other mRNAs in other cell types (Saccomanno et al. 1999; Zhao et al. 2010). No effect of Qk6 depletion is observed on the Qk5 3′ UTR reporter (Fig. 4C), suggesting the possibility that sequences outside the 3′ UTR are required. We hypothesize that in cell types where Qk6 protein is more abundant than Qk5, this regulation may be more pronounced.
The results to this point identify several autoregulatory and cross-isoform regulatory influences that form a network that controls Qk expression and the composition of the different isoforms produced by the gene in myoblasts, in turn controlling large and distinct sets of other genes. Qk5 is the most abundant isoform and is required for all Qk expression at the level of RNA accumulation (Fig. 3F–H) and splicing (Fig. 2), promoting its own and Qk6 expression at low levels (Figs. 3F–H, 4B) but also feeding back negatively on its own expression through a binding site in its 3′ UTR (Fig. 4C,D). Qk6 is less abundant in myoblasts but promotes its own translation (Fig. 4E,F) while inhibiting Qk5 translation (Fig. 4G).
Subtle inhibition of Qk translation reveals homeostatic responses of the Qk network
Evidence supporting the existence of the Qk isoform autoregulatory network has been obtained using siRNA depletions designed to create catastrophic loss of protein, which may quickly overwhelm even a robust homeostatic network. In addition, siRNAs are subject to uncertainties about the extent to which they may target pre-mRNAs in the nucleus (Langlois et al. 2005; Berezhna et al. 2006), complicating interpretation. To test more subtle perturbations of the network and avoid the use of siRNAs, we inhibited translation of all Qk mRNA using a morpholino oligonucleotide to occlude the translational start site shared by all Qk mRNAs and evaluated the expression of Qk protein isoforms (Fig. 5A) and RNAs in isolated chromatin and nucleoplasmic fractions of nuclei and cytoplasmic fractions (Fig. 5; Pandya-Jones et al. 2013). Treatment with the Qk start site morpholino reduced total expression of Qk by ∼50% as compared with the control nontargeting morpholino (Fig. 5A). Fractionation was monitored by the following proteins (Fig. 5A) and nuclear RNA distributions (Supplemental Fig. S5). A panel of qRT–PCR primer pairs that spans key regions of the Qk transcription unit is shown with its position in the branched processing pathway, with Qk6-specific RNAs on top and Qk5-specific RNAs at the bottom (Fig. 5B; see also Figs. 3E, 4A). Upon subtle inhibition of Qk translation, the level of total Qk RNA increases relative to Gapdh RNA in all cell fractions tested (Fig. 5C, left). This response is the opposite of that observed with catastrophic loss of Qk RNA observed with siQk or siQk5 (Fig. 3). However, it is consistent with the observation that Qk5 represses its own expression via a Qk-binding site near the end of its 3′ UTR (Fig. 4C,D). A homeostasis model would predict that lowering Qk5 protein would relieve Qk5-mediated repression of Qk5, which would increase Qk5 RNA levels in order to return to an appropriate level of Qk5 protein, after which repression would be re-established. Catastrophic loss of Qk5 mRNA may prevent this response because a threshold of functional Qk5 may be required to mount it.
Figure 5.

Moderate reduction of total Qk protein results in increased Qk5 RNA processing and decreased Qk6 RNA production. (A) Representative Western blot of whole-cell protein extracts (left) or nuclear extract (right) from C2C12 myoblasts to which either control (ctrl) or Qk morpholino oligomer has been delivered and then probed simultaneously with PanQk, Gapdh, and histone H3 antibodies. Quantitation of the mean infrared signal for total Qk relative to Gapdh (total protein; left) or total Qk relative to histone H3 (nuclear fraction; right) is reported below as percentage protein abundance relative to control ± standard deviation of the mean calculated from three independent biological replicates. (*) P < 0.05. (B) Schematic overview of Qk transcripts and the location of qPCR primer sets used for subsequent measurements. (C) RT-qPCR of RNA extracted from chromatin, nucleoplasmic, or cytoplasmic fractions of C2C12 myoblasts treated with control or Qk morpholino, measuring the abundance of total Qk RNA normalized to Gapdh RNA (left), spliced Qk6 RNA normalized to total Qk RNA (top right), or total Qk5 RNA normalized to total Qk RNA (bottom right). All values are reported as mean fold change relative to control ± standard deviation of the mean from three independent replicates. (**) P < 0.01; (*) P < 0.05. (D) RT-qPCR of RNA extracted from chromatin or nucleoplasmic fractions of C2C12 myoblasts treated with control or Qk morpholino, measuring abundance of spliced Qk5 RNA, unspliced Qk5, polyadenylation readthrough Qk5, or Qk nonsense-mediated decay (NMD) isoform, each normalized to total Qk5 RNA. All values are reported as mean fold change relative to control ± standard deviation of the mean from three independent replicates. (**) P < 0.01.
We next asked how inhibition of Qk translation affects the apportioning of total Qk transcripts in the directions of Qk5 mRNA (Fig. 5C bottom right) and Qk6 mRNA (Fig. 5C, top right) production. In all fractions tested, the relative amount of spliced Qk6 mRNA decreased significantly, whereas the relative amount of spliced Qk5 mRNA increased (Fig. 5C, right). This response to subtle Qk protein depletion shows that alternative splicing has shifted in favor of the Qk5 isoform, a response that will lead to resetting of Qk protein and isoform composition by increasing Qk5 expression.
