

The majority of insects carry maternally inherited intracellular bacteria that are important in their hosts’ biology, ecology, and evolution. Some of these bacterial symbionts cause a reproductive failure known as cytoplasmic incompatibility (CI). In CI, the mating of symbiont-infected males and uninfected females produces few or no daughters. The CI symbiont then spreads and can have a significant impact on the insect host population. Cardinium, a bacterial endosymbiont of the parasitoid wasp Encarsia in the Bacteroidetes, is the only bacterial lineage known to cause CI outside the Alphaproteobacteria, where Wolbachia and another recently discovered CI symbiont reside. Here, we sought insight into the gene expression of a CI-inducing Cardinium strain in its natural host, Encarsia suzannae. Our study provides the first insights into the Cardinium transcriptome and provides support for the hypothesis that Wolbachia and Cardinium target similar host pathways with distinct and largely unrelated sets of genes.
KEYWORDS: Bacteroidetes, Cardinium, cytoplasmic incompatibility, host-microbe interaction, RNA sequencing, endosymbionts, gene expression
ABSTRACT
Cytoplasmic incompatibility (CI) is an intriguing, widespread, symbiont-induced reproductive failure that decreases offspring production of arthropods through crossing incompatibility of infected males with uninfected females or with females infected with a distinct symbiont genotype. For years, the molecular mechanism of CI remained unknown. Recent genomic, proteomic, biochemical, and cell biological studies have contributed to understanding of CI in the alphaproteobacterium Wolbachia and implicate genes associated with the WO prophage. Besides a recently discovered additional lineage of alphaproteobacterial symbionts only moderately related to Wolbachia, Cardinium (Bacteroidetes) is the only other symbiont known to cause CI, and genomic evidence suggests that it has very little homology with Wolbachia and evolved this phenotype independently. Here, we present the first transcriptomic study of the CI Cardinium strain cEper1, in its natural host, Encarsia suzannae, to detect important CI candidates and genes involved in the insect-Cardinium symbiosis. Highly expressed transcripts included genes involved in manipulating ubiquitination, apoptosis, and host DNA. Female-biased genes encoding ribosomal proteins suggest an increase in general translational activity of Cardinium in female wasps. The results confirm previous genomic analyses that indicated that Wolbachia and Cardinium utilize different genes to induce CI, and transcriptome patterns further highlight expression of some common pathways that these bacteria use to interact with the host and potentially cause this enigmatic and fundamental manipulation of host reproduction.
IMPORTANCE The majority of insects carry maternally inherited intracellular bacteria that are important in their hosts’ biology, ecology, and evolution. Some of these bacterial symbionts cause a reproductive failure known as cytoplasmic incompatibility (CI). In CI, the mating of symbiont-infected males and uninfected females produces few or no daughters. The CI symbiont then spreads and can have a significant impact on the insect host population. Cardinium, a bacterial endosymbiont of the parasitoid wasp Encarsia in the Bacteroidetes, is the only bacterial lineage known to cause CI outside the Alphaproteobacteria, where Wolbachia and another recently discovered CI symbiont reside. Here, we sought insight into the gene expression of a CI-inducing Cardinium strain in its natural host, Encarsia suzannae. Our study provides the first insights into the Cardinium transcriptome and provides support for the hypothesis that Wolbachia and Cardinium target similar host pathways with distinct and largely unrelated sets of genes.
INTRODUCTION
Terrestrial arthropods are commonly associated with one or more intracellular, maternally transmitted bacterial symbionts that may profoundly influence their ecology and evolution (1, 2). Strictly maternally inherited symbionts spread in host populations by enhancing the daughter-producing capacity of female hosts relative to uninfected individuals (3). Obligate or “primary” symbionts are generally nutritional mutualists, thus increasing offspring production generally, while facultative or “secondary” symbionts may also benefit their hosts directly (2, 4) or manipulate host reproduction in ways that promote the production or fitness of infected females (5, 6). One of these symbiont-driven host manipulations is cytoplasmic incompatibility (CI). At its simplest, CI symbionts in the male host modify sperm such that only eggs with the same symbiont can “rescue” them, and the embryo develops normally. Conversely, mating between infected males and uninfected females generally results in embryo lethality. By depressing the relative fitness of uninfected females, the CI phenotype leads to an increase in the production of symbiont-infected females in the population (7, 8). CI is caused by three symbiont lineages: Cardinium hertigii, in the Bacteroidetes, and Wolbachia pipientis and a recently discovered clade of symbionts of a coconut beetle, in the Alphaproteobacteria (9). CI-causing Wolbachia is more prevalent among arthropods (~40% compared to ~9% for Cardinium [10, 11]) and has received considerable attention, including analyses of multiple sequenced genomes (12–19) and some elegant cytogenetic studies (20, 21). However, the molecular genetic basis of CI has been unresolved for some time and is just recently beginning to be understood. A recent study of Wolbachia strain wPip in mosquitoes suggests that two adjacent genes may be important: cidB (wPa_0283) and cidA (wPa_0282) (22). Similarly, two genes carried in the WO prophage eukaryotic association module from wMel (cifA [WD0631] and cifB [WD0632]) have also been shown to be able to recapitulate the CI phenotype (23). Homologs of these genes were highly expressed in the ovaries of the parasitoid wasp Nasonia vitripennis by wVitA, another CI Wolbachia strain (23). While a deubiquitylating domain in cidB was hypothesized to be important for CI in one study (22), this domain was not found to be conserved among CI strains in another (23).
Cardinium CI was much more recently discovered than Wolbachia CI (24), and its absence in model arthropod systems (particularly Drosophila and mosquitoes) as well as the minute size of the arthropod hosts in which CI Cardinium has been documented (parasitoid wasps and mites [e.g., references 24 and 25]) has made its study challenging. The first Cardinium CI genome showed only four homologous genes possibly involved in host-cell interaction with CI Wolbachia; these included a putative patatin-like phospholipase, an uncharacterized membrane protein, putative RNA helicase, and a cold shock protein. In spite of this, the cytological appearance of embryo death in CI Cardinium-affected hosts is broadly similar to that caused by CI Wolbachia (26). Further, the functional overlap of some protein families such as ankyrin repeat proteins between these two lineages leaves open the possibility of conserved host targets and functional convergence of the CI mechanism (26, 27).
Here, we sought insight into the highly expressed and sex-specific differentially expressed (DE) genes of the CI-inducing Cardinium strain cEper1 in its natural host, Encarsia suzannae (Hymenoptera: Aphelinidae), with a transcriptome sequencing (RNA-Seq) approach. The genome of cEper1 (27) provided candidate CI genes to evaluate for expression and the opportunity to highlight novel transcripts and hypothetical proteins that may need greater study. Additionally, the recently published genome of an apparently asymptomatic (28) strain of Cardinium in Bemisia tabaci (cBtQ1) (29) allowed us to compare and further posit functions for these genes. Our work represents the first Cardinium expression profile. In general, very few transcriptomes of arthropod endosymbionts have been sequenced until very recently (30–32). This may be because of the high level of technical difficulty of recovering enough RNA from uncultivable bacteria within eukaryotic hosts, combined with costs that have only recently become affordable. The potential value of this approach, however, is illustrated by a recent study in which the transcriptome of a defensive Spiroplasma showed a spiroplasma-encoded toxin, highly expressed only when its Drosophila host was parasitized by nematodes (31). Also, a thorough stage- and sex-specific transcriptomic analysis of Wolbachia closely related to the CI symbiont wMel in Drosophila melanogaster provided the first insights into sex-biased expression by this symbiont (30). Other Wolbachia transcriptome studies examined the role of the mutualist Wolbachia in the native host tissues of the filarial nematode system (32–34), documenting immune system avoidance and ATP biosynthesis (32).
RESULTS
The Cardinium hertigii cEper1 transcriptome—general features.
The Cardinium hertigii cEper1 genome contains 835 predicted protein coding sequences (CDSs), with 782 chromosomal and 53 plasmid (pCher) genes. In our strand-specific RNA sequencing experiment, 445 million reads were generated in total, with an average of 74,094,307 reads per sample. Approximately 1% of the reads mapped to the Cardinium hertigii cEper1 genome (Table 1). More than 60% of the reads mapped were mRNA reads. The mean theoretical redundancy of coverage was 24.7× ± 4.7× and 32.4× ± 7.4× for chromosomal and plasmid genes, respectively. Additionally, the single perfect match coverage was 94.5% for chromosomal and 89.6% for plasmid genes (median values over all replicates). Fifteen potential novel transcripts, of which seven were putative antisense RNAs of annotated genes, were identified (see Table S1 in the supplemental material). There was no differential expression of the novel transcripts in males and females, and the putative functions of these transcripts are unknown.
TABLE 1 .