Finally we examined how morpholino inhibition altered the fraction of Qk5 processing precursors and products in the nucleus as a fraction of total Qk5 RNA (Fig. 5D). Spliced Qk5 (via the Qk5-specific last 3′ splice site) (Fig. 5B) associated with chromatin increases, concomitant with a decrease in unspliced Qk5 RNA. The change in the amount of RNA that reads through the poly(A) site for Qk5 mRNA is reduced but mirrors the reduction in unspliced RNA (Fig. 5D). We could detect no change in the level of the nonsense-mediated decay (NMD) isoform of Qk5 in the nucleus. We conclude that the model for autoregulation of Qk5 explains the consequences of a subtle loss of Qk protein as follows: loss of repression of Qk5 on its own 3′ UTR followed by increased Qk5 expression that both increases overall accumulation of Qk RNA and directs splicing of more Qk5 mRNA. As Qk5 mRNA increases, Qk5 protein levels return and are sufficient to repress Qk5 again via its 3′ UTR. Qk6 levels return more slowly; as Qk5 levels reset, the splicing choice increasingly favors Qk6 mRNA, which is more efficiently translated as Qk6 protein levels are restored.
Cross-isoform Qk regulatory control is conserved in C6 glioma cells
Qk5 is the predominant Qk isoform present in C2C12 myoblasts (Fig. 1B). In contrast, Qk6 and Qk7 are the predominant isoforms in adult mouse brains (Hardy et al. 1996). If the regulatory interactions that we observed in C2C12 myoblasts are at play in other cells, then those with high Qk6 and low Qk5 might be explained as follows. The strong requirement of Qk5 for Qk6 RNA that we observed in myoblasts might be less if the positive translational autoregulation of Qk6 (Fig. 4G,F) was enhanced. Under such conditions, Qk6 might also more efficiently repress the translation of Qk5 mRNA (Fig. 4H). To begin testing this network in other cells, we used quantitative Western blotting to measure Qk5, Qk6, and Qk7 protein abundance (Fig. 6A,B). C2C12 cells have a high Qk5/Qk6 ratio (with Qk7 nearly undetectable) (Figs. 1A, 6A,B). Consistent with previous findings (Hardy et al. 1996), adult mouse optic nerve and cerebellum tissue have distinct Qk5/Qk6/Qk7 compositions and a much lower Qk5/Qk6 ratio (Fig. 6A). In contrast, rat C6 glioma (Benda et al. 1971) and CG4 oligodendrocyte precursor cells (Sharma et al. 2011) have intermediate Qk5/Qk6 ratios, with different amounts of Qk6 or Qk7 (Fig. 6A,B). We chose rat C6 glioma cells as a counterexample to mouse C2C12 cells to test whether the autoregulatory controls observed above operate in cells with lower Qk5/Qk6 ratios.
Figure 6.

Qk cross-isoform regulation is conserved in rat C6 glioma cells. (A) Representative Western blots of whole-cell protein extracts from C2C12 myoblasts, rat C6 glioma cells, rat CG4 oligodendrocyte precursor cells, mouse optic nerve tissue, and mouse cerebellum tissue probed with Qk5 (top), Qk6 (middle), or Qk7 (bottom) in the magenta channel and PanQk in the green channel. (B) Western blot (see A) infrared intensity values for each isoform were normalized to PanQk and used to determine the mean percentage of each protein isoform relative to total (PanQk) in whole-cell protein extracts from C2C12 myoblasts, rat C6 glioma cells, rat CG4 oligodendrocyte precursor cells, mouse optic nerve tissue, and mouse cerebellum sampled in biological triplicate. (C) Representative Western blots of whole-cell protein extracts from rat C6 glioma cells transfected with siNT, siQk5-1, or siQk6/7 and probed simutaneously with either Qk5 and Gapdh (left), Qk6 and Gapdh (middle), or Qk7 and Gapdh (right) antibodies. Quantitation of the mean infrared signal for the Qk isoform relative to Gapdh is reported below as the percentage protein abundance relative to siNT ± standard deviation of the mean calculated from three independent biological replicates. (**) P < 0.01. (D) RT-qPCR of RNA extracted from rat C6 glioma cells described in C measuring Qk5 RNA (left) or Qk6/7 RNA (right) normalized to Mapk3 mRNA and displayed as fold change relative to siNT ± standard deviation. (**) P < 0.01.
We treated C6 glioma cells to deplete either the Qk5 or Qk6/7 isoforms (the siRNA target sequences are conserved in mice and rats). Although overall siRNA-mediated depletion appears less efficient in these cells as compared with C2C12 cells, the loss of Qk6/7 upon depletion of Qk5 is evident (Fig. 6C), indicating that the hierarchical requirement of Qk5 for expression of Qk6/7 RNA (Fig. 6C) and protein (Fig. 6B) is conserved. The magnitude of the reduction of Qk6 protein in C6 glioma cells is less than that observed in C2C12 myoblasts (cf. Fig. 6C and Supplemental Fig. S3A). As in myoblasts, we observed an increase in Qk5 protein under Qk6/7 depletion in C6 glioma cells (Fig. 6D), indicating that the feedback repression of Qk6 on Qk5 (Fig. 3G) is conserved. In contrast to C2C12 cells, where siRNA depletion of Qk6 does not affect Qk5 mRNA, C6 cells suffer a significant reduction of Qk5 RNA under this condition (Fig. 6D), which represents a 2.6-fold increase in translational efficiency in C6 glioma cells (P < 0.01) compared with a nonsignificant ∼1.4-fold in C2C12 myoblasts upon loss of Qk6/7. Due to the higher relative concentration of Qk6 in C6 glioma cells, the translational repression of Qk5 appears more efficient. Taken together, these findings suggest that the Qk autogenous regulatory network operates at various settings in different cells (Fig. 7A), where the Qk5/Qk6/Qk7 composition can vary in a stable way using parallel sets of autoregulatory and cross-regulatory controls. The existence of multiple different stable settings of the Qk5/Qk6/Qk7 composition indicates that the Qk regulatory network is responsive to the influence of other regulatory proteins and RNAs, which may add controls on top of the Qk network to achieve homeostasis at different Qk5/Qk6/Qk7 compositions.