Transcriptome sequencing read statistics, read processing, mapping, and coverage
| Data set statistic |
cEper1 in E. suzannae by sex and replicate |
|||||
|---|---|---|---|---|---|---|
| Female |
Male |
|||||
| 1st | 2nd | 3rd | 1st | 2nd | 3rd | |
| Total no. of reads | 80,080,835 | 73,121,816 | 66,679,042 | 63,836,426 | 81,372,753 | 79,474,970 |
| Trimmed reads, no. (%) | 77,353,052 (96.59) | 70,355,505 (96.22) | 64,410,445 (96.60) | 61,843,438 (96.88) | 78,712,070 (96.73) | 76,899,760 (96.76) |
| Trimmed reads mapped to C. hertigii cEper1 chromosome (genome size, 0.89 Mb), no. (%) |
725,968 (0.94) | 834,050 (1.19) | 693,560 (1.08) | 534,524 (0.86) | 728,725 (0.93) | 651,504 (0.85) |
| mRNA reads mapped to chromosomal genes, no. (%) |
434,696 (59.87) | 577,310 (69.21) | 432,106 (62.30) | 297,994 (55.75) | 474,008 (65.05) | 411,779 (63.20) |
| Reads assigned to genes involved in host-cell interactions,a no. (%) |
10,353 (2.38) | 13,943 (2.42) | 9,728 (2.25) | 8,406 (2.82) | 13,506 (2.85) | 12,816 (3.11) |
| Reads assigned to transporter genes, no. (%) |
23,327 (5.37) | 30,589 (5.30) | 20,910 (4.84) | 17,040 (5.72) | 25,305 (5.34) | 23,663 (5.75) |
| Reads assigned to transposases, no. (%) |
23,102 (5.31) | 31,299 (5.42) | 18,775 (4.34) | 18,508 (6.21) | 30,759 (6.49) | 29,227 (7.10) |
| Theoretical redundancy of coverage |
24.5× | 32.5× | 24.4× | 16.8× | 26.7× | 23.2× |
| Single perfect coverage (C. hertigii cEper1 chromosome), % |
94.32 | 95.87 | 86.32 | 92.48 | 95.39 | 94.65 |
| Trimmed reads mapped to C. hertigii pCher plasmid (genome size, 0.06 Mb), no. (%) |
52,626 (0.07) | 61,874 (0.09) | 31,325 (0.05) | 40, 882 (0.07) | 63,651 (0.08) | 64,554 (0.08) |
| mRNA reads mapped to plasmid genes, no. |
37,892 | 44,723 | 23,215 | 28,795 | 45,146 | 44,688 |
| Theoretical redundancy of coverage |
32.8× | 38.7× | 20.1× | 24.9× | 39.1× | 38.7× |
| Single perfect coverage (C. hertigii pCher plasmid), % |
88.69 | 91.38 | 73.19 | 86.51 | 91.15 | 90.49 |
Including ankyrin repeats, TPRs, and ubiquitin system-interacting genes.
Potential novel cEper1 transcripts and putative antisense RNAs, calculated with ReadXplorer. The parameter “number of read starts” counts the number of reads and filters low-coverage regions with a number of reads below a certain threshold. The threshold used to detect novel transcripts was set to a 10-read minimum. “Coverage increase” indicates the coverage increase in percent from one position to the neighboring one (for the detection of novel transcripts, the threshold was set to a minimum of 50% increase from the start of the transcript to the position before). “Transcript stop” indicates the position at the chromosome where the putative transcript ends. Download TABLE S1, PDF file, 0.03 MB (32.3KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Heat maps show a widely homogenous expression level of chromosomal genes among cEper1 bacteria in male and female replicates (Fig. 1). Among the most highly expressed genes for cEper1 across male and female replicates, many housekeeping genes (chaperones, genes involved in ribosomal machinery, and replication- or transcription-associated genes) were found (Table 2; Tables S2 to S4): for example, RNase P, 60-kDa chaperonin GroEL, and the DNA-directed RNA polymerase subunits beta and beta′. In female replicates, the DNA-directed RNA polymerase subunits, two elongation factors, one polyribonucleotide nucleotidyltransferase, and the signal recognition particle receptor FtsY were more highly expressed than in male replicates. In males, chaperone protein ClpB and one putative sodium-solute symporter appeared more highly expressed than in female replicates, but these differences were not statistically significant. The expression level of plasmid genes revealed some heterogeneity of expression that does not cleanly separate into male or female replicates, perhaps due to the absence of putative transcriptional regulators on the plasmids. In addition, several hypothetical proteins were found among the most highly expressed genes. Interestingly, we also found a high level of expression of transposases: 72 out of 129 transposases (55.8%) were expressed (Table S5), of which 18 were among the 100 most highly expressed genes. Although we cannot determine if these transposases are transpositionally active, the expressed transposases in cEper1 could contribute to genomic recombination, as the cEper1 and the cBtQ1 genomes show signs of substantial genomic rearrangements (29).
FIG 1 .

Gene expression of cEper1 chromosomal (A) and plasmid (B) genes in female (FR) and male (MR) wasp pool replicates. tRNAs and rRNAs were excluded for all analysis. Expression values are given as transformed log values of normalized counts per gene (red, low expression; green, high expression). FR, cEper1 genes in female wasp replicates; MR, cEper1 genes in male wasp replicates.
TABLE 2 .
The 20 most highly expressed cEper1 genes in female and male wasp replicatesd
| Current GenBank locus taga |
Locus tag from reference 27b |
Description | Mean cEper1 rank in sex: |
|
|---|---|---|---|---|
| Males | Females | |||
| AL022_RS03910 | —c | RNase P | 1 | 1 |
| AL022_RS03480 | CAHE_0757 | Hypothetical protein | 2 | 2 |
| AL022_RS01165 | CAHE_0254 | 60-kDa chaperonin GroEL | 3 | 3 |
| AL022_RS00235 | CAHE_0050 | Hypothetical protein | 4 | 4 |
| AL022_RS03100 | CAHE_0677 | Putative DEAD box ATP-dependent RNA helicase | 6 | 5 |
| AL022_RS00080 | CAHE_0016 | Chaperone protein DnaK | 5 | 6 |
| AL022_RS01560 | CAHE_0338 | DNA-directed RNA polymerase subunit beta′ | 18 | 7 |
| AL022_RS01520 | CAHE_0330 | Elongation factor Tu | 12 | 8 |
| AL022_RS03700 | CAHE_0796 | Putative sodium-solute symporter | 7 | 9 |
| AL022_RS01795 | CAHE_0390 | Hypothetical protein | 8 | 10 |
| AL022_RS02130 | CAHE_0458 | Putative phage tail sheath protein Afp4-like | 10 | 11 |
| AL022_RS01565 | CAHE_0339 | DNA-directed RNA polymerase subunit beta | 34 | 12 |
| AL022_RS00330 | CAHE_0069 | Elongation factor G | 21 | 13 |
| AL022_RS01865 | CAHE_0406 | Hypothetical protein | 19 | 14 |
| AL022_RS00520 | CAHE_0112 | Polyribonucleotide nucleotidyltransferase | 20 | 15 |
| AL022_RS02455 | CAHE_0536 | Chaperone protein ClpB | 9 | 16 |
| AL022_RS01915 | CAHE_0417 | Signal recognition particle receptor FtsY | 27 | 17 |
| AL022_RS00835 | CAHE_0182 | Putative chaperone protein Skp | 17 | 18 |
| AL022_RS01425 | CAHE_0315 | Chromosome partitioning protein ParA | 16 | 19 |
| AL022_RS02665 | CAHE_0586 | 30S ribosomal protein S1 | 32 | 20 |
| AL022_RS01815 | CAHE_0394 | Hypothetical protein | 13 | 21 |
| AL022_RS01610 | CAHE_0352 | Putative sodium-solute symporter | 11 | 22 |
| AL022_RS04180 | CAHE_p0065 | Putative transposase | 15 | 34 |
| AL022_RS04095 | CAHE_p0043 | Hypothetical protein | 14 | 37 |
GenBank accession numbers NC_018605.1 and NC_018606.1.
—, not annotated in the work of Penz et al. (27).
The ranking is based on mean normalized counts per gene of female and male wasp replicates.
Classification of expression level of cEper1 genes in female and male Encarsia wasps. Download TABLE S2, XLSX file, 0.1 MB (70.3KB, xlsx) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Thirty cEper1 genes that were the most highly transcribed in male wasps are listed and sorted by fold change from their expression in female wasps. Genes meeting the DE criteria (P < 0.05 and multiple testing correction of FDR of <10%) are highlighted in bold. Download TABLE S3, PDF file, 0.03 MB (34.1KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Thirty cEper1 genes that were the most highly transcribed in female wasps are listed and sorted by the fold change from their expression in male wasps. Genes meeting the DE criteria (P < 0.05 and multiple testing correction of FDR of <10%) are highlighted in bold. Download TABLE S4, PDF file, 0.03 MB (35.4KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Gene expression of transposases in the Cardinium hertigii cEper1 genome. Download TABLE S5, PDF file, 0.1 MB (66.9KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Expressed genes with eukaryotic domains: candidates for host-cell interaction and CI.
Many intracellular bacteria, including symbionts and pathogens, use proteins harboring eukaryotic domains such as ankyrin repeats or tetratricopeptide repeats (TPRs) to interfere with various host-cell functions (35, 36), including ubiquitination (37). Cardinium cEper1 expressed many genes with eukaryotic domains that are candidates for host-cell interaction and/or the CI phenotype (Table 2). Fourteen out of 19 ankyrin repeat proteins identified in the genome were transcribed, with expression levels ranging from low to very high in cEper1 (Table S6). Four moderately or highly expressed ankyrins on the cEper1 chromosome showed high amino acid identity to homologs in cBtQ1 (CAHE_0095, 93%; CAHE_0435, 66%; CAHE_0680, 88%; CAHE_0834, 80%). In contrast, three highly expressed ankyrins located on the pCher plasmid were absent or nonfunctional in cBtQ1. While CAHE_p0007 and CAHE_p0014 have no homologs in cBtQ1, CAHE_p0026 has two pseudogene homologs on the cBtQ1 plasmid (see below). All three tetratricopeptide repeat (TPR) proteins present in the cEper1 Cardinium genome (CAHE_0312, CAHE_0450, and CAHE_0452) were expressed moderately. Other transcripts with eukaryotic domains include CAHE_0028, a gene encoding a putative ubiquitin protease; CAHE_0010, with a WH2 motif and an N-terminal proline-rich domain commonly present in actin binding proteins; and CAHE_0286, a patatin-like phospholipase with high amino acid identity (64%) to homologs from WO prophages in Wolbachia. CAHE_0706 encodes a collagen-like protein that contains collagen triple helix repeats, and CAHE_0677 encodes a putative DEAD box ATP-dependent RNA helicase that was among the most highly expressed genes (Table 2). The DEAD box RNA helicase gene was predicted to be located within an operon together with the cold shock protein CAHE_0676, which is also among the most highly expressed genes (Table 2). CAHE_0676 and CAHE_0677 homologs are found in Amoebophilus and also in Wolbachia (27).