Figure 7.

Models of Qk autogenous regulation. (A) Quantitative rheostat model: Various relative levels of Qk protein isoforms are observed in different cell/tissue types, such as Qk5 high/Qk67 low in C2C12 cells, medium Qk5/medium Qk67 in rat C6 glioma cells, and high Qk67/low Qk5 in adult mouse optic nerve and cerebellum tissues. These different relative protein levels support the observed autoregulatory and cross-isoform regulatory interactions defined in this study, albeit by imposing greater influence (thicker lines) on specific processing steps when their relative concentration is higher (larger font) than in other cell/tissue types. Arrows denote positive regulation, while lines ending in perpendicular line denote negative regulation. (B) Hierarchical model: The Qk gene transcribes a single pre-mRNA that is processed to make mature Qk mRNA isoforms, which are then exported from the nucleus and translated into Qk5 and Qk6 proteins. The NLS unique to the Qk5 C terminus mediates nuclear import; thus, more Qk5 protein is observed in the nucleus. Here Qk5 promotes nuclear accumulation of Qk pre-mRNA and formation of Qk5 mRNA through a splicing-based mechanism. Accumulation of Qk5 protein negatively feeds back to reduce its own levels while also positively feeding back to promote accumulation of Qk6 mRNA. Qk6 protein is localized predominantly in the cytoplasm, negatively regulates the translation of Qk5, and positively regulates its own translation. Dotted lines represent regulatory interactions, while solid lines represent the spatial and temporal flow of genetic information.
Discussion
We initiated this study to understand how RBP family isoforms that recognize the same RNA sequence in different functional contexts (splicing, RNA transport, translation, and decay) are regulated within a single cell so that each of these processes is appropriately supplied with the correct amount of RBP. We chose Qk because all isoforms arise from a single gene, thus avoiding the complexities that exist with other RBP families that express multiple isoforms from each of multiple genes. In the case of Qk, isoform function is largely determined by cellular compartment: Nuclear functions (splicing and nuclear RNA accumulation) are executed by the predominantly nuclear isoform Qk5, whereas cytoplasmic functions (translational control and mRNA decay) are executed by the predominantly cytoplasmic Qk6 (Figs. 1–3; Hardy et al. 1996; Lu et al. 2003). We note that both isoforms appear to have dynamic subpopulations outside of their predominant localization sites and that the two-dimensional imaging method that we used may overestimate nuclear localization due to cytoplasm above and below the nucleus (Fig. 1). Furthermore, the level of natural heterodimerization is unknown and could influence localization and function in ways that we generated artificially (Figs. 1, 2). Homeostasis and control of the levels of each isoform are established through a network of autoregulatory and cross-regulatory controls (Fig. 7) whereby Qk5 is required for its own and Qk6 RNA expression from the nucleus (Fig. 3), and Qk6 activates its own mRNA and represses Qk5 mRNA at the translational level (Fig. 4). The strength of these controls is tunable such that cells express different ratios of Qk5 and Qk6 in a stable characteristic way (Fig. 7).
A division of labor between Qk protein isoforms
We previously identified Qk protein as a regulator of muscle-specific alternative splicing through binding to intronic ACUAA sequence motifs (Hall et al. 2013). Since the Qk5 isoform contains a NLS (Wu et al. 1999), we expected Qk5 to be responsible for splicing. Indeed, Qk5 is both necessary and sufficient for splicing regulation (Figs. 2, 3), whereas Qk6 and Qk7 are dispensable (Fig. 2). Although artificially overexpressed Myc-Qk6 can activate splicing (Fig. 1F), abnormally high nuclear localization after increased expression in cells with already high levels of Qk5 is likely a consequence of heterodimerization with endogenous Qk5 that increases nuclear concentration of Qk dimers (Fig. 1F; Pilotte et al. 2001). A mutant Qk protein containing all of the common Qk sequences but lacking isoform-specific C termini (QkBody) is distributed throughout the nucleus and cytoplasm and suffices for splicing, whereas Qk6 does not (Fig. 2). Careful comparison of the cellular distribution of the QkBody protein with that of Qk6 indicates that the eight Qk6-specific C-terminal amino acids encode a cytoplasmic retention or nuclear export signal (Nakielny and Dreyfuss 1999; Cook and Conti 2010) that increases its cytoplasmic representation as compared with QkBody (Fig. 2G,H). Qk5 appears to control events other than splicing by unknown mechanisms that lead to nuclear RNA accumulation (Figs. 3F–H, 4B), at least a part of which seems independent of splicing per se (Fig. 4B). Qk5 also represses its own expression through a Qk-binding site in its 3′ UTR, possibly through nuclear retention (Larocque et al. 2002; Nabel-Rosen et al. 2002), but the mechanism and subcellular location of this repression are unknown. Qk6 protein is an activator of its own translation (Fig. 4E,F), a function consistent with its predominantly cytoplasmic localization (Zhao et al. 2010). Since these distinct functions correspond with the main subcellular distributions of each isoform and since isoform-specific C-terminal protein sequences seem dedicated only to localization, we suspect that isoform functions are enforced primarily by localization. It follows that all Qk protein elements required to interact with the splicing, translation, export, and decay machineries lie within QkBody and that additional dissection of Qk protein sequences will be necessary to map the sites required for function in each of those processes.