Transcription of Cardinium hertigii cEper1 genes encoding proteins possibly involved in host-cell interactions. Download TABLE S6, PDF file, 0.04 MB (46.5KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In addition to these previously identified candidate host-cell interaction genes, we also identified some novel effector candidates based on our RNA-Seq data. CAHE_0017 (moderately expressed) and CAHE_0267 (highly expressed) share 40% amino acid identity and are putative DNA-interacting proteins belonging to a previously described family of widely spread proteins harboring a MutS domain that affiliate with subfamily MutS8 (InterPro domains IPR007696, IPR027417, and IPR000432) (38, 39). Another highly expressed novel host-cell interaction candidate protein identified here is CAHE_0662, an integral membrane protein harboring an inhibitor of apoptosis-promoting Bax1 domain (InterPro domain IPR006214).
Sex-specific transcription of genes.
With the RNA-Seq approach, 15 differentially expressed (DE) genes were found, of which 12 were upregulated in cEper1 found within females and three in cEper1 in males (Fig. 2; Table 3). DE genes were moderately expressed, except for the DNA-directed RNA polymerase subunits and CAHE_p0026, which were among the most highly expressed genes overall. In females, the DE genes consisted largely of genes involved in transcription and translation: five DE genes encoding ribosomal proteins were upregulated, indicating an increased general translational activity. In males, three DE genes were upregulated, including the previously mentioned CAHE_p0026, a putative RING domain ubiquitin ligase (InterPro domain IPR001841) located on the plasmid pCher, which also contains ankyrin repeats and was identified as a putative CI candidate gene previously (27). A second gene with higher expression in males was CAHE_p0027, a hypothetical protein with a homolog only in cBtQ1 (58% amino acid identity). Although CAHE_p0026 and CAHE_p0027 are both located on the pCher plasmid and adjacent to each other, these two genes are transcribed in opposite directions and are thus not part of an operon. The third gene upregulated in males is CAHE_0544; while the function of this gene is unknown, it contains a putative P-loop containing a nucleoside triphosphate hydrolase domain (InterPro domain IPR027417), and its only homolog is a truncated pseudogene in cBtQ1.
FIG 2 .
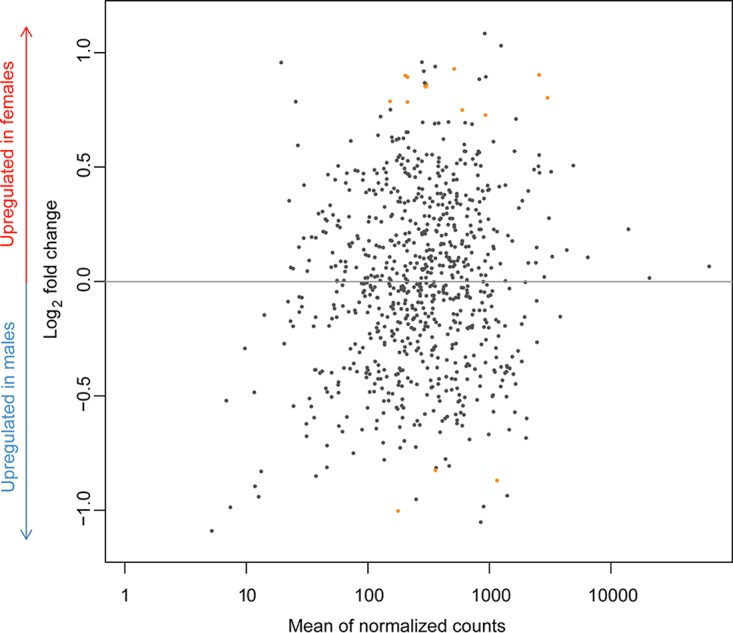
Normalized mean transcription values of cEper1 chromosomal and plasmid genes are plotted against log2 fold change values for each gene. Differentially expressed (DE) genes are highlighted in orange.
TABLE 3 .
List of cEper1 DE genes determined with RNA sequencing and DE calling in DESeqc
| Category and current GenBank locus taga | Locus tag from reference 27b | Description | Mean expression value for cEper1 in: |
Log2 fold change | P value | FDR value | |
|---|---|---|---|---|---|---|---|
| Males | Females | ||||||
| Genes upregulated in cEper1 in females | |||||||
| AL022_RS00605 | CAHE_0131 | 30S ribosomal protein S5 | 353.56 | 673.17 | 0.93 | <0.001 | 0.022 |
| AL022_RS01565 | CAHE_0339 | DNA-directed RNA polymerase subunit beta′ | 1,783.38 | 3,335.95 | 0.90 | <0.001 | 0.009 |
| AL022_RS02585 | CAHE_0565 | Transcription elongation factor GreA | 141.55 | 264.19 | 0.90 | 0.001 | 0.052 |
| AL022_RS00600 | CAHE_0130 | 50S ribosomal protein L30 | 147.84 | 274.74 | 0.89 | <0.001 | 0.052 |
| AL022_RS01110 | Aspartate-tRNA ligase | 214.87 | 390.65 | 0.86 | <0.001 | 0.040 | |
| AL022_RS01125 | CAHE_0242 | Dipeptide transport system permease protein OppC | 212.17 | 384.43 | 0.86 | <0.001 | 0.040 |
| AL022_RS02215 | CAHE_0475 | 30S ribosomal protein S9 | 213.37 | 385.64 | 0.85 | 0.001 | 0.052 |
| AL022_RS01560 | CAHE_0338 | DNA-directed RNA polymerase subunit beta—rpoB | 2,191.78 | 3,822.27 | 0.80 | <0.001 | 0.035 |
| AL022_RS00610 | CAHE_0132 | 50S ribosomal protein L18 | 111.66 | 192.77 | 0.79 | 0.001 | 0.079 |
| AL022_RS03105 | CAHE_0678 | Hypothetical protein | 155.67 | 268.09 | 0.78 | 0.001 | 0.079 |
| AL022_RS00470 | CAHE_0102 | Membrane protein insertase YidC | 446.65 | 750.46 | 0.75 | 0.001 | 0.052 |
| AL022_RS01545 | CAHE_0335 | 50S ribosomal protein L1 | 701.12 | 1,160.67 | 0.73 | 0.001 | 0.057 |
| Genes upregulated in cEper1 in males | |||||||
| AL022_RS02490 | CAHE_0544 | Hypothetical protein | 459.68 | 259.51 | −0.82 | 0.002 | 0.095 |
| AL022_RS04030 | CAHE_p0026 | RING domain-containing protein, ankyrin repeats | 1,491.23 | 816.33 | −0.87 | 0.001 | 0.062 |
| AL022_RS04035 | CAHE_p0027 | Hypothetical protein | 235.71 | 117.67 | −1.00 | 0.001 | 0.052 |
GenBank accession numbers NC_018605.1 and NC_018606.1.
Mean expression values are normalized counts per gene of female and male wasp replicates.
To confirm the accuracy of expression profiles obtained from the transcriptome sequencing experiment, independent validation of DE genes was performed with reverse transcriptase quantitative PCR (RT-qPCR). First, the same replicates used for RNA-Seq (except for one female replicate where no RNA was left after sequencing) were examined. A strong correlation between transcriptional differences (fold changes between male and female replicates) measured by RNA-Seq and RT-qPCR was found (regression P < 0.001; correlation coefficient r = 0.98; Fig. S1), providing strong evidence for the quantitative accuracy of the RNA data set. This analysis revealed the same trends in regulation of all 15 DE genes tested relative to the RNA-Seq experiment. Second, DE genes of two independent replicates of males and females were examined with RT-qPCR to test if these expression patterns are constant and reproducible across samples. Sex-enriched expression was confirmed for 8 out of 15 DE genes (Fig. 3). The three male-specific genes were not confirmed in the independent replicates by RT-qPCR, indicating a diversified expression pattern among samples.
FIG 3 .

RT-qPCR confirmation of cEper1 DE genes. RT-qPCR was done with three replicates per sex which were also used for the RNA-Seq experiment and with two independent replicates per sex. The expression level of DE genes was normalized to that of the two housekeeping genes gyrB and groEL. P values were listed for genes which were confirmed to be differentially expressed (significantly or by trend).
(A) Fold change differences of sex-specific expression patterns detected with RNA-Seq and RT-qPCRs are compared. (B) The correlation of fold change differences between the two methods used is shown. In panels A and B, only replicates that were used for both methods (three male and three female replicates) were included in the calculation. Download FIG S1, TIF file, 2.7 MB (2.7MB, tif) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Cardinium metabolism, transporters, and secretion system.