Our analysis of the broader effects of loss of specific Qk isoforms indicates that the specific regulatory functions identified in reporter tests extend into the transcriptome. We identified sets of mRNAs whose levels significantly increase or decrease upon loss of Qk5 or Qk6 (Supplemental Fig. S3C–E; Supplemental Table 2A–I). In particular, distinct sets of mRNAs that increase in level after loss of Qk5 or Qk6 are enriched for Qk-binding sites identified by CLIP (Fig. 3D; Supplemental Fig. S3G), similar to the Qk5 mRNA itself (Fig. 4C,D). The existence of separate sets of mRNAs whose levels are controlled by only one Qk isoform or the other is further support for the idea that the division of labor between these isoforms, enforced by distinct subcellular localization, is deeply integrated into cell function.
Autoregulation and cross-isoform regulation control Qk isoform composition
Our initial effort to deplete cells of each single isoform of Qk protein in turn was complicated by the strong dependence of Qk6 expression on expression of Qk5 (Fig. 3). We were able to construct cells in which endogenous isoforms were depleted and replaced with a single isoform to confirm that Qk5 is responsible for splicing (Fig. 2A–C). Furthermore, the Qk5 isoform-specific C-terminal tail containing the noncanonical NLS does not contain an essential splicing-specific function: It can be replaced by an N-terminal SV40NLS to promote splicing. In addition, it seems that any manipulation that increases the amount of the common Qk sequences in the nucleus, including recruitment of Qk6 or QkBody by heterodimerization with Qk5, will augment Qk-dependent activation (or repression) of splicing (Figs. 1F,G, 2D–F). In the remaining experiments, using such depleted and reconstructed cells was impractical, and we assigned a function to Qk5 if Qk5 depletion compromised that function but Qk6 depletion did not. If depletion of both Qk5 and Qk6 compromised a function similarly to depletion of Qk6 alone, we tentatively assigned that function to Qk6 because depletion of Qk5 leads indirectly but efficiently to depletion of Qk6. It is important to note the danger in this conclusion because our transcriptome analysis identifies genes whose expression does not fall neatly into two categories, probably because, like Qk, they are regulated by both Qk isoforms to different extents.
Using a combination of endogenous Qk expression measurements (Fig. 3) and artificial reporter constructs (Fig. 4), we found that Qk5 is required for nuclear accumulation of both its own transcripts and those encoding Qk6/7 (Fig. 3). We also provide evidence that Qk6 represses translation of Qk5 mRNA but activates translation of its own mRNA (Fig. 4). These results allowed us to build a model of Qk autogenous and cross-isoform regulation for C2C12 cells (Fig. 7). This control network is more similar to a rheostat than a bistable switch where either Qk5 or Qk6 expression dominates because it supports stable intermediate isoform mixtures from the Qk gene in different cell types (Fig. 7A). The structure of this network is such that the same compartmentalization that constrains the functional role of each isoform is also used to create the autoregulatory and cross-regulatory controls. Qk5 acts on its own RNA and that of Qk6/7 through nuclear processes, whereas Qk6 acts on both its own mRNA and that of Qk5 through translation and decay in the cytoplasm. This network controls both the total amount of Qk protein by combining a strong dependence of Qk6 expression on Qk5 with negative feedback of Qk5 by both Qk5 (at the RNA level) and Qk6 (at the translational level). Autoregulation by RBPs is common and can be imposed at different RNA processing steps, including splicing (Wollerton et al. 2004; Lareau et al. 2007; Ni et al. 2007), polyadenylation (Dai et al. 2012), mRNA stability/decay (Ayala et al. 2011), and translation (de Melo Neto et al. 1995; Wu and Bag 1998). Although cross-paralog regulation exists between RBP family members encoded by separate genes (Boutz et al. 2007b; Spellman et al. 2007; Wang et al. 2012), the Qk regulatory network highlights the role of compartmentalization in the execution and regulation of the division of labor among the Qk isoforms (Fig. 7). Other RBP families may use similar mechanisms for autogenous and cross-isoform control, but, for those encoded by multiple genes, complex control of transcription may provide an added layer of regulation not observed for Qk.
Having described this network using siRNA depletion strategies, we were concerned about two potentially confounding effects. One is that catastrophic loss of mRNA for Qk may compromise the ability of the cell to respond sufficiently to restore homeostasis. We tested the potential of the network to produce a homeostatic response by an orthogonal method of inhibiting expression by using a morpholino to block the translation start site sequences shared by all Qk isoform mRNAs (Fig. 5). This showed that instead of the complete loss of nuclear RNA observed with siRNAs against Qk5 mRNA, translational inhibition allowed cells to respond by increasing RNA levels from the Qk gene (Fig. 5C). This could be due to promotion of nuclear RNA accumulation by Qk5 or be an indirect effect on the rate of Qk transcription, but, in either case, Qk5 mRNA increases, as does Qk5 splicing at the expense of Qk6 splicing, as a fraction of total RNA from the Qk locus. This provides strong evidence that Qk5 helps control the ratio of Qk5 and Qk6 mRNA from the gene through alternative splicing.