Cardinium has highly reduced metabolic capabilities and encodes only two complete biosynthetic pathways (27, 29): the B-vitamin biotin biosynthesis and the lipoate pathway, all genes of which were expressed. The expression level for most of these genes was moderate; only the biotin synthase BioB (CAHE_0559) was highly expressed. Perhaps to compensate for reduced metabolic capabilities, cEper1 encodes 62 transport proteins, all expressed. Four moderately expressed putative nucleotide transport proteins were found in Cardinium: CAHE_0018, CAHE_0158, CAHE_0160, and CAHE_0789, all of which belong to the ATP:ADP antiporter family. cEper1 also expresses a putative S-adenosylmethionine transporter (CAHE_0109, moderate expression), which shows 47% amino acid identity to the functionally characterized homolog from Amoebophilus asiaticus (40) and 93% amino acid identity to a homolog in cBtQ1 (29). The transport system Opp A-F (CAHE_0240 to _0242, _0244, and _0245) and the C4-dicarboxylate transporter DcuAB (CAHE_0645 and _0647) were moderately expressed, indicating a functional import system for oligopeptides, amino acids, and dicarboxylates. Cardinium cEper1 also encodes 12 putative sodium-solute symporters, all expressed. Two of them (CAHE_0796 and CAHE_0352) were among the most highly expressed genes overall. Last, we found transcripts of all 15 genes identified previously as a novel, putative phage-derived protein secretion system (27, 41, 42) (Table S7). Some of these antifeeding prophage (AFP) genes were among the highly expressed genes, including the putative phage tail sheath protein (CAHE_0458) and the AFP11-like phage baseplate protein (CAHE_0037).
Transcription of Cardinium hertigii cEper1 genes encoding proteins of the putative antifeeding prophage-like secretion system. Download TABLE S7, PDF file, 0.03 MB (36.7KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Candidate genes for host interaction and CI.
The Cardinium strain cEper1 expresses a number of genes with eukaryotic domains, signal peptides directing secretion from the bacterial cell, or other genes that are likely involved with host cells, some of them potentially CI candidates, some likely more generally involved in symbiosis. While genomic data show independent evolution of CI in Cardinium and Wolbachia (12–16, 27, 43), the evidence to date suggests convergence in the CI phenotype (26), suggesting that the molecular targets may be similar. Further, some of the patterns of gene expression suggest possible convergence of function between the two lineages and between Cardinium and other symbionts.
Candidates for manipulation of host ubiquitination system.
Recent research suggesting that CI Wolbachia may target the host ubiquitination system, a key regulatory process in eukaryotes (22, 44), makes the presence of highly expressed ubiquitin ligase and protease genes in Cardinium especially intriguing. Caution is warranted, however, since it is not clear that, in Wolbachia (wPip) CI, the rescue is achieved via restoration of ubiquitination, and the region of homology of one of the CI candidate genes across Wolbachia CI strains does not include the ubiquitin protease domain (23). Further, cEper1 does not harbor homologs of Wolbachia cidA and cidB but does express CAHE_p0026, a putative RING domain ubiquitin ligase with ankyrin repeats, located on the plasmid pCher. Many bacterial effector proteins interfering with the host ubiquitin system mimic host structures or motifs, and RING domain proteins have been shown to mimic RING-type E3 ubiquitin ligases (37, 45). The only significant homologs of CAHE_p0026 were found on the Cardinium cBtQ1 plasmid pcBtQ1 (37% and 48% amino acid identity), but both cBtQ1 genes are disrupted by a transposase and are therefore nonfunctional pseudogenes. Interestingly, the Cardinium strain cBtQ1 does not appear to cause CI or another known reproductive manipulation of its host and is considered asymptomatic (28, 29). In addition to the RING domain ubiquitin ligase, CAHE_0028, a gene encoding a putative ubiquitin protease, is also highly expressed. Ubiquitin proteases are present in a few symbiotic and pathogenic bacteria, e.g., in Arsenophonus nasoniae, a male-killer bacterium of Nasonia vitripennis, and in the pathogens Chlamydia trachomatis and Salmonella enterica serovar Typhimurium (37, 46, 47). The ubiquitin protease of Cardinium is conserved in cBtQ1 as well (27, 29) and could work in concert with the ligase in contributing to the CI phenotype or in host manipulation generally.
Candidates for manipulation of host DNA.
The phenotype of both dying CI Wolbachia and CI Cardinium-influenced embryos includes improper condensation of host chromosomes and disrupted cell cycle timing of mitotic divisions (20, 26). Several expressed cEper1 genes are likely to interact with host chromatin. CAHE_0677 encodes a putative DEAD box ATP-dependent RNA helicase and was among the most highly expressed genes. Eukaryotic DEAD box RNA helicases promote mitotic chromosome segregation together with the RNA interference pathway (48). Cardinium cBtQ1, Wolbachia, and the closest relative to Cardinium, the amoeba symbiont Amoebophilus asiaticus, also harbor a DEAD box ATP-dependent RNA helicase gene that is highly similar to cEper1 (98%, 54%, and 52% amino acid identity, respectively). The helicase gene was predicted to be located within an operon together with the cold shock protein CAHE_0676, which is also a highly expressed gene. In addition, two novel putative DNA-interacting candidates were identified from our RNA-Seq data: CAHE_0017 (moderately expressed) and CAHE_0267 (highly expressed) harbor a MutS domain. MutS homologs are predicted to be involved in DNA mismatch repair or recombination and to be critical for replication fidelity and genome stability in both prokaryotes and eukaryotes (38, 39). Defects in the mismatch repair system can lead to meiotic defects. Somewhat surprisingly for bacterial DNA-interacting proteins, both Cardinium proteins harbor a predicted signal peptide, indicating that they are secreted from the bacterial cell. Interestingly, Cardinium cBtQ1 and A. asiaticus have also two genes homologous (with 36% and 92% amino acid identity, respectively) to CAHE_0017 and CAHE_0267. The homologs in cBtQ1 also contain a predicted signal peptide, whereas the A. asiaticus homologs do not.
Other candidates for host manipulation.
Another novel host-cell interaction candidate protein identified here is CAHE_0662, an integral membrane protein harboring an inhibitor of the apoptosis-promoting Bax1 domain (InterPro domain IPR006214), also among the most highly expressed genes (Table 2). Eukaryotic homologs are also known as Golgi antiapoptotic protein (GAAP) or as transmembrane Bax inhibitor motif (TMBIM) family proteins. These homologs localize predominantly to the Golgi apparatus and have been shown to inhibit apoptosis by releasing Ca2+ stored in the endoplasmic reticulum (ER) and the Golgi apparatus (49). CAHE_0662 has homologs in Cardinium cBtQ1 (85% amino acid identity), and more distant homologs (with approximately 40% amino acid identity) can be found in Orientia, Rickettsia, and plant-associated Alphaproteobacteria (49). More generally, nonhomologous putative apoptosis-inhibiting genes have recently been found in the WO prophage eukaryotic association modules in Wolbachia (50), and apoptosis inhibition by Wolbachia has also been implicated in preserving normal ovarian development in a parasitoid wasp system in which the host is dependent on the symbiont (51). The Cardinium GAAP-like protein shows 28% amino acid identity to the human homologs and harbors a number of amino acid residues which have been shown to be essential for function of GAAPs (49, 52). We thus hypothesize that CAHE_0662 is a protein involved in host-cell interaction, perhaps by inhibiting apoptosis of infected host cells.
Two other host-cell interaction candidate genes identified in the cEper1 genome were moderately expressed. CAHE_0010 contains a WH2 motif and a proline-rich domain at the N terminus, often part of actin binding proteins. A homolog of CAHE_0010 was found in cBtQ1 (46% amino acid identity). CAHE_0286, a patatin-like phospholipase, has high amino acid identity (64%) to homologs from WO prophages in Wolbachia. WO prophages appear to play an important role in reproductive manipulation in Wolbachia, as highly sex-specific expression patterns have been detected for WO prophage genes (43, 53, 54). Several proteins associated with WO prophage regions are reproductive manipulation candidates, and there is a concentration of genes with eukaryotic domains in the eukaryotic association module of the WO phage (50). More recently, the involvement of cifA and cifB, two genes in the eukaryotic association module of WO prophage, in CI has been described by LePage and coworkers (23), providing strong evidence for an involvement of WO prophages in Wolbachia CI. Similarly to the recently described cidA and cidB, cEper1 does not harbor homologs of cifA and cifB.
The CI Cardinium transcriptome also highlighted which of hundreds of hypothetical proteins could be important in metabolism or host manipulation. Another putative host-cell interaction protein might be CAHE_0757, which is the second most highly expressed gene in both female and male cEper1. CAHE_0757 has only one hit in GenBank, in cBtQ1; however, this homolog shows only 28% amino acid identity to CAHE_0757. Interestingly, CAHE_0757 is predicted to contain a signal peptide as well.
Ankyrin repeat domain and tetratricopeptide repeat domain proteins.
Ankyrin proteins are significantly enriched in many intracellular host-associated bacteria compared to free-living bacteria and are likely part of an effective interaction system between bacterial and host proteins. Interestingly, CI-inducing Cardinium hertigii, A. asiaticus, and Wolbachia strains are particularly enriched in ankyrin proteins, while they are absent or rare in other Bacteroidetes and mutualistic Wolbachia strains (12, 13, 27, 55, 56). Ankyrin proteins mediate protein-protein interactions in eukaryotes (57), and some intracellular bacteria secrete ankyrin proteins to manipulate host-cell functions (58–62). Ankyrin repeat proteins have long been considered potential CI effectors in Wolbachia spp., and some studies showed sex-specific gene expression of ANK genes (30, 43, 53, 63–65), but their direct role in Wolbachia CI is unclear (65, 66), and targeted studies have generally failed to confirm a direct role of these abundant proteins in Wolbachia CI (65, 66). The ankyrin repeat proteins found in both Wolbachia and Cardinium genomes share no similarity except for the shared ankyrin repeat motif.