Since the quantitative output from the Qk network appears to be set at different Qk5/Qk6/Qk7 ratios in different cell types (Fig. 6A), it must also be sensitive to regulation by other factors. For example, the RBP RBFOX2 reduces Qk7 expression by repressing alternative splicing in human embryonic stem cells (Yeo et al. 2009). Since RbFox2 is an abundant splicing regulator in C2C12 cells (Bland et al. 2010; Singh et al. 2014), this may explain why Qk7 is nearly undetectable (Figs. 1B, 6A). However, this seems not to hold in the mouse cerebellum, where both Qk7 (Fig. 6A) and RbFox2 (Gehman et al. 2012) are abundant. In another example, miR-214-3p, targets regions common to both the Qk6 and Qk7 3′ UTR (van Mil et al. 2012), which may specifically reduce Qk6 and Qk7 (Irie et al. 2016) or total Qk (Shu et al. 2017). There likely are multiple additional means by which Qk isoform ratios can be regulated, but the Qk autoregulatory and cross-regulatory isoform network uncovered here represents the foundational structure on which that regulation is imposed.
Qk isoform ratio regulation, development, and cancer
Qk is a key regulator of development (Ebersole et al. 1996; Li et al. 2003; Justice and Hirschi 2010) and cancer (Novikov et al. 2011; Chen et al. 2012; Zong et al. 2014). Our model predicts that Qk5 expression precedes expression of other Qk isoforms temporally as transcription is up-regulated at the Qk gene (Fig. 7B) in a developmental context. This expectation appears to hold for embryogenesis in Xenopus (Zorn et al. 1997), zebrafish (Radomska et al. 2016), and mice (Ebersole et al. 1996; Hardy et al. 1996). Additionally, early stem/progenitor cells in Drosophila (Nabel-Rosen et al. 1999) and mice (Hardy 1998) express predominantly Qk5 (or its ortholog, HowL, in Drosophila). In mice, qk knockout is embryonic-lethal (Li et al. 2003), and the qkl1 mutant mouse, which appears to lack Qk5 expression due to disruption of Qk5-specific splicing by a point mutation (Cox et al. 1999), is recessive lethal (Shedlovsky et al. 1988). In this mutant, both Qk6 and Qk7 mRNAs are significantly reduced in visceral endoderm dissected from nonviable embryos (Cox et al. 1999; Bohnsack et al. 2006), suggesting that Qk5 is required for efficient Qk6 and Qk7 expression in this tissue also. These observations provide additional evidence in support of our model of Qk autoregulation and suggest its importance in an evolutionarily conserved developmental context.
The extensively studied qkv mouse model of dysmyelination (Sidman et al. 1964) expresses reduced mRNA levels of all Qk isoforms in glia (Lu et al. 2003) and vascular smooth muscle (van der Veer et al. 2013) due to a deletion that includes a portion of the Qk promoter (Ebersole et al. 1996). Based on the models that we present here (Fig. 7), lower Qk5 protein levels in qkv oligodendrocyte precursor cells may be insufficient for robust Qk6 and Qk7 up-regulation that normally occurs around the peak of myelination (Ebersole et al. 1996). Since deletion of qk in oligodendrocytes is lethal (Darbelli et al. 2016), the reduction in Qk6 and Qk7 in qkv oligodendrocytes could be due to reduced expression of Qk5 to a level just sufficient to maintain the viability of the organism. In this view, the imbalance of Qk isoforms in qkv oligodendrocytes results in developmental defects and the tremor phenotype because there is insufficient Qk5 to promote adequate expression of Qk6 and Qk7.
Finally, disruption of tissue-specific Qk isoform expression patterns is also observed in non-small-cell lung cancer (NSCLC) (de Miguel et al. 2016; Sebestyen et al. 2016) and glioblastoma (Jin et al. 2004) patient samples (human Qk is encoded by the QKI gene). Although overall Qk protein expression is reduced, an isoform ratio switch occurs in which healthy lung cells expressing mostly Qk6 transition to a state in NSCLC samples where cells express predominantly Qk5 (de Miguel et al. 2016; Sebestyen et al. 2016). This isoform switch likely shifts QKI function away from cytoplasmic regulation such as translation and decay (Saccomanno et al. 1999; Zhao et al. 2010) toward nuclear processes such as splicing and primary transcript stability. Understanding the relationship of QKI gene function in the context of cancer and other diseases requires evaluation of the specific functions of Qk protein isoforms on their own RNAs as well as on the other pre-mRNAs and mRNAs from the many other genes that QKI regulates.
Materials and Methods
Cell Culture
C2C12 cells were routinely cultured in Dulbecco's modified Eagle medium (DMEM) with high glucose (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies) at 37°C with 5% CO2. For differentiation experiments, C2C12 cells were allowed to reach ∼90% confluency, the medium was changed to DMEM supplemented with 5% horse serum (Life Technologies), and the cells were harvested 72 h after medium change. C6 glioma cells were cultured in F-12K medium (American Type Culture Collection) supplemented with fetal bovine serum at 2.5% and horse serum at 15% as described (Benda et al. 1968) at 37°C with 5% CO2. CG4 oligodendrocyte precursor cells were grown in DMEM with high glucose supplemented with N1 (5 μg/mL transferrin, 100 μM putrescine, 20 nM progesterone, 20 nM selenium), 10 ng/mL biotin, 5 μg/mL insulin, and 30% B104 cell-conditioned medium as described (Sharma et al. 2011) at 37°C with 5% CO2; all supplements were purchased from Sigma-Aldrich.
Additional methods and oligonucleotide sequences are in the Supplemental Material.
RNA-seq and CLIP-seq reads analyzed in this study can be found under Gene Expression Omnibus accession number GSE102615.