Three tetratricopeptide repeat (TPR) genes were moderately expressed (CAHE_0312, CAHE_0450, and CAHE_0452) and may also be important in host manipulation. TPRs often have central roles in vital cell processes in eukaryotes, and they may be directly related to the virulence of bacterial pathogens (35). Proteins containing TPRs can regulate defined cell cycle transitions, for example, the anaphase-promoting complex in eukaryotes (67), and were also found in high numbers in A. asiaticus and in Chlamydiae, Orientia, and nematode Wolbachia genomes (55, 56, 68, 69). Like the ankyrins, the TPR Cardinium genes expressed show no homology to Wolbachia other than the TPR domain.
Cardinium metabolism, transporters, and secretion system.
Like many other intracellular symbionts, the cEper1 Cardinium showed a highly reduced metabolic capability and dependence on a large assortment of transporters. Complete pathways for lipoate and biotin biosynthesis were expressed at moderate levels. Lipoate is a highly conserved sulfur-containing cofactor that is essential for the function of key enzymatic processes. The acquisition and use of lipoate are also associated with bacterial virulence and pathogenesis (70). The expression of a complete biotin biosynthesis pathway is more surprising. The pathway is incomplete in the whitefly Cardinium strain cBtQ1, suggesting that it is not necessary for symbiont metabolism. It also seems unlikely to be required by the host. Biotin is typically ingested by insects (71), and although it may be supplied by symbionts to blood feeders whose diet customarily lacks B vitamins (72), parasitoids like Encarsia wasps that consume whole insects are unlikely to have dietary imbalances. Further, Encarsia suzannae insects cured of their Cardinium symbionts are able to survive and reproduce normally (e.g., references 73 and 74. The role of this vitamin in the wasp-Cardinium interaction is therefore unclear.
We found moderate to high transcription of all 15 genes in the unusual putative phage-derived protein secretion system identified in the Cardinium genome (27, 41). The secretion system is related to the antifeeding prophage (AFP) from Serratia entomophila and to other phage-derived secretion systems (42, 75, 76). While the whitefly Cardinium strain cBtQ1 (29) has a putative type I secretion system, this is absent from cEper1, and no other known protein secretion system was documented (27). In the current study, we found that some AFP genes were among the most highly expressed genes, suggesting a substantial functional role of the AFP apparatus in communication with the host cell. While CI candidate genes in Wolbachia might be secreted by a type IV secretion system, in Cardinium, CI candidate genes may be translocated into the host by the AFP-like protein secretion system. Recent studies have shown that AFP-like genes are not phylum specific but widespread among various bacterial and archaeal lineages (76) and that an AFP homolog of the symbiont Pseudoalteromonas luteoviolacea is responsible for induction of metamorphosis of the tubeworm Hydroides elegans (75). A recent study showed that the Amoebophilus asiaticus AFP-like gene cluster represents a functional contractile secretion system (42); this novel secretion system may contribute structurally to the regular array of tubes visible in electron micrographs of Cardinium (42, 77–79).
Conclusions.
Here, we provide the first insight into gene expression of the CI-causing Cardinium strain cEper1 in its natural host. This bacterium shows very little homology to CI Wolbachia, but Cardinium expression patterns suggest that the two symbionts may target at least some of the same host pathways. In Cardinium cEper1, this includes a highly expressed RING domain ubiquitin ligase potentially targeting the same host pathway as genes that have been implicated in ubiquitin manipulation in CI Wolbachia (22). Other highly expressed Cardinium cEper1 candidates that show functional similarity to Wolbachia genes include a DEAD box ATP-dependent helicase, an apoptosis-inhibiting gene (50), and ankyrin repeat domain protein genes (43, 63). To analyze the role of CI candidates in more detail in future experiments, it would be interesting to look for Cardinium proteins associated with infected male E. suzannae sperm (e.g., reference 44). To better understand the Cardinium-host interaction more generally, it would also be valuable to express Cardinium CI candidate proteins in a heterologous host and then use the recombinant proteins as “bait” to identify interacting host proteins. In addition, comparing the host gene expression data from infected and uninfected hosts, especially perhaps in male and female ovaries and testes, may also provide valuable complementary insights into the molecular basis of CI in this symbiotic system.
MATERIALS AND METHODS
Encarsia suzannae cultures.
Encarsia suzannae (previously known as Encarsia pergandiella [73]) is a parasitoid wasp (Hymenoptera: Aphelinidae) infected with the CI symbiont Cardinium hertigii cEper1. Wasps were collected from their host whiteflies (Bemisia tabaci) in Weslaco, TX, in 2006 (80, 81). The wasps were cultured on whiteflies that were not infected with Rickettsia, on cowpea plants (Vigna unguiculata). Males of E. suzannae develop as hyperparasitoids and were cultivated by providing virgin adult females with late-instar larvae or early-stage pupae of the primary parasitoid Eretmocerus eremicus. Since female E. suzannae insects are primary parasitoids, whitefly nymphs were provided to mated, adult females for female wasp production. Therefore, males and females of E. suzannae were cultivated separately in 50-cm3 cages (27°C, ambient relative humidity).
Male and female wasps were cultivated in four cages per sex. All leaves bearing wasp pupae in one cage were placed in an emergence jar (81) and resulted in 350 to 500 1- to 3-day-old wasps of one sex. Since each wasp weighs approximately 18.68 μg (data not shown), the starting weight of each sample was approximately 6.54 to 9.34 mg, roughly equivalent to the weight of four to six Drosophila melanogaster females (82). At day 3, all wasps in each jar were collected into one tube and subsequently shock frozen at −80°C for 2 min.
RNA extraction, HiSeq 2500 sequencing, and sequencing data analysis.
Total RNA was extracted from all eight single-sex pools. RNA was isolated immediately after wasps were shock frozen. For RNA isolation, the Trizol reagent (Invitrogen) was used, and contaminating genomic DNA was digested with the Turbo DNA-free kit (Ambion) according to the manufacturer’s instructions. After DNase treatment, RNA was dissolved in 15 µl double-distilled water with diethyl pyrocarbonate (ddH2ODEPC), and the complete digestion of DNA was confirmed by PCR with a 16S rRNA gene targeting general bacterial primer panel 27F-1492R (83). The integrity of the purified RNA was verified with an Agilent 2100 bioanalyzer (Agilent Technologies). RNA was stored at −80°C until use. Samples were subjected to standard Illumina library preparation using the NEBNext Ultra RNA library prep kit according to the manufacturer’s instructions. An RNA-Seq test run with different rRNA removal kits revealed that the best cEper1 transcriptome coverage was achieved with the Ribo-Zero Magnetic Gold (epidemiology) kit (Epicentre Biotechnologies; data not shown), so this kit was used. Six double-stranded cDNA libraries, three for each sex, were single end sequenced (50 bp) using an Illumina HiSeq2500 machine at the Vienna Biocenter Core Facilities (VBCF) NGS unit (http://www.vbcf.ac.at). Sequences were quality filtered with mothur (84) using trimming parameters as follows: number of ambiguous bases allowed = 0, minimum length of reads = 30 bp, minimum average quality score allowed over a window of 10 bp = 25, maximum length of homopolymers = 8 bp. Quality-filtered reads were mapped to the Cardinium hertigii chromosome and plasmid, NCBI RefSeq NC_018605.1 and NC_018606.1 (27), respectively, in the Burrows-Wheeler aligner (85). Most genome annotations are based on our original automatic genome annotation of the cEper1 genome using MicrScope/MaGe, which was then verified by manual searches of proteins against Swiss-Prot and UniProt, as well as searches against PFAM and SMART (see reference 27 for details). Read counts per predicted gene were calculated by ReadXplorer (86) and imported in DESeq Bioconductor using the R software environment (87). Gene counts were normalized to size factors of libraries and dispersion estimation. Normalized gene expression values are listed as normalized read counts per gene. An average normalized read count of >1,000 (top 10% of all genes) was considered highly expressed, and a read count of <60 (last 10% of all genes) was considered to show a low expression level. All genes with read counts between 60 and 1,000 were classified as moderately expressed. Differentially expressed (DE) genes between cEper1 in the male and female pool replicates were determined by DESeq using a binomial distribution model (88). Genes were considered DE if P was <0.05 and if multiple testing correction of false discovery rate (FDR) was <10% (89). Putative novel transcripts were detected with the “Transcription Start Site Detection” option in ReadXplorer, considering the number of read starts at the position and the minimal coverage increase from one position to the next (86).
Confirmation of DE genes with RT-qPCR.
For the RT-qPCRs, the first RNA from the same three replicates was used as for the RNA sequencing experiment, except for one female replicate where no RNA was left after sequencing. This replicate was replaced by the fourth biological replicate of RNA extracted from females. Second, four additional independent replicates, which were not used for RNA-Seq, were produced (two from females and two from males) to test if the gene expression patterns were reproducible among samples.
Transcription into cDNA was made with random hexamer primers (RevertAid H Minus First Strand cDNA synthesis kit; Thermo Scientific) and 5 µl RNA. RNA secondary structures were broken up at 65°C for 5 min, and cDNA synthesis was done at 45°C for 60 min after a preincubation at 25°C for 5 min. The reaction was terminated by heating at 70°C for 5 min, and cDNA was stored at −20°C.
Primers targeting DE genes and two housekeeping genes (gyrB and groEL) were designed using Primer 3 (version 0.4.0) (90) and Primer-BLAST (91). In silico specificity screens were done with BLAST. Annealing temperatures were optimized with genomic DNA isolated from E. suzannae infected with cEper1 (Table S8).