Supplementary Material
Acknowledgments
We thank Sean Ryder, Doug Black, Jeremy Sanford, Mariano-Garcia Blanco, Shelton Bradrick, Rhonda Perriman, David Feldheim, Jason Talkish, and Susan Strome for reagents, discussion, and advice. W.S.F. was supported by National Institutes of Health (NIH) predoctoral T32 GM008646 and California Institute of Regenerative Medicine (CIRM) predoctoral fellowship award TG2-01157 and in part through a US–Israel Binational Science Foundation grant to Yael Mandel-Gutfreund and M.A. This work was supported by NIH grant R01-GM040478 to M.A.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.302059.117.
References
- Aberg K, Saetre P, Jareborg N, Jazin E. 2006. Human QKI, a potential regulator of mRNA expression of human oligodendrocyte-related genes involved in schizophrenia. Proc Natl Acad Sci 103: 7482–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D'Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, et al. 2011. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30: 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benda P, Lightbody J, Sato G, Levine L, Sweet W. 1968. Differentiated rat glial cell strain in tissue culture. Science 161: 370–371. [DOI] [PubMed] [Google Scholar]
- Benda P, Someda K, Messer J, Sweet WH. 1971. Morphological and immunochemical studies of rat glial tumors and clonal strains propagated in culture. J Neurosurg 34: 310–323. [DOI] [PubMed] [Google Scholar]
- Berezhna SY, Supekova L, Supek F, Schultz PG, Deniz AA. 2006. siRNA in human cells selectively localizes to target RNA sites. Proc Natl Acad Sci 103: 7682–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuck C, Qu S, Fagg WS, Ares M Jr, Williamson JR. 2012. Structural analysis of the Quaking homodimerization interface. J Mol Biol 423: 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland CS, Wang ET, Vu A, David MP, Castle JC, Johnson JM, Burge CB, Cooper TA. 2010. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res 38: 7651–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack BL, Lai L, Northrop JL, Justice MJ, Hirschi KK. 2006. Visceral endoderm function is regulated by quaking and required for vascular development. Genesis 44: 93–104. [DOI] [PubMed] [Google Scholar]
- Boutz PL CG, Stoilov P, Black DL. 2007a. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev 21: 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL SP, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares M Jr, Black DL. 2007b. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 21: 1636–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres JF, Screaton GR, Krainer AR. 1998. A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev 12: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Zhu J, Manche L, Huber E, Krainer AR, Caceres JF. 2002. Nuclear export and retention signals in the RS domain of SR proteins. Mol Cell Biol 22: 6871–6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. 2012. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75: 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheever A, Ceman S. 2009. Phosphorylation of FMRP inhibits association with Dicer. RNA 15: 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AJ, Paik JH, Zhang H, Shukla SA, Mortensen R, Hu J, Ying H, Hu B, Hurt J, Farny N, et al. 2012. STAR RNA-binding protein Quaking suppresses cancer via stabilization of specific miRNA. Genes Dev 26: 1459–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AG, Conti E. 2010. Nuclear export complexes in the frame. Curr Opin Struct Biol 20: 247–252. [DOI] [PubMed] [Google Scholar]
- Cox RD, Hugill A, Shedlovsky A, Noveroske JK, Best S, Justice MJ, Lehrach H, Dove WF. 1999. Contrasting effects of ENU induced embryonic lethal mutations of the quaking gene. Genomics 57: 333–341. [DOI] [PubMed] [Google Scholar]
- Dai W, Zhang G, Makeyev EV. 2012. RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res 40: 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damianov A, Black DL. 2010. Autoregulation of Fox protein expression to produce dominant negative splicing factors. RNA 16: 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbelli L, Vogel G, Almazan G, Richard S. 2016. Quaking regulates neurofascin 155 expression for myelin and axoglial junction maintenance. J Neurosci 36: 4106–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin RG, Shiue L, Prins J, de Boer HC, Singh A, Fagg WS, van Gils JM, Duijs JM, Katzman S, Kraaijeveld AO, et al. 2016. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat Commun 7: 10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo Neto OP, Standart N, Martins de Sa C. 1995. Autoregulation of poly(A)-binding protein synthesis in vitro. Nucleic Acids Res 23: 2198–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Miguel FJ, Pajares MJ, Martinez-Terroba E, Ajona D, Morales X, Sharma RD, Pardo FJ, Rouzaut A, Rubio A, Montuenga LM, et al. 2016. A large-scale analysis of alternative splicing reveals a key role of QKI in lung cancer. Mol Oncol 10: 1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z, Kole R. 1991. Selection of splice sites in pre-mRNAs with short internal exons. Mol Cell Biol 11: 6075–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole TA, Chen Q, Justice MJ, Artzt K. 1996. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat Genet 12: 260–265. [DOI] [PubMed] [Google Scholar]
- Fu XD, Ares M Jr. 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15: 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau A, Richard S. 2005. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol 12: 691–698. [DOI] [PubMed] [Google Scholar]
- Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Ares M Jr, Mody I, Black DL. 2011. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet 43: 706–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Meera P, Stoilov P, Shiue L, O'Brien JE, Meisler MH, Ares M Jr, Otis TS, Black DL. 2012. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev 26: 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MP, Nagel RJ, Fagg WS, Shiue L, Cline MS, Perriman RJ, Donohue JP, Ares M Jr. 2013. Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA 19: 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada N, Ito H, Iwamoto I, Mizuno M, Morishita R, Inaguma Y, Kawamoto S, Tabata H, Nagata K. 2013. Biochemical and morphological characterization of A2BP1 in neuronal tissue. J Neurosci Res 91: 1303–1311. [DOI] [PubMed] [Google Scholar]