Primer sequences, amplicon length, and annealing temperature of primers targeting cEper1 DE genes and two housekeeping genes (gyrB and groEL). Annealing temperatures were optimized with genomic DNA isolated from a culture of E. suzannae infected with Cardinium hertigii cEper1. Download TABLE S8, PDF file, 0.03 MB (32.1KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RT-qPCR was performed according to the MIQE guidelines (92), listed in Table S9. Each qPCR mixture was pipetted in duplicate with Brilliant III SYBR Green qPCR low-ROX master mix, according to the manufacturer’s instructions (Agilent Technologies). All primers were used at a final concentration of 250 nM. All reactions were performed with an initial denaturation step at 95°C (3 min), followed by 40 cycles of 95°C for 5 s and annealing for 20 s with a fluorescence measurement at the last step of each cycle. A melting curve, ranging from 70°C to 90°C, with fluorescence measurements at 1°C intervals, was done after all real-time PCRs, to determine the specificity of the reaction. qPCRs were performed using a Stratagene Mx3000P real-time PCR system (Agilent Technologies).
MIQE guidelines for RT-qPCR assays. Download TABLE S9, PDF file, 0.1 MB (63.2KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
For inhibition testing and to evaluate the efficiencies of the DE and housekeeping gene PCR assays, standard curves were pipetted with purified PCR products of cDNA samples, which were adjusted to 1 ng/µl with a Qubit 2.0 fluorometer (Life Technologies). Negative-control and reverse transcriptase (RT)-minus controls (reverse transcription reaction without addition of reverse transcriptase) were used.
For the mRNA quantitation, 0.1 ng cDNA was used as the template in each qPCR. Data were analyzed using Mx300P MxPro software (Stratagene), and relative quantitation was performed with the comparative threshold cycle (CT) method. Values were normalized using two housekeeping genes (gyrB and groEL). Significant differences in bacterial gene expression between male and female cEper1-positive pool replicates were determined with the Welch two-sample t test in R (version 3.2.0) with genes being considered DE if P was <0.05.
Sequence analysis of highly expressed and differentially expressed genes.
Highly expressed and differentially expressed genes were analyzed for the presence of functional domains using InterPro (93) and PFAM (94). The presence of transmembrane helices and signal peptides was checked with the TMHMM server 2.0 (95) and SignalP 4.0 (96), respectively.
Accession number(s).
cEper1 sequencing data were deposited at the NCBI Sequence Read Archive under accession no. PRJEB13864.
ACKNOWLEDGMENTS
We acknowledge Sarah and Seth Bordenstein and John. F. Beckmann for sharing unpublished data.
This study was funded by a joint international project to S.S.-E. and M.S.H. between the NSF (IOS-1256905) and the Austrian Science Fund FWF (I 1251-B25).
REFERENCES
- 1.Duron O, Bouchon D, Boutin S, Bellamy L, Zhou LQ, Engelstadter J, Hurst GD. 2008. The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol 6:27. doi: 10.1186/1741-7007-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moran NA, McCutcheon JP, Nakabachi A. 2008. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- 3.Bull J. 1983. Evolution of sex determining mechanisms. Benjamin/Cummings Publishing, Menlo Park, CA. [Google Scholar]
- 4.Oliver KM, Degnan PH, Hunter MS, Moran NA. 2009. Bacteriophages encode factors required for protection in a symbiotic mutualism. Science 325:992–994. doi: 10.1126/science.1174463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Neill SL, Hoffmann AA, Werren JH (ed). 1997. Influential passengers—inherited microorganisms and arthropod reproduction. Oxford University Press, New York, NY. [Google Scholar]
- 6.Zchori-Fein E, Bourtzis K (ed). 2011. Manipulative tenants—bacteria associated with arthropods. CRC Press, Boca Raton, FL. [Google Scholar]
- 7.Werren JH, Baldo L, Clark ME. 2008. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol 6:741–751. doi: 10.1038/nrmicro1969. [DOI] [PubMed] [Google Scholar]
- 8.Poinsot D, Charlat S, Merçot H. 2003. On the mechanism of Wolbachia-induced cytoplasmic incompatibility: confronting the models with the facts. Bioessays 25:259–265. doi: 10.1002/bies.10234. [DOI] [PubMed] [Google Scholar]
- 9.Takano SI, Tuda M, Takasu K, Furuya N, Imamura Y, Kim S, Tashiro K, Iiyama K, Tavares M, Amaral AC. 2017. Unique clade of alphaproteobacterial endosymbionts induces complete cytoplasmic incompatibility in the coconut beetle. Proc Natl Acad Sci U S A 114:6110–6115. doi: 10.1073/pnas.1618094114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zug R, Hammerstein P. 2012. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One 7:e38544. doi: 10.1371/journal.pone.0038544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Russell JA, Funaro CF, Giraldo YM, Goldman-Huertas B, Suh D, Kronauer DJ, Moreau CS, Pierce NE. 2012. A veritable menagerie of heritable bacteria from ants, butterflies, and beyond: broad molecular surveys and a systematic review. PLoS One 7:e51027. doi: 10.1371/journal.pone.0051027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klasson L, Walker T, Sebaihia M, Sanders MJ, Quail MA, Lord A, Sanders S, Earl J, O’Neill SL, Thomson N, Sinkins SP, Parkhill J. 2008. Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol Biol Evol 25:1877–1887. doi: 10.1093/molbev/msn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klasson L, Westberg J, Sapountzis P, Näslund K, Lutnaes Y, Darby AC, Veneti Z, Chen LM, Braig HR, Garrett R, Bourtzis K, Andersson SGE. 2009. The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc Natl Acad Sci U S A 106:5725–5730. doi: 10.1073/pnas.0810753106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newton ILG, Clark ME, Kent BN, Bordenstein SR, Qu JX, Richards S, Kelkar YD, Werren JH. 2016. Comparative genomics of two closely related Wolbachia with different reproductive effects on hosts. Genome Biol Evol 8:1526–1542. doi: 10.1093/gbe/evw096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sutton ER, Harris SR, Parkhill J, Sinkins SP. 2014. Comparative genome analysis of Wolbachia strain wAu. BMC Genomics 15:928. doi: 10.1186/1471-2164-15-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu M, Sun LV, Vamathevan J, Riegler M, Deboy R, Brownlie JC, McGraw EA, Martin W, Esser C, Ahmadinejad N, Wiegand C, Madupu R, Beanan MJ, Brinkac LM, Daugherty SC, Durkin AS, Kolonay JF, Nelson WC, Mohamoud Y, Lee P, Berry K, Young MB, Utterback T, Weidman J, Nierman WC, Paulsen IT, Nelson KE, Tettelin H, O’Neill SL, Eisen JA. 2004. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol 2:E69. doi: 10.1371/journal.pbio.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellegaard KM, Klasson L, Näslund K, Bourtzis K, Andersson SG. 2013. Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genet 9:e1003381. doi: 10.1371/journal.pgen.1003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metcalf JA, Jo M, Bordenstein SR, Jaenike J, Bordenstein SR. 2014. Recent genome reduction of Wolbachia in Drosophila recens targets phage WO and narrows candidates for reproductive parasitism. PeerJ 2:e529. doi: 10.7717/peerj.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinto SB, Stainton K, Harris S, Kambris Z, Sutton ER, Bonsall MB, Parkhill J, Sinkins SP. 2013. Transcriptional regulation of Culex pipiens mosquitoes by Wolbachia influences cytoplasmic incompatibility. PLoS Pathog 9:e1003647. doi: 10.1371/journal.ppat.1003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serbus LR, Casper-Lindley C, Landmann F, Sullivan W. 2008. The genetics and cell biology of Wolbachia-host interactions. Annu Rev Genet 42:683–707. doi: 10.1146/annurev.genet.41.110306.130354. [DOI] [PubMed] [Google Scholar]
- 21.Tram U, Sullivan W. 2002. Role of delayed nuclear envelope breakdown and mitosis in Wolbachia-induced cytoplasmic incompatibility. Science 296:1124–1126. doi: 10.1126/science.1070536. [DOI] [PubMed] [Google Scholar]
- 22.Beckmann JF, Ronau JA, Hochstrasser M. 2017. A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility. Nat Microbiol 2:17007. doi: 10.1038/nmicrobiol.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LePage DP, Metcalf JA, Bordenstein SR, On JM, Perlmutter JI, Shropshire JD, Layton EM, Funkhouser-Jones LJ, Beckmann JF, Bordenstein SR. 2017. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 543:243–247. doi: 10.1038/nature21391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter MS, Perlman SJ, Kelly SE. 2003. A bacterial symbiont in the Bacteroidetes induces cytoplasmic incompatibility in the parasitoid wasp Encarsia pergandiella. Proc Biol Sci 270:2185–2190. doi: 10.1098/rspb.2003.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gotoh T, Noda H, Ito S. 2007. Cardinium symbionts cause cytoplasmic incompatibility in spider mites. Heredity 98:13–20. doi: 10.1038/sj.hdy.6800881. [DOI] [PubMed] [Google Scholar]
- 26.Gebiola M, Giorgini M, Kelly SE, Doremus MR, Ferree PM, Hunter MS. 2017. Cytological analysis of cytoplasmic incompatibility induced by Cardinium suggests convergent evolution with its distant cousin Wolbachia. Proc Biol Sci 284:20171433. doi: 10.1098/rspb.2017.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penz T, Schmitz-Esser S, Kelly SE, Cass BN, Müller A, Woyke T, Malfatti SA, Hunter MS, Horn M. 2012. Comparative genomics suggests an independent origin of cytoplasmic incompatibility in Cardinium hertigii. PLoS Genet 8:e1003012. doi: 10.1371/journal.pgen.1003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang YW, Liu LY, Zhang HL, Jiang DF, Chu D. 2014. Competitive ability and fitness differences between two introduced populations of the invasive whitefly Bemisia tabaci Q in China. PLoS One 9:e100423. doi: 10.1371/journal.pone.0100423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santos-Garcia D, Rollat-Farnier PA, Beitia F, Zchori-Fein E, Vavre F, Mouton L, Moya A, Latorre A, Silva FJ. 2014. The genome of Cardinium cBtQ1 provides insights into genome reduction, symbiont motility, and its settlement in Bemisia tabaci. Genome Biol Evol 6:1013–1030. doi: 10.1093/gbe/evu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutzwiller F, Carmo CR, Miller DE, Rice DW, Newton ILG, Hawley RS, Teixeira L, Bergman CM. 2015. Dynamics of Wolbachia pipientis gene expression across the Drosophila melanogaster life cycle. G3 5:2843–2856. doi: 10.1534/g3.115.021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamilton PT, Leong JS, Koop BF, Perlman SJ. 2014. Transcriptional responses in a Drosophila defensive symbiosis. Mol Ecol 23:1558–1570. doi: 10.1111/mec.12603. [DOI] [PubMed] [Google Scholar]
- 32.Darby AC, Armstrong SD, Bah GS, Kaur G, Hughes MA, Kay SM, Koldkjær P, Rainbow L, Radford AD, Blaxter ML, Tanya VN, Trees AJ, Cordaux R, Wastling JM, Makepeace BL. 2012. Analysis of gene expression from the Wolbachia genome of a filarial nematode supports both metabolic and defensive roles within the symbiosis. Genome Res 22:2467–2477. doi: 10.1101/gr.138420.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao RU, Huang Y, Abubucker S, Heinz M, Crosby SD, Mitreva M, Weil GJ. 2012. Effects of doxycycline on gene expression in Wolbachia and Brugia malayi adult female worms in vivo. J Biomed Sci 19:21. doi: 10.1186/1423-0127-19-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luck AN, Evans CC, Riggs MD, Foster JM, Moorhead AR, Slatko BE, Michalski ML. 2014. Concurrent transcriptional profiling of Dirofilaria immitis and its Wolbachia endosymbiont throughout the nematode life cycle reveals coordinated gene expression. BMC Genomics 15:1041. doi: 10.1186/1471-2164-15-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cerveny L, Straskova A, Dankova V, Hartlova A, Ceckova M, Staud F, Stulik J. 2013. Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect Immun 81:629–635. doi: 10.1128/IAI.01035-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jernigan KK, Bordenstein SR. 2014. Ankyrin domains across the tree of life. PeerJ 2:e264. doi: 10.7717/peerj.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Y, Zhu YQ. 2015. Diversity of bacterial manipulation of the host ubiquitin pathways. Cell Microbiol 17:26–34. doi: 10.1111/cmi.12384. [DOI] [PubMed] [Google Scholar]
- 38.Ogata H, Ray J, Toyoda K, Sandaa RA, Nagasaki K, Bratbak G, Claverie JM. 2011. Two new subfamilies of DNA mismatch repair proteins (MutS) specifically abundant in the marine environment. ISME J 5:1143–1151. doi: 10.1038/ismej.2010.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Z, Nei M, Ma H. 2007. The origins and early evolution of DNA mismatch repair genes—multiple horizontal gene transfers and co-evolution. Nucleic Acids Res 35:7591–7603. doi: 10.1093/nar/gkm921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haferkamp I, Penz T, Geier M, Ast M, Mushak T, Horn M, Schmitz-Esser S. 2013. The endosymbiont Amoebophilus asiaticus encodes an S-adenosylmethionine carrier that compensates for its missing methylation cycle. J Bacteriol 195:3183–3192. doi: 10.1128/JB.00195-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Penz T, Horn M, Schmitz-Esser S. 2010. The genome of the amoeba symbiont “Candidatus Amoebophilus asiaticus” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. Virulence 1:541–545. doi: 10.4161/viru.1.6.13800. [DOI] [PubMed] [Google Scholar]
- 42.Böck D, Medeiros JM, Tsao HF, Penz T, Weiss GL, Aistleitner K, Horn M, Pilhofer M. 2017. In situ architecture, function, and evolution of a contractile injection system. Science 357:713–717. doi: 10.1126/science.aan7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker T, Klasson L, Sebaihia M, Sanders MJ, Thomson NR, Parkhill J, Sinkins SP. 2007. Ankyrin repeat domain-encoding genes in the wPip strain of Wolbachia from the Culex pipiens group. BMC Biol 5:39. doi: 10.1186/1741-7007-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beckmann JF, Fallon AM. 2013. Detection of the Wolbachia protein WPIP0282 in mosquito spermathecae: implications for cytoplasmic incompatibility. Insect Biochem Mol Biol 43:867–878. doi: 10.1016/j.ibmb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perrett CA, Lin DY, Zhou D. 2011. Interactions of bacterial proteins with host eukaryotic ubiquitin pathways. Front Microbiol 2:143. doi: 10.3389/fmicb.2011.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rytkönen A, Holden DW. 2007. Bacterial interference of ubiquitination and deubiquitination. Cell Host Microbe 1:13–22. doi: 10.1016/j.chom.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilkes TE, Darby AC, Choi JH, Colbourne JK, Werren JH, Hurst GD. 2010. The draft genome sequence of Arsenophonus nasoniae, son-killer bacterium of Nasonia vitripennis, reveals genes associated with virulence and symbiosis. Insect Mol Biol 19(Suppl 1):59–73. doi: 10.1111/j.1365-2583.2009.00963.x. [DOI] [PubMed] [Google Scholar]
- 48.Pek JW, Kai T. 2011. DEAD box RNA helicase Belle/DDX3 and the RNA interference pathway promote mitotic chromosome segregation. Proc Natl Acad Sci U S A 108:12007–12012. doi: 10.1073/pnas.1106245108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carrara G, Saraiva N, Parsons M, Byrne B, Prole DL, Taylor CW, Smith GL. 2015. Golgi anti-apoptotic proteins are highly conserved ion channels that affect apoptosis and cell migration. J Biol Chem 290:11785–11801. doi: 10.1074/jbc.M115.637306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bordenstein SR, Bordenstein SR. 2016. Eukaryotic association module in phage WO genomes from Wolbachia. Nat Commun 7:13155. doi: 10.1038/ncomms13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pannebakker BA, Loppin B, Elemans CP, Humblot L, Vavre F. 2007. Parasitic inhibition of cell death facilitates symbiosis. Proc Natl Acad Sci U S A 104:213–215. doi: 10.1073/pnas.0607845104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang Y, Bruni R, Kloss B, Assur Z, Kloppmann E, Rost B, Hendrickson WA, Liu Q. 2014. Structural basis for a pH-sensitive calcium leak across membranes. Science 344:1131–1135. doi: 10.1126/science.1252043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang GH, Niu LM, Ma GC, Xiao JH, Huang DW. 2014. Large proportion of genes in one cryptic WO prophage genome are actively and sex-specifically transcribed in a fig wasp species. BMC Genomics 15:893. doi: 10.1186/1471-2164-15-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanogo YO, Dobson SL. 2006. WO bacteriophage transcription in Wolbachia-infected Culex pipiens. Insect Biochem Mol Biol 36:80–85. doi: 10.1016/j.ibmb.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 55.Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N, Bhattacharyya A, Kapatral V, Kumar S, Posfai J, Vincze T, Ingram J, Moran L, Lapidus A, Omelchenko M, Kyrpides N, Ghedin E, Wang S, Goltsman E, Joukov V, Ostrovskaya O, Tsukerman K, Mazur M, Comb D, Koonin E, Slatko B. 2005. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol 3:e121. doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmitz-Esser S, Tischler P, Arnold R, Montanaro J, Wagner M, Rattei T, Horn M. 2010. The genome of the amoeba symbiont “Candidatus Amoebophilus asiaticus” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J Bacteriol 192:1045–1057. doi: 10.1128/JB.01379-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, Mahajan A, Tsai MD. 2006. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45:15168–15178. doi: 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]
- 58.Voth DE. 2011. ThANKs for the repeat: intracellular pathogens exploit a common eukaryotic domain. Cell Logist 1:128–132. doi: 10.4161/cl.1.4.18738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pan X, Lührmann A, Satoh A, Laskowski-Arce MA, Roy CR. 2008. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science 320:1651–1654. doi: 10.1126/science.1158160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.VieBrock L, Evans SM, Beyer AR, Larson CL, Beare PA, Ge H, Singh S, Rodino KG, Heinzen RA, Richards AL, Carlyon JA. 2014. Orientia tsutsugamushi ankyrin repeat-containing protein family members are type 1 secretion system substrates that traffic to the host cell endoplasmic reticulum. Front Cell Infect Microbiol 4:186. doi: 10.3389/fcimb.2014.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beyer AR, VieBrock L, Rodino KG, Miller DP, Tegels BK, Marconi RT, Carlyon JA. 2015. Orientia tsutsugamushi strain Ikeda ankyrin repeat-containing proteins recruit SCF1 ubiquitin ligase machinery via poxvirus-like F-box motifs. J Bacteriol 197:3097–3109. doi: 10.1128/JB.00276-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Min CK, Kwon YJ, Ha NY, Cho BA, Kim JM, Kwon EK, Kim YS, Choi MS, Kim IS, Cho NH. 2014. Multiple Orientia tsutsugamushi ankyrin repeat proteins interact with SCF1 ubiquitin ligase complex and eukaryotic elongation factor 1 alpha. PLoS One 9:e105652. doi: 10.1371/journal.pone.0105652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sinkins SP, Walker T, Lynd AR, Steven AR, Makepeace BL, Godfray HCJ, Parkhill J. 2005. Wolbachia variability and host effects on crossing type in Culex mosquitoes. Nature 436:257–260. doi: 10.1038/nature03629. [DOI] [PubMed] [Google Scholar]