- Hardy RJ. 1998. QKI expression is regulated during neuron-glial cell fate decisions. J Neurosci Res 54: 46–57. [DOI] [PubMed] [Google Scholar]
- Hardy RJ, Loushin CL, Friedrich VL Jr, Chen Q, Ebersole TA, Lazzarini RA, Artzt K. 1996. Neural cell type-specific expression of QKI proteins is altered in quakingviable mutant mice. J Neurosci 16: 7941–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, Tajnik M, Konig J, Ule J. 2014. iCLIP: protein–RNA interactions at nucleotide resolution. Methods 65: 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie K, Tsujimura K, Nakashima H, Nakashima K. 2016. MicroRNA-214 promotes dendritic development by targeting the schizophrenia-associated gene quaking (Qki). J Biol Chem 291: 13891–13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA, Nelson DL, Moses K, Warren ST. 2004. Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci 7: 113–117. [DOI] [PubMed] [Google Scholar]
- Justice MJ, Hirschi KK. 2010. The role of quaking in mammalian embryonic development. Adv Exp Med Biol 693: 82–92. [DOI] [PubMed] [Google Scholar]
- Kawai T, Fan J, Mazan-Mamczarz K, Gorospe M. 2004. Global mRNA stabilization preferentially linked to translational repression during the endoplasmic reticulum stress response. Mol Cell Biol 24: 6773–6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD. 2007. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 8: 533–543. [DOI] [PubMed] [Google Scholar]
- Koizumi J, Okamoto Y, Onogi H, Mayeda A, Krainer AR, Hagiwara M. 1999. The subcellular localization of SF2/ASF is regulated by direct interaction with SR protein kinases (SRPKs). J Biol Chem 274: 11125–11131. [DOI] [PubMed] [Google Scholar]
- Kondo T, Furuta T, Mitsunaga K, Ebersole TA, Shichiri M, Wu J, Artzt K, Yamamura K, Abe K. 1999. Genomic organization and expression analysis of the mouse qkI locus. Mamm Genome 10: 662–669. [DOI] [PubMed] [Google Scholar]
- Langlois MA, Boniface C, Wang G, Alluin J, Salvaterra PM, Puymirat J, Rossi JJ, Lee NS. 2005. Cytoplasmic and nuclear retained DMPK mRNAs are targets for RNA interference in myotonic dystrophy cells. J Biol Chem 280: 16949–16954. [DOI] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. 2007. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446: 926–929. [DOI] [PubMed] [Google Scholar]
- Larocque D, Pilotte J, Chen T, Cloutier F, Massie B, Pedraza L, Couture R, Lasko P, Almazan G, Richard S. 2002. Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron 36: 815–829. [DOI] [PubMed] [Google Scholar]
- Larocque D, Galarneau A, Liu H-N, Scott M, Almazan G, Richard S. 2005. Protection of p27Kip1 mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat Neurosci 8: 27–33. [DOI] [PubMed] [Google Scholar]
- Lee JA, Damianov A, Lin CH, Fontes M, Parikshak NN, Anderson ES, Geschwind DH, Black DL, Martin KC. 2016. Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron 89: 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Tang ZZ, Black DL. 2009. An inducible change in Fox-1/A2BP1 splicing modulates the alternative splicing of downstream neuronal target exons. Genes Dev 23: 2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Li D, Feng Y. 2000. Destabilization and mislocalization of myelin basic protein mRNAs in quaking dysmyelination lacking the QKI RNA-binding proteins. J Neurosci 20: 4944–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Takakura N, Oike Y, Imanaka T, Araki K, Suda T, Kaname T, Kondo T, Abe K, Yamamura K. 2003. Defective smooth muscle development in qkI-deficient mice. Dev Growth Differ 45: 449–462. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Zhang Y, Ku L, Wang H, Ahmadian A, Feng Y. 2003. The quakingviable mutation affects qkI mRNA expression specifically in myelin-producing cells of the nervous system. Nucleic Acids Res 31: 4616–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, Maniatis T. 2007. The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 27: 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandler MD, Ku L, Feng Y. 2014. A cytoplasmic quaking I isoform regulates the hnRNP F/H-dependent alternative splicing pathway in myelinating glia. Nucleic Acids Res 42: 7319–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel-Rosen H, Dorevitch N, Reuveny A, Volk T. 1999. The balance between two isoforms of the Drosophila RNA-binding protein how controls tendon cell differentiation. Mol Cell 4: 573–584. [DOI] [PubMed] [Google Scholar]
- Nabel-Rosen H, Volohonsky G, Reuveny A, Zaidel-Bar R, Volk T. 2002. Two isoforms of the Drosophila RNA binding protein, how, act in opposing directions to regulate tendon cell differentiation. Dev Cell 2: 183–193. [DOI] [PubMed] [Google Scholar]
- Nakielny S, Dreyfuss G. 1999. Transport of proteins and RNAs in and out of the nucleus. Cell 99: 677–690. [DOI] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikov L, Park JW, Chen H, Klerman H, Jalloh AS, Gamble MJ. 2011. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol Cell Biol 31: 4244–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, Bhatt DM, Lin CH, Tong AJ, Smale ST, Black DL. 2013. Splicing kinetics and transcript release from the chromatin compartment limit the rate of Lipid A-induced gene expression. RNA 19: 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz I, Kosti I, Ares M Jr, Cline M, Mandel-Gutfreund Y. 2014. RBPmap: a Web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res 42: W361–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilotte J, Larocque D, Richard S. 2001. Nuclear translocation controlled by alternatively spliced isoforms inactivates the QUAKING apoptotic inducer. Genes Dev 15: 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomska KJ, Sager J, Farnsworth B, Tellgren-Roth A, Tuveri G, Peuckert C, Kettunen P, Jazin E, Emilsson LS. 2016. Characterization and expression of the zebrafish qki paralogs. PLoS One 11: e0146155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscigno RF, Garcia-Blanco MA. 1995. SR proteins escort the U4/U6.U5 tri-snRNP to the spliceosome. RNA 1: 692–706. [PMC free article] [PubMed] [Google Scholar]