- 64.Papafotiou G, Oehler S, Savakis C, Bourtzis K. 2011. Regulation of Wolbachia ankyrin domain encoding genes in Drosophila gonads. Res Microbiol 162:764–772. doi: 10.1016/j.resmic.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 65.Duron O, Boureux A, Echaubard P, Berthomieu A, Berticat C, Fort P, Weill M. 2007. Variability and expression of ankyrin domain genes in Wolbachia variants infecting the mosquito Culex pipiens. J Bacteriol 189:4442–4448. doi: 10.1128/JB.00142-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamada R, Iturbe-Ormaetxe I, Brownlie JC, O’Neill SL. 2011. Functional test of the influence of Wolbachia genes on cytoplasmic incompatibility expression in Drosophila melanogaster. Insect Mol Biol 20:75–85. doi: 10.1111/j.1365-2583.2010.01042.x. [DOI] [PubMed] [Google Scholar]
- 67.Schreiber A, Stengel F, Zhang Z, Enchev RI, Kong EH, Morris EP, Robinson CV, da Fonseca PC, Barford D. 2011. Structural basis for the subunit assembly of the anaphase-promoting complex. Nature 470:227–232. doi: 10.1038/nature09756. [DOI] [PubMed] [Google Scholar]
- 68.Cho NH, Kim HR, Lee JH, Kim SY, Kim J, Cha S, Kim SY, Darby AC, Fuxelius HH, Yin J, Kim JH, Kim J, Lee SJ, Koh YS, Jang WJ, Park KH, Andersson SGE, Choi MS, Kim IS. 2007. The Orientia tsutsugamushi genome reveals massive proliferation of conjugative type IV secretion system and host-cell interaction genes. Proc Natl Acad Sci U S A 104:7981–7986. doi: 10.1073/pnas.0611553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Domman D, Collingro A, Lagkouvardos I, Gehre L, Weinmaier T, Rattei T, Subtil A, Horn M. 2014. Massive expansion of ubiquitination-related gene families within the Chlamydiae. Mol Biol Evol 31:2890–2904. doi: 10.1093/molbev/msu227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spalding MD, Prigge ST. 2010. Lipoic acid metabolism in microbial pathogens. Microbiol Mol Biol Rev 74:200–228. doi: 10.1128/MMBR.00008-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lipke H, Fraenkel G. 1956. Insect nutrition. Annu Rev Entomol 1:17–44. doi: 10.1146/annurev.en.01.010156.000313. [DOI] [Google Scholar]
- 72.Nikoh N, Hosokawa T, Moriyama M, Oshima K, Hattori M, Fukatsu T. 2014. Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc Natl Acad Sci U S A 111:10257–10262. doi: 10.1073/pnas.1409284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gebiola M, Monti MM, Johnson RC, Woolley JB, Hunter MS, Giorgini M, Pedata PA. 2016. A revision of the Encarsia pergandiella species complex (Hymenoptera: Aphelinidae) shows cryptic diversity in parasitoids of whitefly pests. Syst Entomol 42:31–59. doi: 10.1111/syen.12187. [DOI] [Google Scholar]
- 74.Gebiola M, Kelly SE, Hammerstein P, Giorgini M, Hunter MS. 2016. ‘Darwin’s corollary’ and cytoplasmic incompatibility induced by Cardinium may contribute to speciation in Encarsia wasps (Hymenoptera: Aphelinidae). Evolution 70:2447–2458. doi: 10.1111/evo.13037. [DOI] [PubMed] [Google Scholar]
- 75.Shikuma NJ, Pilhofer M, Weiss GL, Hadfield MG, Jensen GJ, Newman DK. 2014. Marine tubeworm metamorphosis induced by arrays of bacterial phage tail-like structures. Science 343:529–533. doi: 10.1126/science.1246794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sarris PF, Ladoukakis ED, Panopoulos NJ, Scoulica EV. 2014. A phage tail-derived element with wide distribution among both prokaryotic domains: a comparative genomic and phylogenetic study. Genome Biol Evol 6:1739–1747. doi: 10.1093/gbe/evu136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zchori-Fein E, Perlman SJ, Kelly SE, Katzir N, Hunter MS. 2004. Characterization of a “Bacteroidetes” symbiont in Encarsia wasps (Hymenoptera: Aphelinidae): proposal of “Candidatus Cardinium hertigii.” Int J Syst Evol Microbiol 54:961–968. doi: 10.1099/ijs.0.02957-0. [DOI] [PubMed] [Google Scholar]
- 78.Bigliardi E, Sacchi L, Genchi M, Alma A, Pajoro M, Daffonchio D, Marzorati M, Avanzati AM. 2006. Ultrastructure of a novel Cardinium sp. symbiont in Scaphoideus titanus (Hemiptera: Cicadellidae). Tissue Cell 38:257–261. doi: 10.1016/j.tice.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 79.Zchori-Fein E, Gottlieb Y, Kelly SE, Brown JK, Wilson JM, Karr TL, Hunter MS. 2001. A newly discovered bacterium associated with parthenogenesis and a change in host selection behavior in parasitoid wasps. Proc Natl Acad Sci U S A 98:12555–12560. doi: 10.1073/pnas.221467498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perlman SJ, Kelly SE, Hunter MS. 2008. Population biology of cytoplasmic incompatibility: maintenance and spread of Cardinium symbionts in a parasitic wasp. Genetics 178:1003–1011. doi: 10.1534/genetics.107.083071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harris LR, Kelly SE, Hunter MS, Perlman SJ. 2010. Population dynamics and rapid spread of Cardinium, a bacterial endosymbiont causing cytoplasmic incompatibility in Encarsia pergandiella (Hymenoptera: Aphelinidae). Heredity 104:239–246. doi: 10.1038/hdy.2009.130. [DOI] [PubMed] [Google Scholar]
- 82.Katz AJ, Young SSY. 1975. Selection for high adult body-weight in Drosophila populations with different structures. Genetics 81:163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–147. In Stackebrandt E, Goodfellow M (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Chichester, United Kingdom. [Google Scholar]
- 84.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hilker R, Stadermann KB, Doppmeier D, Kalinowski J, Stoye J, Straube J, Winnebald J, Goesmann A. 2014. ReadXplorer—visualization and analysis of mapped sequences. Bioinformatics 30:2247–2254. doi: 10.1093/bioinformatics/btu205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.R Development Core Team 2008. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 88.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc B Stat Methodol 57:289–300. [Google Scholar]
- 90.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3—new capabilities and interfaces. Nucleic Acids Res 40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. 2012. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 93.Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, Sangrador-Vegas A, Scheremetjew M, Rato C, Yong SY, Bateman A, Punta M, Attwood TK, Sigrist CJ, Redaschi N, Rivoire C, Xenarios I, Kahn D, Guyot D, Bork P, Letunic I, Gough J, Oates M, Haft D, Huang H, Natale DA, Wu CH, Orengo C, Sillitoe I, Mi H, Thomas PD, Finn RD. 2015. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res 43:D213–D221. doi: 10.1093/nar/gku1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 96.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Potential novel cEper1 transcripts and putative antisense RNAs, calculated with ReadXplorer. The parameter “number of read starts” counts the number of reads and filters low-coverage regions with a number of reads below a certain threshold. The threshold used to detect novel transcripts was set to a 10-read minimum. “Coverage increase” indicates the coverage increase in percent from one position to the neighboring one (for the detection of novel transcripts, the threshold was set to a minimum of 50% increase from the start of the transcript to the position before). “Transcript stop” indicates the position at the chromosome where the putative transcript ends. Download TABLE S1, PDF file, 0.03 MB (32.3KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Classification of expression level of cEper1 genes in female and male Encarsia wasps. Download TABLE S2, XLSX file, 0.1 MB (70.3KB, xlsx) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Thirty cEper1 genes that were the most highly transcribed in male wasps are listed and sorted by fold change from their expression in female wasps. Genes meeting the DE criteria (P < 0.05 and multiple testing correction of FDR of <10%) are highlighted in bold. Download TABLE S3, PDF file, 0.03 MB (34.1KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Thirty cEper1 genes that were the most highly transcribed in female wasps are listed and sorted by the fold change from their expression in male wasps. Genes meeting the DE criteria (P < 0.05 and multiple testing correction of FDR of <10%) are highlighted in bold. Download TABLE S4, PDF file, 0.03 MB (35.4KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Gene expression of transposases in the Cardinium hertigii cEper1 genome. Download TABLE S5, PDF file, 0.1 MB (66.9KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Transcription of Cardinium hertigii cEper1 genes encoding proteins possibly involved in host-cell interactions. Download TABLE S6, PDF file, 0.04 MB (46.5KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Fold change differences of sex-specific expression patterns detected with RNA-Seq and RT-qPCRs are compared. (B) The correlation of fold change differences between the two methods used is shown. In panels A and B, only replicates that were used for both methods (three male and three female replicates) were included in the calculation. Download FIG S1, TIF file, 2.7 MB (2.7MB, tif) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Transcription of Cardinium hertigii cEper1 genes encoding proteins of the putative antifeeding prophage-like secretion system. Download TABLE S7, PDF file, 0.03 MB (36.7KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primer sequences, amplicon length, and annealing temperature of primers targeting cEper1 DE genes and two housekeeping genes (gyrB and groEL). Annealing temperatures were optimized with genomic DNA isolated from a culture of E. suzannae infected with Cardinium hertigii cEper1. Download TABLE S8, PDF file, 0.03 MB (32.1KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
MIQE guidelines for RT-qPCR assays. Download TABLE S9, PDF file, 0.1 MB (63.2KB, pdf) .
Copyright © 2017 Mann et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.