- Ryder SP, Williamson JR. 2004. Specificity of the STAR/GSG domain protein Qk1: implications for the regulation of myelination. RNA 10: 1449–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccomanno L, Loushin C, Jan E, Punkay E, Artzt K, Goodwin EB. 1999. The STAR protein QKI-6 is a translational repressor. Proc Natl Acad Sci 96: 12605–12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, Gray NK, Beckmann K, Caceres JF. 2004. A novel role for shuttling SR proteins in mRNA translation. Genes Dev 18: 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebestyen E, Singh B, Minana B, Pages A, Mateo F, Pujana MA, Valcarcel J, Eyras E. 2016. Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res 26: 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Maris C, Allain FH, Black DL. 2011. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Mol Cell 41: 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlovsky A, King TR, Dove WF. 1988. Saturation germ line mutagenesis of the murine t region including a lethal allele at the quaking locus. Proc Natl Acad Sci 85: 180–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu P, Fu H, Zhao X, Wu C, Ruan X, Zeng Y, Liu W, Wang M, Hou L, Chen P, et al. 2017. MicroRNA-214 modulates neural progenitor cell differentiation by targeting Quaking during cerebral cortex development. Sci Rep 7: 8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidman R, Dickie M, Appel S. 1964. Mutant mice (quaking and jimpy) with deficient myelination in the central nervous system. Science (New York, NY) 144: 309–311. [DOI] [PubMed] [Google Scholar]
- Singh RK, Xia Z, Bland CS, Kalsotra A, Scavuzzo MA, Curk T, Ule J, Li W, Cooper TA. 2014. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol Cell 55: 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman R, Llorian M, Smith CWJ. 2007. Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol Cell 27: 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhang Z, Sinha R, Karni R, Krainer AR. 2010. SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat Struct Mol Biol 17: 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplova M, Hafner M, Teplov D, Essig K, Tuschl T, Patel DJ. 2013. Structure-function studies of STAR family Quaking proteins bound to their in vivo RNA target sites. Genes Dev 27: 928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL. 2005. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol Cell Biol 25: 10005–10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veer EP, de Bruin RG, Kraaijeveld AO, de Vries MR, Bot I, Pera T, Segers FM, Trompet S, van Gils JM, Roeten MK, et al. 2013. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ Res 113: 1065–1075. [DOI] [PubMed] [Google Scholar]
- van Mil A, Grundmann S, Goumans MJ, Lei Z, Oerlemans MI, Jaksani S, Doevendans PA, Sluijter JP. 2012. MicroRNA-214 inhibits angiogenesis by targeting Quaking and reducing angiogenic growth factor release. Cardiovasc Res 93: 655–665. [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. 2016. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13: 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S, Steitz JA. 2007. AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell 128: 1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. 2012. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150: 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Vogel G, Yu Z, Richard S. 2013. The QKI-5 and QKI-6 RNA binding proteins regulate the expression of microRNA 7 in glial cells. Mol Cell Biol 33: 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW. 2004. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell 13: 91–100. [DOI] [PubMed] [Google Scholar]
- Wu J, Bag J. 1998. Negative control of the poly(A)-binding protein mRNA translation is mediated by the adenine-rich region of its 5′-untranslated region. J Biol Chem 273: 34535–34542. [DOI] [PubMed] [Google Scholar]
- Wu JI, Reed RB, Grabowski PJ, Artzt K. 2002. Function of quaking in myelination: regulation of alternative splicing. Proc Natl Acad Sci 99: 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zhou L, Tonissen K, Tee R, Artzt K. 1999. The Quaking I-5 protein (QKI-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J Biol Chem 274: 29202–29210. [DOI] [PubMed] [Google Scholar]
- Yaffe D, Saxel O. 1977. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270: 725–727. [DOI] [PubMed] [Google Scholar]
- Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. 2009. An RNA code for the FOX2 splicing regulator revealed by mapping RNA–protein interactions in stem cells. Nat Struct Mol Biol 16: 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zearfoss NR, Clingman CC, Farley BM, McCoig LM, Ryder SP. 2011. Quaking regulates Hnrnpa1 expression through its 3′ UTR in oligodendrocyte precursor cells. PLoS Genet 7: e1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Mandler MD, Yi H, Feng Y. 2010. Quaking I controls a unique cytoplasmic pathway that regulates alternative splicing of myelin-associated glycoprotein. Proc Natl Acad Sci 107: 19061–19066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong FY, Fu X, Wei WJ, Luo YG, Heiner M, Cao LJ, Fang Z, Fang R, Lu D, Ji H, et al. 2014. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet 10: e1004289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn AM, Grow M, Patterson KD, Ebersole TA, Chen Q, Artzt K, Krieg PA. 1997. Remarkable sequence conservation of transcripts encoding amphibian and mammalian homologues of quaking, a KH domain RNA-binding protein. Gene 188: 199–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.